

Abstract
Objective:
Aberrant expression of ten-eleven translocation 1 (TET1) plays a critical role in tumor development and progression. We systematically summarized the latest research progress on the role and mechanisms of TET1 in cancer biology.
Data Sources:
Relevant articles published in English from 1980 to April 2016 were selected from the PubMed database. The terms “ten-eleven translocation 1,” “5mC,” “5hmC,” “microRNA,” “hypoxia,” and “embryonic stem cell” were used for the search.
Study Selection:
Articles focusing on the role and mechanism of TET1 in tumor were reviewed, including clinical and basic research articles.
Results:
TET proteins, the key enzymes converting 5-methylcytosine to 5-hydroxymethylcytosine, play vital roles in DNA demethylation regulation. Recent studies have shown that loss of TET1 is associated with tumorigenesis and can be used as a potential biomarker for cancer therapy, which indicates that TET1 serves as tumor suppressor gene. Moreover, besides its dioxygenase activity, TET1 could induce epithelial-mesenchymal transition and act as a coactivator to regulate gene transcription, such as developmental regulator in embryonic stem cells (ESCs) and hypoxia-responsive gene in cancer. The regulation of TET1 is also correlated with microRNA in a posttranscriptional modification process. Hence, it is complex but critical to comprehend the mechanisms of TET1 in the biology of ESCs and cancer.
Conclusions:
TET1 not only serves as a demethylation enzyme but also plays multiple roles during tumorigenesis and progression. More studies should be carried out to elucidate the exact mechanisms of TET1 and its associations with cancer before considering it as a therapeutic tool.
Keywords: 5-hydroxymethylcytosine, 5-methylcytosine, Embryonic Stem Cells, Hypoxia, MicroRNA, Ten-eleven Translocation Proteins
INTRODUCTION
Epigenetics refers to inherited changes in gene expression but not altering the DNA sequence. Aberrant DNA methylation is one of the critical events in epigenetics and also plays an important role during tumorigenesis.[1] DNA methylation at the fifth carbon (5-methylcytosine [5mC]) is a covalent biochemical modification, which mostly is located at the position of cytosine-phosphate-guanine (CpG) dinucleotides.[2] This process is catalyzed by DNA methyltransferases (DNMTs) in the presence of a methyl donor S-adenosylmethionine.[3] DNA methylation has a pivotal effect on gene expression stability and always occurs in repetitive genomic regions that are associated with gene transcriptional repression and genomic stability.[1,4] During tumorigenesis, the pattern of DNA methylation is broken which exhibits global hypomethylation and regional aberrant hypermethylation coexisting. These disturb genomic stability and lead to aberrant regulation of gene expression [Figure 1].[5]
Figure 1.
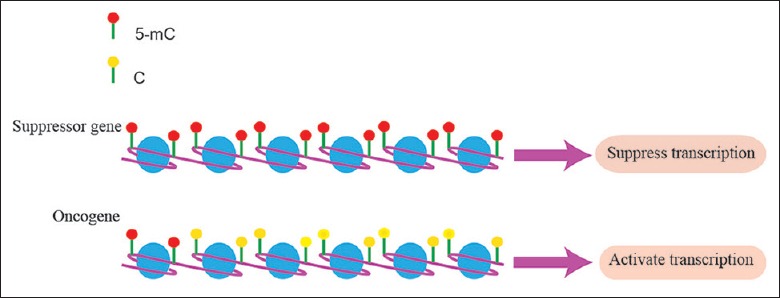
Aberrant DNA methylation regulates gene expression. During tumorigenesis, the pattern of DNA methylation is disturbed. The promoter of tumor suppressor genes always tends to be hypermethylated, resulting in the suppression of gene transcription. However, hypomethylation exists in the oncogene's promoter, which finally leads to aberrant gene expression.
It was not until 2009 that 5-hydroxymethylcytosine (5hmC) as an intermediate base in the process of DNA demethylation was unambiguously identified.[6] At the same time, it was reported that ten-eleven translocation (TET) proteins, the mammalian paralogs of trypanosome proteins J-binding protein 1 (JBP1) and JBP2 containing three members (TET1, TET2, and TET3), could convert 5mC to 5hmC depending on their dioxygenase activity.[7] These findings opened a new era of understanding mammalian epigenetics and proposed 5hmC as the “sixth” base, which is stable and ubiquitous in human genome, as well as 5mC.[8] In a variety of human malignancies, it is reported that the abnormal expression of TET proteins is always accompanied by a decrease in 5hmC.[9,10,11] In this review, we discussed the function of TET1 and its role in tumorigenesis.
STRUCTURE OF TEN-ELEVEN TRANSLOCATION PROTEINS
Among the three TET protein members, TET1 was the first to be identified as a mixed lineage leukemia (MLL) translocation partner gene in patients with acute myeloid leukemia harboring t(10;11)(q22;q23).[12] The TET protein family, as a subfamily of 2-oxoglutarate (2-OG) oxygenase catalyzers, includes a catalytic domain (CD) that harbors a double-stranded β-helix fold and a cysteine-rich domain. The dioxygenase activity of the CD is dependent on Fe2+ and 2-OG. At the amino terminal of TET1 and TET3, there is a CXXC-type zinc finger domain that can bind DNA at the CpG site.[13] However, different from that of TET1 and TET3, the CXXC domain of TET2 is encoded by a distinct gene, IDAX [Figure 2].[7,13]
Figure 2.
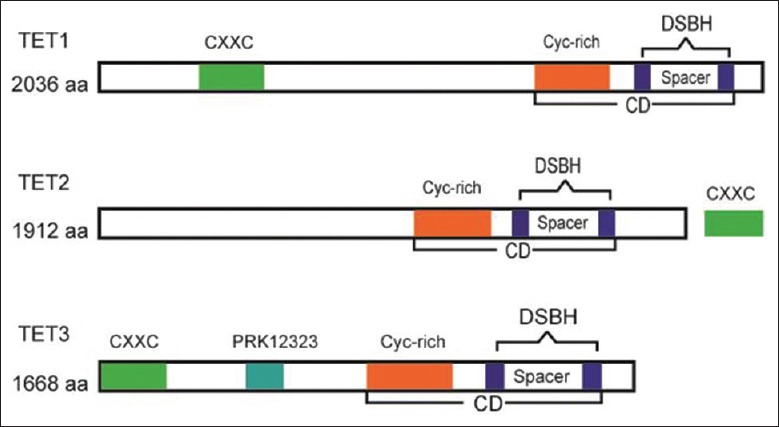
Structure of TET proteins. TETs include a CD that harbors a DSBH domain and a Cys-rich domain. At the amino terminal of TET1 and TET3, there is a CXXC-type zinc finger domain. The CXXC domain of TET2 is encoded by a distinct gene, IDAX. TET: Ten-eleven translocation; CD: Catalytic domain; DSBH: Double-stranded β-helix; Cys-rich: Cysteine-rich.
It is reported that the CD of TET1 plays a key role in DNA demethylation, but the function of the CXXC domain is underestimated.[14] Recent studies have reported that overexpression of TET1-CD causes global DNA demethylation while overexpression of TET1 full-length (TET1-FL) showed only mild DNA demethylation. These different results may be related to the preference of the CXXC domain, which targets nonmethylated CpG-dense regions to limit TET1 demethylation activities because of the rare nature of the substrate 5mC. Once the TET1 protein is overexpressed, the excess proteins may target the hypermethylation region and the less CpG-dense region to induce mild DNA demethylation.[15,16] Therefore, the demethylation activity of TET1 is associated with the genomic status of methylation. However, the exact site of TET1 during demethylation is still unclear.
TEN-ELEVEN TRANSLOCATION 1 AND DNA DEMETHYLATION
Ten-eleven translocation 1 and active demethylation
There are two pathways involved in DNA demethylation: one is replication-dependent demethylation, known as “passive demethylation,” and the other is TET enzyme-induced “active demethylation.” Through the function of dioxygenase activity, TETs could iteratively oxidize 5mC to generate 5hmC, and then 5-formylcytosine (5fC) and 5-carboxylcytosine (5CaC). On the other hand, thymine-DNA glycosylases can either decarboxylase 5fC or decarboxylase 5caC and replace it with an unmodified cytosine through DNA base excision repair enzymes, which finally complete a cycle of dynamic cytosine removal [Figure 3].[14,17,18]
Figure 3.
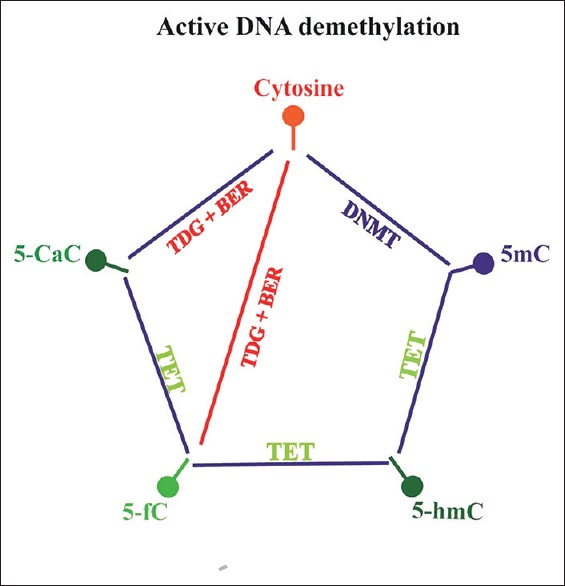
DNA methylation cycle. DNMTs catalyze cytosine to generate 5mC. TET proteins can iteratively oxidize 5mC to 5hmC, 5fC, and 5CaC. TDG can decarboxylase either 5fC or 5caC and replace it with an unmodified cytosine through BER, which finally completes a cycle of dynamic cytosine removal. DNMTs: DNA methyltransferases; TET: Ten-eleven translocation; 5mC: 5-methylcytosine; 5hmC: 5-hydroxymethylcytosine; 5fC: 5-formylcytosine; 5caC: 5-carboxylcytosine; TDG: Thymine DNA glycosylase; BER: Base excision repair. This figure was modified based on the work of Shen et al.
Ten-eleven translocation 1 and hypomethylation status
Apart from induced DNA active demethylation, TET1 could prevent methylation spreading through maintenance of DNA hypomethylation status.[19] It is well established that CpG dinucleotide islands (CGIs) are referred to a dense CpG content that generally lacks DNA methylation while the rest tend to be hypermethylated including the dispersed CpGs in gene coding regions and the CpG-rich repetitive heterochromatin regions.[20] Through the CXXC binding domain, TET1 prefers to bind hypomethylated, high CpG-dense regions (such as CGIs, gene promoters) to prevent DNMT activity and inhibit DNA methylation.[21] TET1 also binds to dispersed CpGs, which are always methylated in euchromatin and catalyze 5mC to 5hmC, limiting the accessibility of DNMTs or methyl-binding proteins (MBPs).[20] Indeed, the overexpression of TET1-FL increased 5hmC accumulation, especially at the edge of hypomethylated CGIs, while depletion of TET1 increased the spreading of DNA methylation from the edges of methylated regions into hypomethylated CGIs. Meanwhile, overexpression of TET1-FL reduced the DNA methylation levels in dispersed hypermethylated CGIs and showed mild DNA demethylation in genome.[19] These results illustrated that besides demethylation activity, TET1 could facilitate gene expression through maintenance of DNA hypomethylation status at their targets [Figure 4].
Figure 4.
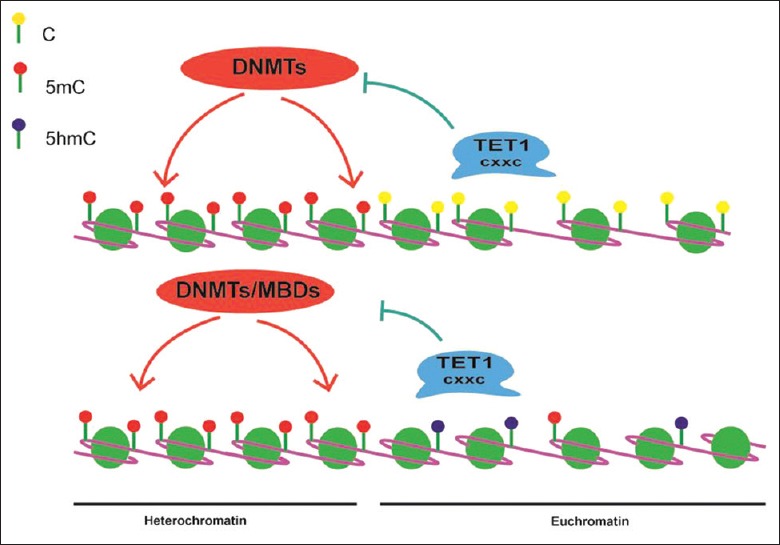
TET1 maintains hypomethylation status. Through the CXXC domain, TET1 binds to hypomethylated, high CpG-dense regions to prevent DNMT's activity and resist methylation. TET1 also binds to dispersed CpGs, limiting the accessibility of DNMTs or MBDs. MBDs: Methyl-binding proteins; TET1: Ten-eleven translocation 1; CpG: Cytosine-phosphate-guanine; DNMTs: DNA methyltransferases.
Distinct catalytic activity between ten-eleven translocation proteins
Although TET proteins act as dioxygenase to modulate DNA methylation status, each TET has a different function in building genome-wide 5mC and 5hmC pattern. In a model of pluripotent embryonic carcinoma cell, depletion of TET1 contributed to widespread 5hmC reduction while TET2 and TET3 depletion decreased 5hmC at a subset of TET1 targets. These results showed the redundant roles of TET proteins in modulating the methylation pattern.[22]
There was also a tendency to target the loci of TET1, which has prominent affinity for high CpG density promoters, while TET2, especially at low CpG density promoters, tended to maintain 5hmC.[15,23] It is evident that 5hmC was enriched in distal regulatory regions and gene bodies, such as enhancers, exons, and introns.[24,25] When depletion of TET2 or TET3 took place unexpectedly, there was an accumulation of 5hmC, particularly in introns of highly expressed genes.[23] One explanation for this is that in the demethylation cascade reaction, TET2 and TET3 may be mainly involved in the formation of downstream cytosine intermediates (5fC and 5CaC). Once the function of TET2 or TET3 is disrupted, 5hmC accumulated in many regions of the genome. Given that 5hmC mainly accumulates in introns, these results might indicate that 5hmC removal from introns is partly due to the demethylation function of TET2 and TET3.[13,15,23]
Although the TET protein family is essential for genomic methylation pattern, the exact function of each TET protein in regulating the 5hmC level is still unclear. It is also ambiguous how each TET protein recognizes its target region and specifically binds to the DNA sequence. The impact of this process on gene splicing fidelity and the rate of gene transcription are still elusive.
5-HYDROXYMETHYLCYTOSINE IN HUMAN SOLID TUMOR
Similar to 5mC, a depletion of genomic 5hmC has been widely reported in cancerous tissues of the prostate, breast, and colon compared with normal tissues.[16] Loss of 5hmC content has been further confirmed in a broad spectrum of solid tumors, such as in cancerous tissues of colon/rectum,[8,26] stomach,[26] parathyroid,[27] liver,[9,28,29] lung, pancreas, breast, and prostate.[9,29] In solid cancers, the low level of 5hmC is associated with poor prognostic outcomes and the reduction may be one of the causes for the high cumulative recurrence in hepatocellular cancer.[28,30] Many studies have reported that the reduction of 5hmC in solid tumors is mainly due to the loss of TET1 expression.[26,31] However, downregulation of TET1 is not always accompanied by depleted 5hmC content. It is likely that the level of 5hmC is affected by multiple mechanisms, including suppression of TET1 and overexpression of DNMT.[32]
5hmC not only serves as an intermediate base during DNA demethylation but also plays a potential role in epigenetic regulation. Previous studies had shown that 5hmC could interact with proteins such as the methyl-CpG-binding domain protein 3, Uhrf1, and MeCP2 and finally influence the chromatin structure and gene expression.[33] Kafer et al. had reported that 5hmC could localize to DNA damage sites and promote genome stability.[34] This finding may explain the relationship between low 5hmC levels and cancer. However, little is known about the mechanism of 5hmC recognition and the reader of 5hmC.
REGULATION OF TEN-ELEVEN TRANSLOCATION 1 BY MICRORNA
MicroRNAs (miRNAs) are small noncoding RNAs, only 20–22 nucleotide long, that regulate gene expression at the posttranscriptional level and have important roles in tumor formation and progression.[35] Zhang et al. had reported that TET1 was the direct target of miR-29a through 3’-untranslated region.[36] Further studies have confirmed that the miR-29 family regulated active DNA demethylation through direct interaction with TET1 in diverse solid tumors.[37,38] Lin et al. had shown that a negative feedback of miR-29-TET1 was involved in hepatocellular cancer development.[39] Moreover, miR-29b could be induced by mitogen-activated protein kinase (MAPK)-driven ETS1 expression and might lead to TET1 downregulation, which results in the change of epigenetic modification.[40]
In addition to the miR-29 family, there are other miRNAs involved in TET1 regulation. MiR-26a can target TET1 to inhibit its expression, which is accompanied by a change of 5hmC levels.[41] TET1 was also found to be the target of miR-767 whose aberrant activation contributed to decreased TET1 activity during tumorigenesis.[42] In hepatocellular carcinoma, miR-494 was upregulated and repressed TET1 expression, which led to tumor vascular invasion.[43] Furthermore, TET1 was the direct target of miR-520b. In hepatocellular cancer, miR-520b repressed cell proliferation by decreasing the expression of TET1.[44] However, in this process, TET1 may act as an oncogene, which is different from the observation of previous studies.
It is not just that TET1 could be regulated by miRNAs but TET1 could also regulate the miRNA level through its demethylation activity.[39,45] Hence, it is complex and critical to comprehend the regulation mechanisms that orchestrate transcriptional regulation and posttranscriptional regulation during tumor development and progression.
MULTIPLE FUNCTIONS OF TEN-ELEVEN TRANSLOCATION 1
Ten-eleven translocation 1 in embryonic stem cells
During early embryonic development, genome-wide DNA demethylation has been observed, as well as hypomethylated housekeeping and developmental gene promoters.[22,46] Many works have proven that TET1 is highly expressed in embryonic stem cells (ESCs) and responsible for 5hmC maintenance.[47,48,49] TET1 is also the key enzyme to maintain ESC and induce pluripotent stem cells, which shows its essential role in stemness circuits.[50] TET1 depletion would repress the expression of pluripotency factors Oct4, Nanog, and Sox2 through its demethylation activity and finally lead to ESCs differentiation.[49] Furthermore, Neri et al. showed that TET1 was also regulated by the factors Oct3/4, Nanog, and Myc.[48] Thus, the integration of TET1 into the pluripotency transcriptional network may help understand the molecular regulation of ESC development.
Besides promoting gene expression, TET1 also contributes to transcriptional repression in ESCs. In mouse ESCs, TET1 could repress the expression of developmental regulators and somatic lineage differentiation genes, which are polycomb-targeted genes, through the recruitment of Polycomb repressive complex 2 (PRC2).[15] Genome-wide analyses in ESCs have shown that 5hmC is enriched at regulatory elements such as gene bodies, enhancers, and promoters.[51]5hmC was especially located at the start sites of genes whose promoters are marked by H3K27me3 and H3K4me3.[47] H3K27me3 is the key modification in PRC2-regulated developmental regulators, which are associated with low expression. The hypomethylation status made sure that TET1 could interact with PRC2 and form a PRC2 complex to repress gene expression.[52] Further study showed that TET1 was downregulated by PRC2 through H3K27me3 histone mark.[48] However, the functional interaction between TET1 and PRC2 may be indirect because of the failure to coimmunoprecipitate TET1 with PRC2.[15,24] Thus, the mechanistic interplay between Tet1 and PRC2 is still unclear. Moreover, coimmunoprecipitation uncovered that TET1 also bound with Sin3a at nonhydroxymethylated transcription start sites to form a repressive complex.[52] These studies suggest that TET1 contributes to epigenetic plasticity throughout development and differentiation.
Dual functions of ten-eleven translocation 1 during tumorigenesis
Ten-eleven translocation 1 suppresses tumorigenesis
Decreased expression of TET1 has been reported in multiple malignancies.[26,53,54] Although depletions, frame shifts, and nonsense/missense somatic mutations of TET2 were found in myelodysplastic syndromes, myeloproliferative neoplasms, and leukemia,[55,56,57] loss-of-function mutations of TET1 are rarely reported in hematological system diseases as well as in solid tumors.[7,46] Besides mutations, decreased expression of TET1 had been reported in a variety of solid cancers, which was accompanied by a reduction of global 5hmC and accumulation of aberrant DNA methylation.[9,10,11,30,53,58] These suggested that the change of TET1 in tumor tissues made a great contribution to carcinogenesis and tumor progression.
Recent studies have shown that TET1 was significantly decreased in colorectal tumor tissues compared with normal tissues.[53] Neri et al. reported that TET1 silencing was correlated with the proliferation of cancer cells and promoted the growth of tumor xenografts even at later stages. This process was due to the dioxygenase activity of TET1. Through binding to the DKK gene inhibitors of the WNT signaling, TET1 could maintain the hypomethylation of target genes. Loss of TET1 led to the inactivation of the inhibitors of the WNT pathway by hypermethylation, resulting in a cascade activation of the WNT pathway and finally increased cellular proliferation.[59]
Ichimura et al. reported that TET1 was silenced by DNA methylation, which was an early event in CpG island methylator phenotype (CIMP) carcinogenesis and more frequent with CIMP-positive (CIMP-P) than CIMP-negative patients. CIMP-P is a subgroup of colorectal cancer (CRC) that shows multiple methylated CpG islands.[54,60] Therefore, CIMP is a critical molecular feature in the regulation of specific gene expression, particularly transcription factor, and also presents characteristic manifestations during tumor initiation and progression.[61,62] In CIMP-mediated tumorigenesis, TET1 was inactivated through hypermethylation even in precancerous tissue, which was associated with a large number of methylated genes and less metastasis.[54] Given that CIMP could be used as a biomarker in response to therapy and treatment outcomes, TET1 methylation may be used as a novel target in cancer treatment.[62]
Furthermore, Wu et al. showed that suppression of TET1 expression was essential in KRAS-induced tumor transformation. KRAS protein is a member of the RAS proteins family, which is indispensable for cell proliferation, differentiation, and survival.[63,64] Hyperactive RAS will drive cellular transformation and promote cell proliferation through the activation of RAF–MEK–ERK and phosphatidylinositol-3-kinase–AKT cascades.[65] Studies have shown that ERK pathway activation would silence the tumor suppressor genes by hypermethylating their promoters through repressed TET1 expression[63] and elevated DNMT transcription.[64,66] These processes finally led to malignant transformation and promoted cell proliferation. On restoration of the TET1 level in KRAS-transformed cells, tumor suppressor genes were reactivated and colony formation was inhibited.[63] Therefore, TET1-dependent demethylation plays an important role in KRAS-mediated transformation.
In addition to promoting cell proliferation, decreased expression of TET1 also facilitated cell invasion and cancer metastasis in certain tumors through its dioxygenase and DNA-binding activities.[9,11,58,67] Hsu et al. reported that TET1 inhibited tumor invasion in prostate and breast cancers through maintenance of the level of tissue inhibitors of metalloproteinase (TIMP) family proteins 2 and 3. TIMPs are the endogenous regulators of the matrix metalloproteinase (MMP) proteins.[11,68] MMPs are key players in the regulation of cell invasion.[69] TIMP proteins are the inhibitors of MMPs through interacting with MMPs or decreasing the level of MMPs and finally alleviating cell invasion.[11,68,69] Through binding to CpG-rich regions of TIMP2/3, TET1 facilitated the expression of TIMP2/3 by its dioxygenase activity through repression of DNA methylation and suppressed tumor progression and invasion.[11]
TET1 regulated the ability of tumor invasion also through the high mobility group AT-hook2 (HMGA2)/TET1/homeobox A9 (HOXA9) signaling pathway and was associated with poor prognosis in breast cancer.[10,11] HMGA2 is a chromatin remodeling factor that is highly expressed in ESCs and can regulate the expression of transcriptional enhancers.[70] Compared with normal somatic cells, HMGA2 is overexpressed in most malignant tumors and promotes tumor invasion and metastasis.[71] In gene expression array analysis, knocking down HMGA2 increased TET1 expression in breast cancer. Through its demethylation activity, TET1 bound to the promoter of HOXA genes, inhibited their methylation, and then stimulated their expressions, which finally led to diminished growth and migration of cancer cells in mouse xenografts. Furthermore, TET1 could also demethylate itself by binding to its own promoter region and then enhance its expression level. Previous studies had shown that HMGA2 was an independent marker of poor prognosis in triple-negative breast cancers.[72] Further analysis of survival showed that patients with HMGA2 low expression and TET1/HOXA9/7 high expression had a fair overall survival.[10] Further study showed that TET1 could inhibit migration and invasion in lung carcinoma.[67]
All the above results illustrate that TET1 could act as a tumor suppressor gene through its demethylation activity or maintenance of its target genes’ hypomethylated status. These data also indicate that TET1 might have different functions in different organs.
Ten-eleven translocation 1 promotes tumorigenesis
Although many studies have reported that TET1 played an important role in the suppression of malignant transformation, some studies have shown its oncogenic effects on the development of tumors. Huang et al. reported that TET1 played an essential oncogenic role in MLL-rearranged leukemia.[73] In MLL-rearranged leukemia, MLL-fusion proteins target TET1, which was significantly upregulated, leading to the accumulation of global 5hmC level. In addition, in vitro and in vivo functional studies showed that TET1 coregulated with MLL fusion proteins to prompt oncogenic target gene expression including HOXA9, myeloid ecotropic viral integration 1/pre-B-cell leukemia homeobox3 genes.[73] Hence, TET1 plays an indispensable oncogenic role in MLL-rearranged leukemia.
Not only in hematological malignancies but also in solid cancers, TET1 could drive tumor malignancy depending on or independent of its demethylation activity. Indeed, the length of CD is only a small part of the whole length of TET1. There may be other functions of TET1 that regulate the growth and development of cells.[74] Recent advances have shown that TET1 is involved in the change of hypoxia reaction and participates in tumor progression.[75] Hypoxia microenvironment is a pathophysiologic outcome of tumor progression because of inadequate supply of oxygen. Tumor hypoxia would lead to tumor proliferation, change of cell behavior, metastasis, and epithelial-mesenchymal transition (EMT).[76] Hence, tumor hypoxia is a negative prognostic factor for tumor progression and hypoxia-activated prodrugs may provide a new way to target tumor therapy.[77]
When exposed to hypoxia microenvironment, the conversion of 5mC into 5hmC was deregulated by the aberrant TET1, resulting in breast tumor-initiating cell (BTIC) properties. As a response of hypoxia, TET1 was upregulated and increased genomic DNA hydroxymethylation drove tumor necrosis factor-alpha (TNF-α) expression. The expression of TNF-α would activate its downstream p38-MAPK effector pathway, which was an essential pathway for building BTIC properties and EMT promotion.[78] During tumor growth, the hypoxic microenvironment is created, which induces malignant progression such as angiogenesis, aggressive tumor invasion, and proliferation. Hypoxia-inducible factors (HIFs), which are response to hypoxia, are central molecules mediated by transcriptional regulation.[76] A growing number of studies have demonstrated that hypoxia increased genomic 5hmC levels, particularly in hypoxia-responsive genes.[76,79] Tsai et al. showed that TET1 was regulated by hypoxia/HIF-2, which then regulated the expression of hypoxia-responsive genes in CRC. On depletion of TET1, hypoxia-induced EMT was mitigated. Using RNA-seq and 5hmC-seq, the insulin-induced gene 1 (INSIG1), one of the genes involved in cholesterol metabolic process, was identified as a TET1 target gene, which contributed to hypoxia-induced EMT. Independent of its enzymatic activity, TET1 could act as a coactivator to enhance INSIG1 transcription activity by interacting with HIF-2α through the CXXC domain. Overexpression of the TET1 catalytically inactive mutant, hypoxia-induced EMT was rescued in TET1 knockdown cells, which further confirmed that TET1 could serve as a transcription coactivator in addition to its demethylation function.[80] Hence, TET1 contributes to hypoxia-inducible tumorigenesis and regulates the expression of the target genes. Further investigation may focus on the function of TET1 in a hypoxia microenvironment and its role in EMT in malignancy. It is necessary to comprehensively evaluate the correlation between TET1 and cancer.
Altogether, these studies have shown that TET1 regulates gene expression in a multilayered manner, which includes acting as transcription factor and regulating the 5hmC level. Therefore, the exact role and the underlying molecular mechanism of TET1 during cancer development and progression are complicated and need to be further investigated in more detail.
CONCLUSIONS
Since 2009, the TET protein family has been recognized as a demethylation enzyme, whose expression level and regulatory mechanism have been widely discussed. Altered expression levels of TET1 and 5hmC have been found in a broad range of cancers. Considering the multiple functions of TET1, its exact role is still unclear and needs more investigation. Furthermore, the ways of deregulation of TET1 in different cancers are complicated and we still need more evidence before TET1 can be used as a therapeutic tool to treat cancers.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Li-Min Chen
REFERENCES
- 1.Jaenisch R, Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 2.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412–7. doi: 10.1073/pnas.0510310103. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saito Y, Kanai Y, Nakagawa T, Sakamoto M, Saito H, Ishii H, et al. Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int J Cancer. 2003;105:527–32. doi: 10.1002/ijc.11127. doi: 10.1002/ijc.11127. [DOI] [PubMed] [Google Scholar]
- 4.Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res. 1988;48:1159–61. [PubMed] [Google Scholar]
- 5.Dawson MA, Kouzarides T. Cancer epigenetics: From mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30. doi: 10.1126/science.1169786. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–7. doi: 10.1182/blood-2009-03-210039. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang LT, Zhang LJ, Zhang JJ, Ye XX, Xie AM, Chen LY, et al. Quantification of the sixth DNA base 5-hydroxymethylcytosine in colorectal cancer tissue and C-26 cell line. Bioanalysis. 2013;5:839–45. doi: 10.4155/bio.13.28. doi: 10.4155/bio.13.28. [DOI] [PubMed] [Google Scholar]
- 9.Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32:663–9. doi: 10.1038/onc.2012.67. doi: 10.1038/onc.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun M, Song CX, Huang H, Frankenberger CA, Sankarasharma D, Gomes S, et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci U S A. 2013;110:9920–5. doi: 10.1073/pnas.1305172110. doi: 10.1073/pnas.1305172110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu CH, Peng KL, Kang ML, Chen YR, Yang YC, Tsai CH, et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012;2:568–79. doi: 10.1016/j.celrep.2012.08.030. doi: 10.1016/j.celrep.2012.08.030. [DOI] [PubMed] [Google Scholar]
- 12.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003;17:637–41. doi: 10.1038/sj.leu.2402834. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 13.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–9. doi: 10.1038/nature12750. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Zhang X, Clark E, Mulcahey M, Huang S, Shi YG. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010;20:1390–3. doi: 10.1038/cr.2010.156. doi: 10.1038/cr.2010.156. [DOI] [PubMed] [Google Scholar]
- 15.Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–93. doi: 10.1038/nature09934. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haffner MC, Chaux A, Meeker AK, Esopi DM, Gerber J, Pellakuru LG, et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget. 2011;2:627–37. doi: 10.18632/oncotarget.316. doi: 10.18632/oncotarget.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. doi: 10.1126/science.1210597. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen L, Wu H, Diep D, Yamaguchi S, D’Alessio AC, Fung HL, et al. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153:692–706. doi: 10.1016/j.cell.2013.04.002. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin C, Lu Y, Jelinek J, Liang S, Estecio MR, Barton MC, et al. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 2014;42:6956–71. doi: 10.1093/nar/gku372. doi: 10.1093/nar/gku372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451–64. doi: 10.1016/j.molcel.2011.04.005. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Illingworth RS, Bird AP. CpG islands – ‘A rough guide’. FEBS Lett. 2009;583:1713–20. doi: 10.1016/j.febslet.2009.04.012. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Kang J, Lienhard M, Pastor WA, Chawla A, Novotny M, Tsagaratou A, et al. Simultaneous deletion of the methylcytosine oxidases Tet1 and Tet3 increases transcriptome variability in early embryogenesis. Proc Natl Acad Sci U S A. 2015;112:E4236–45. doi: 10.1073/pnas.1510510112. doi: 10.1073/pnas.1510510112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Putiri EL, Tiedemann RL, Thompson JJ, Liu C, Ho T, Choi JH, et al. Distinct and overlapping control of 5-methylcytosine and 5-hydroxymethylcytosine by the TET proteins in human cancer cells. Genome Biol. 2014;15:R81. doi: 10.1186/gb-2014-15-6-r81. doi: 10.1186/gb-2014-15-6-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–8. doi: 10.1038/nature10066. doi: 10.1038/naturne10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kudo Y, Tateishi K, Yamamoto K, Yamamoto S, Asaoka Y, Ijichi H, et al. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012;103:670–6. doi: 10.1111/j.1349-7006.2012.02213.x. doi: 10.1111/j.1349-7006.2012.02213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barazeghi E, Gill AJ, Sidhu S, Norlén O, Dina R, Palazzo FF, et al. 5-hydroxymethylcytosine discriminates between parathyroid adenoma and carcinoma. Clin Epigenetics. 2016;8:31. doi: 10.1186/s13148-016-0197-2. doi: 10.1186/s13148-016-0197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen ML, Shen F, Huang W, Qi JH, Wang Y, Feng YQ, et al. Quantification of 5-methylcytosine and 5-hydroxymethylcytosine in genomic DNA from hepatocellular carcinoma tissues by capillary hydrophilic-interaction liquid chromatography/quadrupole TOF mass spectrometry. Clin Chem. 2013;59:824–32. doi: 10.1373/clinchem.2012.193938. doi: 10.1373/clinchem.2012.193938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin SG, Jiang Y, Qiu R, Rauch TA, Wang Y, Schackert G, et al. 5-hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 2011;71:7360–5. doi: 10.1158/0008-5472.CAN-11-2023. doi: 10.1158/0008-5472.CAN-11-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsai KW, Li GC, Chen CH, Yeh MH, Huang JS, Tseng HH, et al. Reduction of global 5-hydroxymethylcytosine is a poor prognostic factor in breast cancer patients, especially for an ER/PR-negative subtype. Breast Cancer Res Treat. 2015;153:219–34. doi: 10.1007/s10549-015-3525-x. doi: 10.1007/s10549-015-3525-x. [DOI] [PubMed] [Google Scholar]
- 31.Du C, Kurabe N, Matsushima Y, Suzuki M, Kahyo T, Ohnishi I, et al. Robust quantitative assessments of cytosine modifications and changes in the expressions of related enzymes in gastric cancer. Gastric Cancer. 2015;18:516–25. doi: 10.1007/s10120-014-0409-4. doi: 10.1007/s10120-014-0409-4. [DOI] [PubMed] [Google Scholar]
- 32.Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, et al. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: Coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291–8. doi: 10.1093/nar/27.11.2291. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–30. doi: 10.1016/j.cell.2012.11.022. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kafer GR, Li X, Horii T, Suetake I, Tajima S, Hatada I, et al. 5-hydroxymethylcytosine marks sites of DNA damage and promotes genome stability. Cell Rep. 2016;14:1283–92. doi: 10.1016/j.celrep.2016.01.035. doi: 10.1016/j.celrep.2016.01.035. [DOI] [PubMed] [Google Scholar]
- 35.Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11:849–64. doi: 10.1038/nrc3166. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang P, Huang B, Xu X, Sessa WC. Ten-eleven translocation (Tet) and thymine DNA glycosylase (TDG), components of the demethylation pathway, are direct targets of miRNA-29a. Biochem Biophys Res Commun. 2013;437:368–73. doi: 10.1016/j.bbrc.2013.06.082. doi: 10.1016/j.bbrc.2013.06.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobsen A, Silber J, Harinath G, Huse JT, Schultz N, Sander C. Analysis of microRNA-target interactions across diverse cancer types. Nat Struct Mol Biol. 2013;20:1325–32. doi: 10.1038/nsmb.2678. doi: 10.1038/nsmb.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shinden Y, Iguchi T, Akiyoshi S, Ueo H, Ueda M, Hirata H, et al. MiR-29b is an indicator of prognosis in breast cancer patients. Mol Clin Oncol. 2015;3:919–23. doi: 10.3892/mco.2015.565. doi: 10.3892/mco.2015.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin LL, Wang W, Hu Z, Wang LW, Chang J, Qian H. Erratum to: Negative feedback of miR-29 family TET1 involves in hepatocellular cancer. Med Oncol. 2015;32:39. doi: 10.1007/s12032-014-0437-2. doi: 10.1007/s12032-014-0437-2. [DOI] [PubMed] [Google Scholar]
- 40.Taylor MA, Wappett M, Delpuech O, Brown H, Chresta CM. Enhanced MAPK signaling drives ETS1-mediated induction of miR-29b leading to downregulation of TET1 and changes in epigenetic modifications in a subset of lung SCC. Oncogene. 2016 doi: 10.1038/onc.2015.499. doi: 10.1038/onc.2015.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu X, Jin L, Wang X, Luo A, Hu J, Zheng X, et al. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc Natl Acad Sci U S A. 2013;110:17892–7. doi: 10.1073/pnas.1317397110. doi: 10.1073/pnas.1317397110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loriot A, Van Tongelen A, Blanco J, Klaessens S, Cannuyer J, van Baren N, et al. A novel cancer-germline transcript carrying pro-metastatic miR-105 and TET-targeting miR-767 induced by DNA hypomethylation in tumors. Epigenetics. 2014;9:1163–71. doi: 10.4161/epi.29628. doi: 10.4161/epi.29628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chuang KH, Whitney-Miller CL, Chu CY, Zhou Z, Dokus MK, Schmit S, et al. MicroRNA-494 is a master epigenetic regulator of multiple invasion-suppressor microRNAs by targeting ten eleven translocation 1 in invasive human hepatocellular carcinoma tumors. Hepatology. 2015;62:466–80. doi: 10.1002/hep.27816. doi: 10.1002/hep.27816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W, Lu Z, Gao Y, Ye L, Song T, Zhang X. MiR-520b suppresses proliferation of hepatoma cells through targeting ten-eleven translocation 1 (TET1) mRNA. Biochem Biophys Res Commun. 2015;460:793–8. doi: 10.1016/j.bbrc.2015.03.108. doi: 10.1016/j.bbrc.2015.03.108. [DOI] [PubMed] [Google Scholar]
- 45.Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C, et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. 2013;154:311–24. doi: 10.1016/j.cell.2013.06.026. doi: 10.1016/j.cell.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rawluszko-Wieczorek AA, Siera A, Jagodzinski PP. TET proteins in cancer: Current ‘state of the art’. Crit Rev Oncol Hematol. 2015;96:425–36. doi: 10.1016/j.critrevonc.2015.07.008. doi: 10.1016/j.critrevonc.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 47.Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–7. doi: 10.1038/nature10102. doi: 10.1038/natur10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neri F, Incarnato D, Krepelova A, Dettori D, Rapelli S, Maldotti M, et al. TET1 is controlled by pluripotency-associated factors in ESCs and downmodulated by PRC2 in differentiated cells and tissues. Nucleic Acids Res. 2015;43:6814–26. doi: 10.1093/nar/gkv392. doi: 10.1093/nar/gkv392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33. doi: 10.1038/nature09303. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell. 2011;9:166–75. doi: 10.1016/j.stem.2011.07.010. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu H, D’Alessio AC, Ito S, Wang Z, Cui K, Zhao K, et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–84. doi: 10.1101/gad.2036011. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neri F, Incarnato D, Krepelova A, Rapelli S, Pagnani A, Zecchina R, et al. Genome-wide analysis identifies a functional association of Tet1 and polycomb repressive complex2 in mouse embryonic stem cells. Genome Biol. 2013;14:R91. doi: 10.1186/gb-2013-14-8-r91. doi: 10.1186/gb-2013-14-8-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rawluszko-Wieczorek AA, Siera A, Horbacka K, Horst N, Krokowicz P, Jagodzinski PP. Clinical significance of DNA methylation mRNA levels of TET family members in colorectal cancer. J Cancer Res Clin Oncol. 2015;141:1379–92. doi: 10.1007/s00432-014-1901-2. doi: 10.1007/s00432-014-1901-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ichimura N, Shinjo K, An B, Shimizu Y, Yamao K, Ohka F, et al. Aberrant TET1 methylation closely associated with CpG island methylator phenotype in colorectal cancer. Cancer Prev Res (Phila) 2015;8:702–11. doi: 10.1158/1940-6207.CAPR-14-0306. doi: 10.1158/1940-6207.CAPR-14-0306. [DOI] [PubMed] [Google Scholar]
- 55.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301. doi: 10.1056/NEJMoa0810069. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 56.Kameda T, Shide K, Yamaji T, Kamiunten A, Sekine M, Hidaka T, et al. Gene expression profiling of loss of TET2 and/or JAK2V617F mutant hematopoietic stem cells from mouse models of myeloproliferative neoplasms. Genom Data. 2015;4:102–8. doi: 10.1016/j.gdata.2015.04.002. doi: 10.1016/j.gdata.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shide K, Kameda T, Shimoda H, Yamaji T, Abe H, Kamiunten A, et al. TET2 is essential for survival and hematopoietic stem cell homeostasis. Leukemia. 2012;26:2216–23. doi: 10.1038/leu.2012.94. doi: 10.1038/leu.2012.94. [DOI] [PubMed] [Google Scholar]
- 58.Yang L, Yu SJ, Hong Q, Yang Y, Shao ZM. Reduced expression of TET1, TET2, TET3 and TDG mRNAs are associated with poor prognosis of patients with early breast cancer. PLoS One. 2015;10:e0133896. doi: 10.1371/journal.pone.0133896. doi: 10.1371/journal.pone.0133896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neri F, Dettori D, Incarnato D, Krepelova A, Rapelli S, Maldotti M, et al. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene. 2015;34:4168–76. doi: 10.1038/onc.2014.356. doi: 10.1038/onc.2014.356. [DOI] [PubMed] [Google Scholar]
- 60.van Rijnsoever M, Grieu F, Elsaleh H, Joseph D, Iacopetta B. Characterisation of colorectal cancers showing hypermethylation at multiple CpG islands. Gut. 2002;51:797–802. doi: 10.1136/gut.51.6.797. doi: 10.1136/gut.51.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–6. doi: 10.1073/pnas.96.15.8681. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williamson JS, Harris DA, Beynon J, Jenkins GJ. Review of the development of DNA methylation as a marker of response to neoadjuvant therapy and outcomes in rectal cancer. Clin Epigenetics. 2015;7:70. doi: 10.1186/s13148-015-0111-3. doi: 10.1186/s13148-015-0111-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu BK, Brenner C. Suppression of TET1-dependent DNA demethylation is essential for KRAS-mediated transformation. Cell Rep. 2014;9:1827–40. doi: 10.1016/j.celrep.2014.10.063. doi: 10.1016/j.celrep.2014.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang HC, Cho CY, Hung WC. Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA methyltransferase 3b-induced promoter methylation. Cancer Res. 2006;66:8413–20. doi: 10.1158/0008-5472.CAN-06-0685. doi: 10.1158/0008-5472.CAN-06-0685. [DOI] [PubMed] [Google Scholar]
- 65.Greig RG, Koestler TP, Trainer DL, Corwin SP, Miles L, Kline T, et al. Tumorigenic and metastatic properties of “normal” and ras-transfected NIH/3T3 cells. Proc Natl Acad Sci U S A. 1985;82:3698–701. doi: 10.1073/pnas.82.11.3698. doi: 10.1073/pnas.82.11.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bakin AV, Curran T. Role of DNA 5-methylcytosine transferase in cell transformation by fos. Science. 1999;283:387–90. doi: 10.1126/science.283.5400.387. doi: 10.1126/science.283.5400.387. [DOI] [PubMed] [Google Scholar]
- 67.Park SJ, Lee BR, Kim HS, Ji YR, Sung YH, ShikChoi K, et al. Inhibition of migration and invasion by Tet-1 overexpression in human lung carcinoma H460 cells. Oncol Res. 2016;23:89–98. doi: 10.3727/096504015X14496932933539. doi: 10.3727/096504015X14496932933539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bourboulia D, Stetler-Stevenson WG. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin Cancer Biol. 2010;20:161–8. doi: 10.1016/j.semcancer.2010.05.002. doi: 10.1016/j.semcancer.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murphy G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011;12:233. doi: 10.1186/gb-2011-12-11-233. doi: 10.1186/gb-2011-12-11-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ozturk N, Singh I, Mehta A, Braun T, Barreto G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front Cell Dev Biol. 2014;2:5. doi: 10.3389/fcell.2014.00005. doi: 10.3389/fcell.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- 72.Wend P, Runke S, Wend K, Anchondo B, Yesayan M, Jardon M, et al. WNT10B/ß-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol Med. 2013;5:264–79. doi: 10.1002/emmm.201201320. doi: 10.1002/emmm.201201320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang H, Jiang X, Li Z, Li Y, Song CX, He C, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci U S A. 2013;110:11994–9. doi: 10.1073/pnas.1310656110. doi: 10.1073/pnas.1310656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, Cheng AW, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24:310–23. doi: 10.1016/j.devcel.2012.12.015. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kao SH, Wu KJ, Lee WH. Hypoxia, epithelial-mesenchymal transition, and TET-mediated epigenetic changes. J Clin Med. 2016;5(pii):E24. doi: 10.3390/jcm5020024. doi: 10.3390/jcm5020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nagaraju GP, Bramhachari PV, Raghu G, El-Rayes BF. Hypoxia inducible factor-1a: Its role in colorectal carcinogenesis and metastasis. Cancer Lett. 2015;366:11–8. doi: 10.1016/j.canlet.2015.06.005. doi: 10.1016/j.canlet.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 77.Bryant JL, Meredith SL, Williams KJ, White A. Targeting hypoxia in the treatment of small cell lung cancer. Lung Cancer. 2014;86:126–32. doi: 10.1016/j.lungcan.2014.08.003. doi: 10.1016/j.lungcan.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 78.Wu MZ, Chen SF, Nieh S, Benner C, Ger LP, Jan CI, et al. Hypoxia drives breast tumor malignancy through a TET-TNFa-p38-MAPK signaling axis. Cancer Res. 2015;75:3912–24. doi: 10.1158/0008-5472.CAN-14-3208. doi: 10.1158/0008-5472.CAN-14-3208. [DOI] [PubMed] [Google Scholar]
- 79.Mariani CJ, Vasanthakumar A, Madzo J, Yesilkanal A, Bhagat T, Yu Y, et al. TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep. 2014;7:1343–52. doi: 10.1016/j.celrep.2014.04.040. doi: 10.1016/j.celrep.2014.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsai YP, Chen HF, Chen SY, Cheng WC, Wang HW, Shen ZJ, et al. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014;15:513. doi: 10.1186/s13059-014-0513-0. doi: 10.1186/s13059-014-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]