

ABSTRACT
Voltage-gated potassium channels are important regulators of electrical excitation in many tissues, with Kv1.2 standing out as an essential contributor in the CNS. Genetic deletion of Kv1.2 invariably leads to early lethality in mice. In humans, mutations affecting Kv1.2 function are linked to epileptic encephalopathy and movement disorders. We have demonstrated that Kv1.2 is subject to a unique regulatory mechanism in which repetitive stimulation leads to dramatic potentiation of current. In this study, we explore the properties and molecular determinants of this use-dependent potentiation/activation. First, we examine how alterations in duty cycle (depolarization and repolarization/recovery times) affect the onset and extent of use-dependent activation. Also, we use trains of repetitive depolarizations to test the effects of a variety of Thr252 (S2-S3 linker) mutations on use-dependent activation. Substitutions of Thr with some sterically similar amino acids (Ser, Val, and Met, but not Cys) retain use-dependent activation, while bulky or charged amino acid substitutions eliminate use-dependence. Introduction of Thr at the equivalent position in other Kv1 channels (1.1, 1.3, 1.4), was not sufficient to transfer the phenotype. We hypothesize that use-dependent activation of Kv1.2 channels is mediated by an extrinsic regulator that binds preferentially to the channel closed state, with Thr252 being necessary but not sufficient for this interaction to alter channel function. These findings extend the conclusions of our recent demonstration of use-dependent activation of Kv1.2-containing channels in hippocampal neurons, by adding new details about the molecular mechanism underlying this effect.
KEYWORDS: epilepsy, Kv1.2, Potassium channel, voltage-dependent gating
Introduction
Voltage gated potassium (Kv) channels play an essential role in the regulation of threshold and morphology of action potentials in virtually all excitable tissues.1,2 The Kv channel family comprises a large number of genes, and further diversity of Kv channel function and regulation is achieved by heteromeric assembly of channel subunits.3-6 Within the Kv1 family, a small number of studies have reported that Kv1.2 subunits are subject to a unique regulatory mechanism described as ‘use-dependent activation’.7,8 In the context of cellular electrical excitation, the most relevant consequence of use-dependent activation is that rapid, repetitive trains of depolarizations may generate significant potentiation of a repolarizing K+ current that may alter threshold properties or terminate bursts of action potentials.8 The physiological function of use-dependent activation has not yet been established, however it is clear that Kv1.2 channels play a vital role in CNS function as genetic deletion of Kv1.2 causes complete lethality in mice due to generalized seizures.9 Furthermore, mutations of Kv1.2 in humans have been linked to severe consequences including epileptic encephalopathy, intellectual disability and episodic ataxia.10,11 Acquired antibodies against Kv1.2 can also trigger limbic encephalitis and Morvan's syndrome.12,13
Previous studies have proposed that use-dependence of Kv1.2 is conferred by an extrinsic regulatory factor,7 based on various lines of evidence. Firstly, this feature exhibits marked cell-to-cell variability in cells transfected with Kv1.2, suggesting that the primary sequence of the channel itself does not encode everything required for use-dependence.7,8 Secondly, use-dependence dissipates over time when recording in the whole-cell or inside-out patch modes, but not in the lifetime of perforated patch recordings.7 This observation is consistent with loss of an inhibitory regulator (possibly by diffusion out of the cell and into the recording pipette). These observations point toward an extrinsic regulator that modifies Kv1.2 channel gating. Our most recent study has demonstrated this regulatory mechanism in isolated hippocampal neurons, extending previous observations in various mammalian cell lines.8
To further explore use-dependent activation of Kv1.2, we have investigated the duty cycle (frequency and pulse-length) dependence and the essential sequence determinants of this regulatory process. We find that this regulatory mechanism exhibits state-dependence, as the rate of onset of potentiation is accelerated as the time spent in the open state is increased. Use-dependent activation depends strongly on a Thr residue present in the S2–3 linker at position 252, but introduction of a Thr at the equivalent position in other Kv1 channels was not sufficient to re-introduce use-dependence. Furthermore, substitution of small, uncharged residues for Thr in Kv1.2 preserved use-dependence. Therefore, use-dependent activation seems to depend on the geometry and the precise properties of the sidechains in the S2-S3 linker rather than a specific chemical interaction (such as H-bonding) or modification (e.g. phosphorylation) of the Thr. Taken together, we propose that use-dependent activation of Kv1.2 channels is mediated by an extrinsic regulatory molecule with state-dependent affinity for the channel.
Results
Model for Kv1.2 regulation
To provide some context for this study, we will first outline the fundamental features of Kv1.2 activation gating being investigated, as well as a conceptual model that frames our interpretation. The current record in Figure 1A illustrates use-dependent activation/potentiation of WT Kv1.2 channels in response to a train of depolarizing pulses. The first pulse elicits very little current, but subsequent pulses elicit larger and larger currents. The extent of this phenomenon is highly variable in cells expressing Kv1.2, as described in previous studies, and is absent in recordings from other closely related Kv1 channels.14 This behavior can be completely eliminated with a point mutation in the S2-S3 linker (T252R, Fig. 1B).
Figure 1.

(A) Hypothetical model for interpretation of use-dependent activation of Kv1.2 channels. (A,B) Traces in the upper panels illustrate behavior of Kv1.2 and Kv1.2[T252R] channels in response to repetitive 10 ms depolarizations between −80 mV and +60 mV, with a pulse-to-pulse interval of 50 ms. (Lower panels) The gating scheme depicts a proposed model depicting an extrinsic ‘soluble gating regulator’ (SGR) which binds Kv1.2 preferentially in the closed state (potentially in the S2-S3 linker). Upon depolarization, binding affinity for the channel is reduced, causing it to unbind, leading to disinhibition of channels. In Kv1.2[T252R] channels, we propose that binding of the SGR is reduced or abolished, causing channels to largely dwell in the potentiated state (unbound, lower tier of gating scheme).
Various experimental findings (see introduction) have led us to conceive of a hypothetical mechanism for use-dependent activation, dependent on an extrinsic regulatory subunit that may bind Kv1.2 and modulate its gating properties (Fig. 1A, lower panel). We have used the term ‘soluble gating regulator’ (‘SGR’) to describe this regulatory partner of Kv1.2. We speculate that this regulatory subunit interacts preferentially when channels are closed, and hinders channel opening upon depolarization (slowing activation kinetics, and shifting the voltage-dependence of activation). Upon channel opening, interaction with this extrinsic factor is lost or altered leading to ‘disinhibition’ of the channel (moving to the lower tier of the gating scheme). Upon repolarization, sufficient time will allow the ‘SGR’ to bind channels, causing them to again gate slowly. However, channels that have failed to re-associate with the regulatory subunit will remain in a potentiated state, and re-open quickly in a subsequent depolarization. In this way, rapid repetitive stimuli will cause channels to accumulate in the potentiated tier of the model, allowing for larger currents to be generated later in the train of depolarizations.
Previous work has shown that substitution of Thr252 for Arg eliminates use-dependent activation.7 In the context of this model, we speculate that this mutation alters the binding site for the ‘SGR’, thereby weakening its interaction with the channel and causing channels to preferentially occupy the potentiated (unbound) tier of the model (Fig. 1B, lower panel highlighted by blue arrow). Kv1.2[T252R] channels are thus much less susceptible to the effects of the regulatory subunit and exhibit little use-dependence (Fig. 1B).
Duty-cycle dependence of Kv1.2 potentiation
A prediction of this model is that use-dependent activation/potentiation will depend on the frequency and duration of channel stimulation and recovery intervals. For these experiments, we restricted our analysis to cells that exhibited prominent use-dependent activation (>80 %), and normalized currents during each repetitive pulse to the peak current observed during a long depolarization to +60 mV. To explore duty cycle dependence, we first varied the test pulse duration, while maintaining a 10 Hz stimulus frequency (thereby varying the recovery interval between each depolarization, Figure 2A). Brief depolarizations were not sufficient to elicit the maximal amount of current in Kv1.2 channels (for example, 5 ms depolarizations spaced by 95 ms resting intervals). In contrast, as the length of the depolarization stimulus was increased, full current potentiation could be achieved with fewer sweeps (for example, 80 ms depolarizations spaced by 20 ms resting intervals). These observations are consistent with channel opening being required for potentiation/disinhibition.
Figure 2.

Frequency and pulse duration effects on use-dependent activation. Cells transfected with Kv1.2 exhibiting prominent use-dependent activation were subjected to repetitive depolarizing steps from −80 mV to + 60 mV, with variable depolarization or resting intervals. Peak current in each pulse was normalized to the peak current observed during a long pulse to +60 mV. In panel (A), the depolarization duration was altered while maintaining a constant pulse frequency of 10 Hz. In panel (B), the interpulse interval was altered while maintaining a constant 10 ms depolarization duration.
Subsequently, we tested the effects of varying the interpulse interval, while maintaining a consistent depolarizing pulse duration (10 ms), thereby changing the frequency of stimulation (Fig. 2B). Longer recovery intervals elicited submaximal Kv1.2 potentiation whereas shorter (≤50 ms) recovery intervals elicited full potentiation (Fig. 2B). These observations are consistent with the notion that longer resting intervals increase the fraction of channels that can re-associate with the ‘SGR’. In the gating scheme presented in Figure 1A, this would correspond to channels moving from the lower ‘potentiated/unbound’ gating tier, to the upper gating tier. Taken together, these data are consistent with the prediction that increased channel stimulation (either by long pulses, or more frequent stimulation) relieves Kv1.2 from the influence of an inhibitory regulatory factor.
Structure-function analysis of the S2-S3 linker region
Previous work highlighted the importance of Kv1.2 residue Thr252 for use-dependent activation.7 Numerous mutations of Thr252 and 2 upstream phenylalanines (Phe251 and Phe250), have been reported to eliminate use-dependent activation.7 To extend previous investigation of the molecular determinants of this regulatory mechanism, we tested the effects of additional Thr252 mutations, including the isosteric Thr252Val substitution, on channel responses to high-frequency stimulation (Fig. 3A). We also highlight that we have adopted a different stimulation protocol than that used in previously published studies of Thr252 mutants, opting for a sequence of repetitive 10 ms depolarizations delivered at 20 Hz. This protocol enables more detailed quantification of the extent of use-dependent potentiation, based on the percent change in current between the 1st and 100th pulse in a train. Also, this parameter (% use-dependent activation) allows us to illustrate the cell-to-cell variability that is inherent in this regulatory mechanism. In previous studies, potentiation of Kv1.2 was examined with a sequence of 2 significantly longer (∼400 ms) pulses, and cells were classified as ‘slow’ (i.e. sensitive to the use-dependent regulatory mechanism) or ‘fast’ (i.e., insensitive) based on whether the initial depolarization altered the kinetics of the second test pulse.
Figure 3.

Effects of S2-S3 linker mutations on use-dependent activation. Use-dependent activation of a panel of S2-S3 linker mutations was quantified using trains of repetitive depolarizations (20 Hz) from −80 mV (40 ms) to +60 mV (10 ms), as described in Figure 1A. Each data point represents the extent of use-dependent activation observed in a single cell. Prominent use-dependence was observed with Thr, Val, Ser, or Met in position 252 (n = 15–26 for each construct).
Overall, we observed that smaller, uncharged substitutions including Ser, Val and (to a lesser extent) Met, are permissive for use-dependent activation, while bulkier or charged amino acids eliminated the effect. Importantly, the Thr252Val substitution exhibited use-dependent activation that was indistinguishable from WT Kv1.2 channels, strongly indicating that phosphorylation of Thr252 does not regulate use-dependence. Also noteworthy was that despite its steric similarity to Ser, Cys substitution at residue 252 did not preserve use-dependence.
An important difference from previously published work was that Asp and Glu substitutions for Thr252 largely eliminated use-dependent activation, although previously reported to preserve channel potentiation.7 We suspect that this discrepancy is due to different methods of measuring this regulatory mechanism (as highlighted above). Specifically, we have observed that introduction of either Asp or Glu intrinsically slows the kinetics of Kv1.2 activation, and this may have led to their classification as ‘slow’ (sensitive to use-dependent activation). However, using the repetitive pulse protocol, we observed very little pulse-to-pulse potentiation.
We also attempted to transplant use-dependent activation from Kv1.2 into several other Kv1 family channels. Replacement of the native Lys of Kv1.5 with Thr (at the Thr252 equivalent position) has been previously studied, and was not sufficient to restore use-dependent potentiation.7 We extended these experiments by introducing a Thr at the equivalent position in Kv1.1, Kv1.3 and ΔN19-Kv1.4 (Fig. 4). Similar to findings in Kv1.5, Thr substitution in the S2-S3 linker was not sufficient to reintroduce use-dependence, indicating that there are additional channel elements that influence use-dependent activation.
Figure 4.
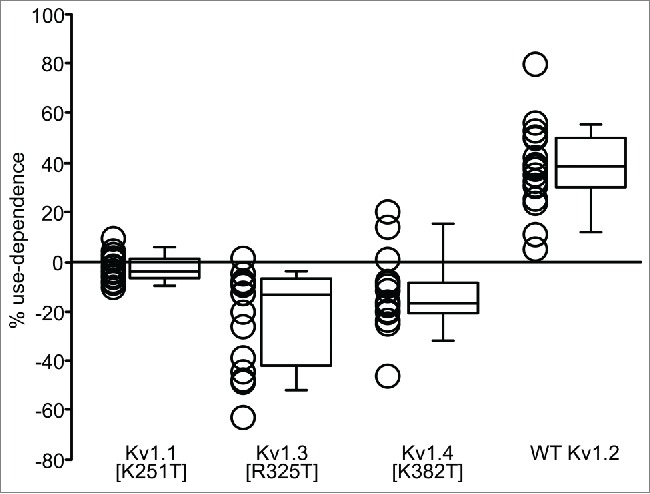
Substitution of Thr at the 252 equivalent position of Kv1.2 into use-dependent activation insensitive channels. Use-dependent activation of indicated mutants of Kv1.1, 1.3, and 1.4 (ΔN19 background to eliminate N-type inactivation), was measured using repetitive 10 ms depolarizations (20 Hz) from −80 mV to +60 mV, as described in previous Figures. In each of these channels a Thr (present in the Kv1.2 S2-S3 linker at position Thr252) was substituted at the equivalent position, and mutant channels were expressed in ltk- fibroblasts. Kv1.2 Thr252 is necessary for use-dependent activation, however introduction of this residue in either Kv1.1, Kv1.3, or Kv1.4 was not sufficient to transfer this use-dependent property (n = 14–20 for each construct).
Effects of S2-S3 linker mutations on surface expression
We investigated whether there was a relationship between channel expression and use-dependence of Kv1.2, by measuring current density and cell surface expression (by western blot) of the same panel of Thr252 mutants (Fig. 5). These experiments were motivated by a lingering uncertainty that large channel expression might overwhelm the expression of an endogenous negative regulator, leading to a stoichiometric mismatch in which many channels would be unbound and therefore appear ‘potentiated’. In some cases (i.e. Thr252Cys, Thr252Lys and Thr252Arg), increased current density was observed together with weaker use-dependence. However, changes in current density relative to WT Kv1.2 were not especially large, and some channels with little or no use-dependent activation (i.e., Thr252His and Thr252Phe) had comparable current density to WT Kv1.2 (Fig. 5C). This finding was corroborated by western blot analysis, in which we quantified the ratio of the intracellular core-glycosylated band (highlighted by ‘IC’ in Figure 5A) and the more heavily glycosylated band corresponding to the cell surface fraction (highlighted by ‘CS’).15 Overall, there were no marked differences in surface expression between strongly use-dependent mutants and those that are resistant to the regulatory mechanism (Fig. 5A, B). One noteworthy S2-S3 linker mutation was Phe251Arg, which abolished use-dependence while dramatically weakening current density and cell surface expression. Overall, these findings suggest that mutations that alter use-dependent activation of Kv1.2 do so by influencing channel interaction with a regulatory factor, rather than a secondary consequence of dramatically increased channel expression leading to stoichiometric mismatch with an extrinsic regulatory subunit.
Figure 5.
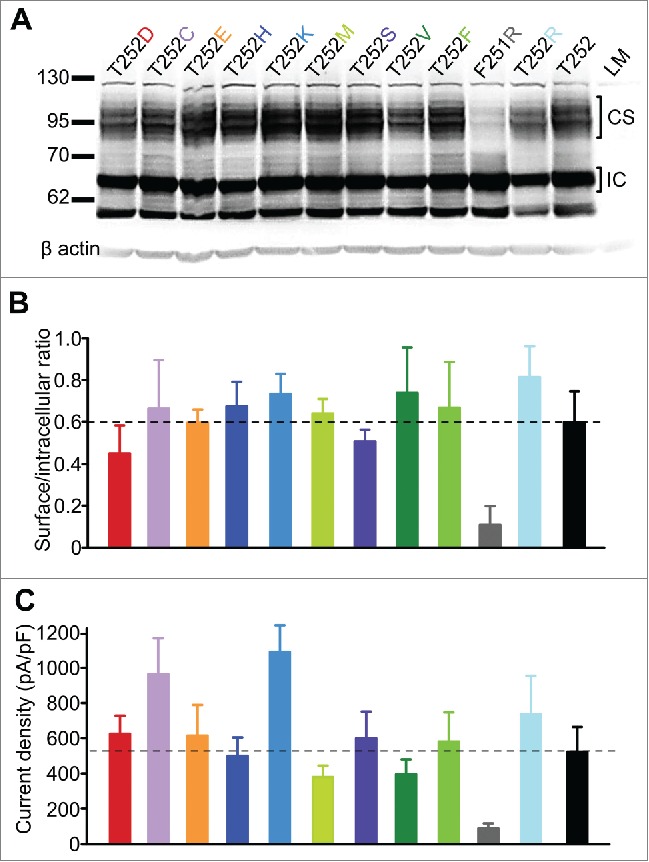
Cell surface expression and current density for a panel of different Kv1.2 S2-S3 linker mutants. (A) Representative western blot of a panel of S2-S3 linker mutants of Kv1.2. Kv1.2channels typically runs as multiple bands on the gel, characterized by a heavier ˜95 kDa glycosylated cell surface band (highlighted by ‘CS’) and a smaller ˜67 kDa intracellular ‘core’ glycosylated band (highlighted by ‘IC’). (B) Densitometry was used to measure the ratio of the cell surface band to the intracellular band (small ratio indicates weak surface expression, n = 3). No significant difference was detected between different mutations, with the exception of Kv1.2[F251R] where relative surface expression was reduced. (C) Current density at 10 mV was measured for cells expressing the panel of Kv1.2 mutant channels. In panels B and C, the dashed horizontal line is a reference for comparison to WT Kv1.2 levels.
Although channel expression cannot account for the loss of use-dependent activation in certain mutants, we have observed a relationship between peak current and use-dependence among Kv1.2 channel mutants with strong use-dependence. For example, in cells transfected with WT Kv1.2, the most prominent use-dependence tended to appear in cells with smaller currents (Fig. 6). This feature was most dramatic in cells co-transfected with Kv1.2 and a pool of Kv1.2 siRNA oligonucleotides that diminished channel expression. Most of the siRNA-treated cells had relatively small currents with very prominent use-dependence (Fig. 6A). Similar observations were made in other Kv1.2 mutants that retained sensitivity to use-dependent activation (Fig. 6B, C). This trend further supports our conclusion that use-dependent activation is mediated by an extrinsic regulator, and the relative abundance of channel and regulator within a cell determines the strength of use-dependence. If there are less Kv1.2 channels on the membrane, it is more likely that the SGR will be able to bind all of the channels, and that the cell will exhibit strong use-dependent activation. If there are many Kv1.2 channels, however, there may be a stoichiometric mismatch leading to markedly less use-dependent activation.
Figure 6.
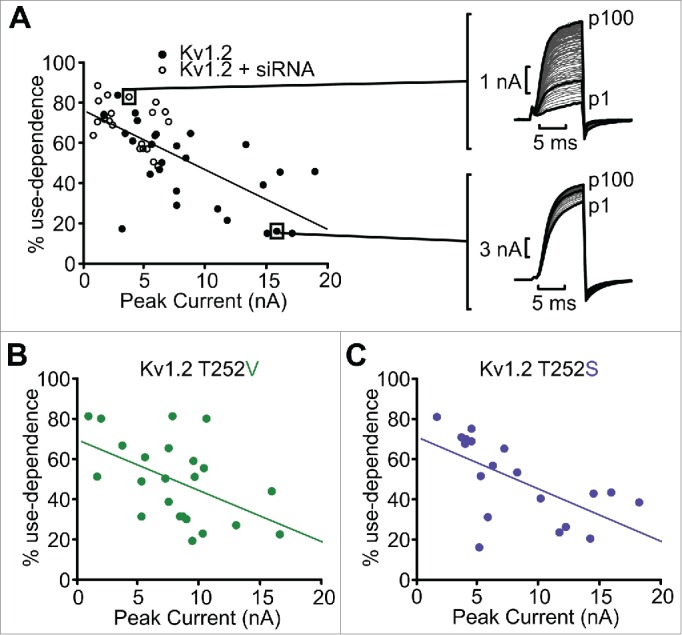
Comparison of peak current to use-dependent activation in Kv1.2, Kv1.2[T252S] and Kv1.2[T252V]. (A) Relationship between peak current and % use-dependent activation, with each data point representing the current magnitude and use-dependence for a single cell. Data points distinguish between cells transfected with Kv1.2 (black circles) vs. cells transfected with Kv1.2 plus siRNA oligonucleotides (white circles). Exemplar traces illustrating pronounced use-dependence in a cell with small currents (top, co-transfected with siRNA), and weak use-dependence in a cell with large currents (bottom) are shown. Similar data were collected for Thr252 mutants that retain sensitivity to use-dependent activation: (B) Kv1.2[T252V], and (C) Kv1.2[T252S].
Discussion
Use-dependent activation/potentiation is a rarely observed property of Kv channels, but exerts a prominent influence on Kv1.2 subunits in homomeric or heteromeric channel assemblies.14 In this study, our objective was to outline and explore our hypothesis that this regulatory mechanism arises from a state-dependent interaction between Kv1.2 subunits and an as yet unidentified extrinsic regulatory factor ('SGR'). The model that we prefer is that a binding site for the regulator exists when Kv1.2 channels are in a closed confirmation, with the interaction becoming much weaker in open channels (Fig. 1A). During a train of depolarizations, channels that have opened (and unbound from the regulator) will accumulate in a ‘potentiated’ state, so long as the interpulse interval is sufficiently short to allow little restoration of the interaction. In addition to the molecular details, we are very excited by the observation of this phenomenon in neurons, as it lays the foundation for investigation of a mechanism for activity-dependent alteration of neuronal excitability.8 Also, our report that this regulatory phenomenon persists in heteromeric channels with even a single Kv1.2 subunit indicates that Kv1.2 may act as an adaptor protein that can harness this regulatory mechanism into channels with mixed subunit composition.8
The dynamic range and potential physiological impact of use-dependent activation imparted by Kv1.2 is important to recognize. Although there is considerable cell-to-cell variability of use-dependent activation (Fig. 3), cells with particularly prominent use-dependent activation can exhibit current increases of nearly 100-fold during a 20Hz train of depolarizations (10 ms pulses, 20 Hz)8. This provides a mechanism through which neurons might dynamically modulate action potential firing, leading to burst termination or silencing of overactive neurons. Use-dependent activation of Kv1.2 channels has not yet been linked to a specific physiological outcome, but complete lethality of Kv1.2 knockout models9 suggest the paramount importance of the channel, and our work has demonstrated that this use-dependent activation is a unique feature of Kv1.2 subunits. Ongoing investigation will be directed toward uncovering the physiological importance of this regulatory mechanism, as well as how it is linked to other mechanisms of Kv channel regulation. For Kv1.2 in particular, some of these regulatory mechanisms include phosphorylation of Ser440/Ser441,16 activity-dependent redistribution of Kv1.2 mediated by the sigma-1 receptor,17 and endocytosis triggered by repetitive action potential firing.18 Kv1.2 gating is also modulated by glycosylation,15 tyrosine phosphorylation,19 and auxiliary Kvβ-subunits.20 Understanding the relative/collective importance of these regulatory mechanisms, and how they are recruited into heteromeric Kv1 channel complexes, will help to define the physiological role of use-dependent activation of Kv1.2.
Materials and methods
Constructs and expression
Kv1 channel cDNAs were expressed using the pcDNA3.1(−) vector (Invitrogen). Mutations were introduced with overlap extension PCR and subcloned into the EcoRI and HindIII sites in the pcDNA3.1(−) MCS using standard methods. Constructs were all verified by diagnostic restriction digests and Sanger sequencing (Genewiz, Inc.).
Mouse ltk- fibroblast cells were maintained in culture in a 5% CO2 incubator at 37°C in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were transfected with channel cDNAs using jetPRIME transfection reagent (Polyplus) and 12–24 hours later were split onto sterile glass coverslips. Cells were co-transfected with fluorescent protein cDNA to allow identification of cells for recording by epifluorescence. Recordings were done 1–3 d following transfection. For siRNA-mediated knockdown of Kv1.2 expression, cells were transfected with 50 nM siRNA pool targeting Kv1.2 (M-091038, Dharmacon), Kv1.2 cDNA and GFP cDNA using Dharmacon Duo transfection reagent. Cells were split onto sterile coverslips 48 hours later, and recorded 12–24 hours after plating.
Electrophysiology
Patch pipettes were manufactured from soda lime capillary glass (Fisher), using a Sutter P-97 puller. When filled with standard recording solutions, pipettes had resistances of 1–2.5 MΩ. Recordings were filtered at 5 kHz, sampled at 10 kHz, with manual capacitance compensation and series resistance compensation between 70–90%, and stored directly on a computer hard drive using Clampex software (Molecular Devices). Bath solution had the following composition: 135 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and was adjusted to pH 7.4 with KOH. Pipette solution had the following composition: 135 mM KCl, 5 mM K-EGTA, 10 mM HEPES and was adjusted to pH 7.2 using KOH. Chemicals were purchased from Sigma-Aldrich or Fisher. Throughout the text, data are presented as mean ± s.e.m.
Western blot analysis
Cell lysates from transfected ltk- fibroblasts were harvested in RIPA buffer 3 d following transfection, separated using 10 % SDS-PAGE gels, and transferred to nitrocellulose membranes using standard methods. Kv1.2 was detected using a monoclonal mouse Kv1.2 antibody (NeuroMAb, clone #K14/16 75–008), HRP-conjugated goat anti-mouse antibody (Applied Biological Materials, Vancouver, Canada, SH023), and SuperSignal West Femto Max Sensitivity Substrate (Thermo Scientific). Chemiluminescence was detected using a FluorChem SP gel imager (Alpha Innotech).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank David Fedida's lab (UBC), for providing some of the mutant constructs used in Figure 3.
Funding
Operating costs of this work were supported by a Human Frontiers in Science Program Young Investigator award to H.T.K. (RGY-0081). V.A.B. received a CIHR CGS-M award and an NSERC Undergraduate Student Research Award, and financial support from the University of British Columbia M.D./Ph.D. program. H.T.K. received salary support as a Michael Smith Foundation for Health Research Scholar, and a CIHR New Investigator.
References
- [1].Shen W, Hernandez-Lopez S, Tkatch T, Held JE, Surmeier DJ. Kv1.2-containing K+ channels regulate subthreshold excitability of striatal medium spiny neurons. J. Neurophysiol 2004; 91:1337-49; PMID:13679409; http://dx.doi.org/ 10.1152/jn.00414.2003 [DOI] [PubMed] [Google Scholar]
- [2].Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron 2008; 58:387-400; PMID:18466749; http://dx.doi.org/ 10.1016/j.neuron.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Po S, Roberds S, Snyders DJ, Tamkun MM, Bennett PB. Heteromultimeric assembly of human potassium channels. Molecular basis of a transient outward current? Circ Res 1993; 72:1326-36; PMID:8495559; http://dx.doi.org/ 10.1161/01.RES.72.6.1326 [DOI] [PubMed] [Google Scholar]
- [4].Sheng M, Liao YJ, Jan YN, Jan LY. Presynaptic A-current based on heteromultimeric K+ channels detected in vivo. Nature 1993; 365:72-5; PMID:8361540; http://dx.doi.org/ 10.1038/365072a0 [DOI] [PubMed] [Google Scholar]
- [5].Wang H, Kunkel DD, Martin TM, Schwartzkroin PA, Tempel BL. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature 1993; 365:75-9; PMID:8361541; http://dx.doi.org/ 10.1038/365075a0 [DOI] [PubMed] [Google Scholar]
- [6].Plane F, Johnson R, Kerr P, Wiehler W, Thorneloe K, Ishii K, Chen T, Cole W. Heteromultimeric Kv1 channels contribute to myogenic control of arterial diameter. Circ Res 2005; 96:216-24; PMID:15618540; http://dx.doi.org/ 10.1161/01.RES.0000154070.06421.25 [DOI] [PubMed] [Google Scholar]
- [7].Rezazadeh S, Kurata HT, Claydon TW, Kehl SJ, Fedida D. An Activation Gating Switch in Kv1.2 Is Localized to a Threonine Residue in the S2-S3 Linker. Biophys J 2007; 93:4173-86; PMID:17766348; http://dx.doi.org/ 10.1529/biophysj.107.116160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Baronas VA, McGuinness BR, Brigidi GS, Gomm Kolisko RN, Vilin YY, Kim RY, Lynn FC, Bamji SX, Yang R, Kurata HT. Use-dependent activation of neuronal Kv1.2 channel complexes. J Neurosci 2015; 35:3515-24; PMID:25716850; http://dx.doi.org/ 10.1523/JNEUROSCI.4518-13.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brew HM, Gittelman JX, Silverstein RS, Hanks TD, Demas VP, Robinson LC, Robbins CA, McKee-Johnson J, Chiu SY, Messing A et al.. Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J Neurophysiol 2007; 98:1501-25; PMID:17634333; http://dx.doi.org/ 10.1152/jn.00640.2006 [DOI] [PubMed] [Google Scholar]
- [10].Syrbe S, Hedrich UB, Riesch E, Djemie T, Muller S, Moller RS, Maher B, Hernandez-Hernandez L, Synofzik M, Caglayan HS et al.. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet 2015; 47(4):393-9; PMID:25751627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, Roder JC. A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem 2010; 285:32160-73; PMID:20696761; http://dx.doi.org/ 10.1074/jbc.M110.153676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Buckley C, Oger J, Clover L, Tuzun E, Carpenter K, Jackson M, Vincent A. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73-8; PMID:11456313; http://dx.doi.org/ 10.1002/ana.1097 [DOI] [PubMed] [Google Scholar]
- [13].Kleopa KA, Elman LB, Lang B, Vincent A, Scherer SS. Neuromyotonia and limbic encephalitis sera target mature shaker-type K+ channels: subunit specificity correlates with clinical manifestations. Brain 2006; 129:1570-84; PMID:16613892; http://dx.doi.org/ 10.1093/brain/awl084 [DOI] [PubMed] [Google Scholar]
- [14].Baronas VA, McGuinness BR, Brigidi GS, Gomm Kolisko RN, Vilin YY, Kim RY, Lynn FC, Bamji SX, Yang R, Kurata HT. Use-dependent activation of neuronal Kv1.2 channel complexes. J Neurosci 2015; 35:3515-24; PMID:25716850; http://dx.doi.org/ 10.1523/JNEUROSCI.4518-13.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fujita T, Utsunomiya I, Ren J, Matsushita Y, Kawai M, Sasaki S, Hoshi K, Miyatake T, Taguchi K. Glycosylation and cell surface expression of Kv1.2 potassium channel are regulated by determinants in the pore region. Neurochem Res 2006; 31:589-96; PMID:16770729; http://dx.doi.org/ 10.1007/s11064-006-9056-4 [DOI] [PubMed] [Google Scholar]
- [16].Yang JW, Vacher H, Park KS, Clark E, Trimmer JS. Trafficking-dependent phosphorylation of Kv1.2 regulates voltage-gated potassium channel cell surface expression. Proc Natl Acad Sci U S A 2007; 104:20055-60; PMID:18056633; http://dx.doi.org/ 10.1073/pnas.0708574104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kourrich S, Hayashi T, Chuang JY, Tsai SY, Su TP, Bonci A. Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell 2013; 152:236-47; PMID:23332758; http://dx.doi.org/ 10.1016/j.cell.2012.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hyun JH, Eom K, Lee KH, Ho WK, Lee SH. Activity-dependent downregulation of D-Type K+ channel subunit Kv1.2 in rat hippocampal CA3 pyramidal neurons. J Physiol 2013; 591:5525-40; PMID:23981714; http://dx.doi.org/ 10.1113/jphysiol.2013.259002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tsai W, Morielli AD, Cachero TG, Peralta EG. Receptor protein tyrosine phosphatase alpha participates in the M1 muscarinic acetylcholine receptor-dependent regulation of Kv1.2 channel activity. EMBO J 1999; 18:109-18; PMID:9878055; http://dx.doi.org/ 10.1093/emboj/18.1.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Accili EA, Kiehn J, Yang Q, Wang Z, Brown AM, Wible BA. Separable kvbeta subunit domains alter expression and gating of potassium channels. J Biol Chem 1997; 272:25824-31; PMID:9325312; http://dx.doi.org/ 10.1074/jbc.272.41.25824 [DOI] [PubMed] [Google Scholar]