

ABSTRACT
Nine different voltage-gated sodium channel isoforms are responsible for inducing and propagating action potentials in the mammalian nervous system. The Nav1.7 channel isoform plays an important role in conducting nociceptive signals. Specific mutations of this isoform may impair gating behavior of the channel resulting in several pain syndromes. In addition to channel mutations, similar or opposite changes in gating may be produced by spider and scorpion toxins binding to different parts of the voltage-gated sodium channel. In the present study, we analyzed the effects of the α-scorpion toxin OD1 and 2 synthetic toxin analogs on the gating properties of the Nav1.7 sodium channel. All toxins potently inhibited channel inactivation, however, both toxin analogs showed substantially increased potency by more than one order of magnitude when compared with that of wild-type OD1. The decay phase of the whole-cell Na+ current was substantially slower in the presence of toxins than in their absence. Single-channel recordings in the presence of the toxins revealed that Na+ current inactivation slowed due to prolonged flickering of the channel between open and closed states. Our findings support the voltage-sensor trapping model of α-scorpion toxin action, in which the toxin prevents a conformational change in the domain IV voltage sensor that normally leads to fast channel inactivation.
Keywords: gating, Nav1.7, patch clamp, scorpion toxin, voltage-gated sodium channel
Introduction
The Nav1.7 voltage-gated sodium channel (VGSC) is encoded by SCN9A; expressed in sensory and sympathetic neurons, Schwann and endocrine cells; and plays a key role in generating and propagating action potentials in the central and peripheral nervous systems.1,2 Nav1.7 channel mutations may result in gain- or loss-of-function, creating pathological conditions such as over-sensitivity or loss of sensitivity to pain, respectively. For example, the S241T and A1746G mutations, which have been linked to inherited or primary erythromelalgia, result in a shift in voltage-dependence of activation to more negative potentials that makes cells hyper-excitable.3,4
Certain toxins from animal venoms can cause similar effects to those caused by mutations. For instance, some VGSC-targeting spider and scorpion β-toxins frequently bind to receptor site 4 (on domain II), where they can shift voltage-dependence of activation to more negative potentials (‘activators’) or more positive potentials (‘inhibitors’).5-7 Toxins targeting site 3 (on domain IV), such as spider toxins, δ-conotoxins and α-scorpion toxins, typically slow inactivation, which prolongs the decay phase of the whole-cell Na+ current.8-11 Interestingly, some toxins appear to target sites 3 and 4 simultaneously, affecting fast inactivation and steady-state activation. A classical example is spider toxin ProTx-II, which has been shown to interact with DII and DIV on Nav1.7, although the affinities are substantially different.12-13 Another example is the recently characterized scorpion α-toxin OD1, which potently inhibits mammalian Nav1.4, Nav1.6 and Nav1.7 inactivation and results in a hyperpolarizing shift in the steady-state voltage-dependence of Nav1.4 and Nav1.6 activation.14-16
Using a high-throughput fluorescence-based assay, we recently identified OD1 analogs with altered Nav1.4, Nav1.6 and Nav1.7 selectivity, and significantly improved potency at Nav1.7.16 This assay relies on membrane potential-sensitive dyes to monitor Nav activation in the presence of sub-threshold concentrations of the site 2-targeting alkaloid veratridine, and facilitates rapid, qualitative and quantitative analysis of VGSC modulators. However, the lack of control over the membrane potential in this assay precludes any detailed, mechanistic VGSC studies at the molecular level.
Here, we report a detailed analysis of how mutations of OD1 affect the toxin's interaction with the Nav1.7 VGSC.
Results
Concentration-dependent effect of OD1 and analogs on sodium current inactivation
Step depolarization of CHO cells stably expressing human Nav1.7 channels from −100 mV to 0 mV in the absence of toxins elicited the whole-cell inward Na+ current with fast onset, followed by an exponential decay with time constants τ1 = 0.77 ± 0.03 ms and τ2 = 6.0 ± 0.4 ms (n = 6). Applying OD1 and its analogs remarkably prolonged the current decay phase in a concentration-dependent manner (Fig. 1A). This effect was quantified by the ratio of the current measured at an interval of 1.5 ms from the current peak to the peak value (Fig. 1B).
Figure 1.
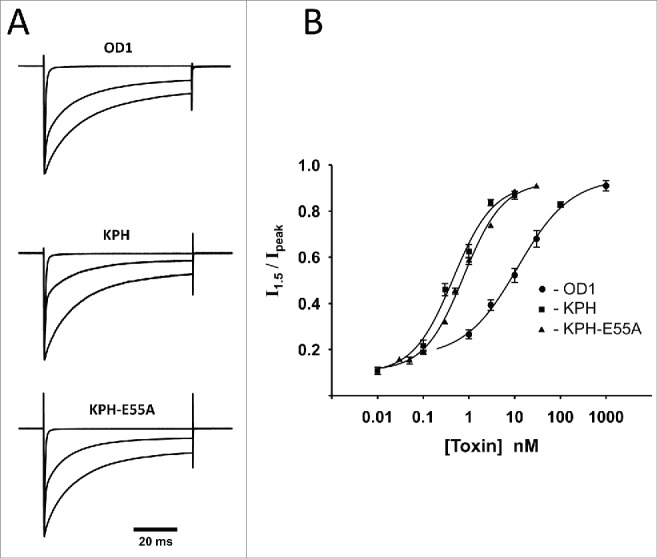
Wild-type OD1 and its analogs KPH and KPH-E55 slow the decay phase of whole-cell Na+ current in a concentration-dependent manner. (A) Three sets of normalized current traces represent responses to step depolarization from a holding potential of −100 mV to 0 mV. Each set consists of a trace in the absence of the toxin (upper fast declining current), at approximately EC50 concentration (middle trace) and saturating concentration. The concentrations were as follows: OD1: 0, 10 nM, 300 nM; KPH: 0, 300 pM, 10 nM; and KPH-E55A: 0, 300 pM, 10 nM. (B) Concentration–dependence of change in inactivation rate of whole-cell Na+ currents. The ratio of whole-cell value taken at 1.5 ms after the peak occurrence to the peak current was taken as an estimate of inactivation rate. Experimental points were fitted with a logistic equation with the following parameters: OD1: EC50 = 12 ± 3 nM, n = 0.7 ± 0.2; KPH: EC50 = 0.4 ± 0.1 nM, n = 1.0 ± 0.3; and KPH-E55A: EC50 = 0.77 ± 0.08 nM, n = 1.0 ± 0.1.
The potency of the OD1 analogs was substantially higher than that of wild-type OD1, exceeding it approximately 30 times for OD1-KPH and approximately 16 times for OD1-KPH-E55A (Fig. 1B). Effects of the toxins on inactivation kinetics were completely eliminated by toxin washout or upon applying strong depolarizing pre-pulses (up to +190 mV) in the presence of OD1-KPH (Fig. 2). The degree of current recovery depended on the amplitude of depolarization, and was estimated by fitting the experimental data to a logistic function yielding a midpoint value of +127 ± 1 mV (n = 4) (Fig. 2B). Pre-pulses to potentials more positive than +190 mV were detrimental to the cell membrane and were not examined.
Figure 2.
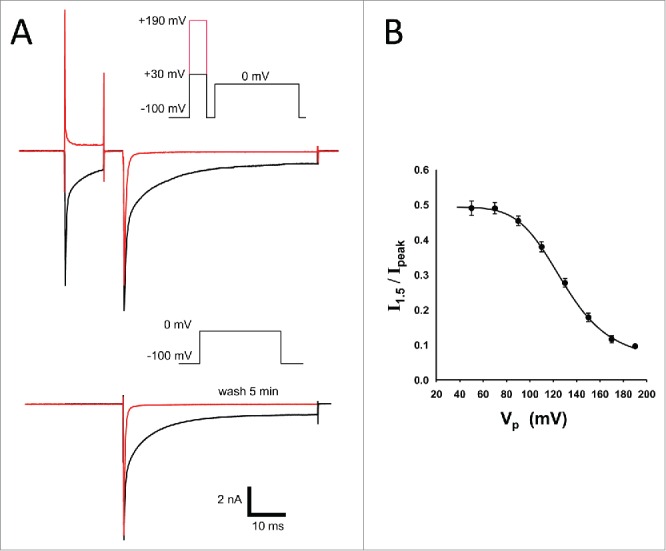
Recovery of Na+ current from toxin action by strong depolarization and washout. (A) Whole-cell Na+ current elicited by membrane depolarization to 0 mV in the presence of 500 pM KPH (black traces). Strong depolarizing pre-pulse to +190 mV restores inactivation rate (red trace on top inset). Na+ currents showed fast and complete recovery from toxin action within 5 min of washout (red trace on the bottom inset). Similar results were obtained for OD1 and KPH-E55A. (B) Voltage-dependence of toxin displacement by depolarizing pre-pulse in the presence of 500 pM KPH (n = 4).
Effects of OD1 and analogs on Nav1.7 channel gating
We analyzed the effects of OD1 and its analogs on Nav1.7 voltage-dependence of activation and inactivation. Current–voltage (I–V) relationships in the absence and presence of toxins are shown in Figure 3A. Half-activation voltage (V0.5) for Nav1.7 was −29.3 ± 0.3 mV (n = 14) in the absence of toxin, and leftward shifted to more negative potentials in the presence of 10 nM wild-type OD1 (V0.5 = −35.8 ± 0.3 mV, n = 7), 500 pM OD1-KPH (V0.5 = −37.9 ± 0.2 mV, n = 5) or 1 nM OD1-KPH-E55A (V0.5 = −39.7 ± 0.2 mV, n = 7), respectively. The minor, but statistically significant, shift caused by wild-type OD1 is in agreement with earlier observations by Maertens et al.,15 who observed a −3 mV shift.
Figure 3.
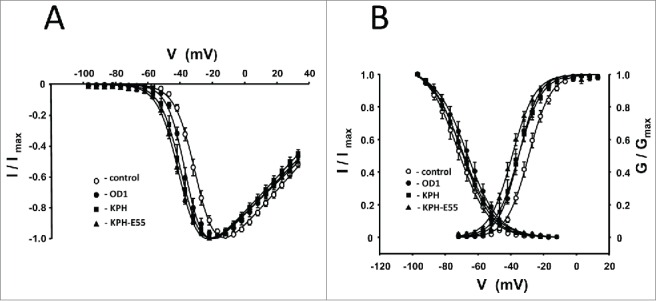
(A) Current-voltage relationships for OD1 and its analogs cause shifts in the current–voltage relationship for Nav1.7-mediated conductance to more negative potentials. (B) Voltage-dependence of steady-state fast inactivation and activation of Nav1.7 channels in the presence of OD1 and its analogs (closed shapes). Fast inactivation was induced by 200 ms pre-pulses followed by a 50 ms test pulse to 0 mV. Open circles represent experimental values obtained in the absence of a toxin. Solid lines are fits of experimental points with Boltzmann functions. Parameters of fitting are given in the Table 1. Toxin concentrations were as follows: OD1, 10 nM; KPH, 500 pM; and KPH-E55A, 1 nM.
Along with I–V changes, the toxins caused similar shifts in the voltage-dependence of activation (Fig. 3B, Table 1). The resulting V0.5 shifts are minor and, at least for wild-type OD1, substantially smaller than the negative shifts observed on Nav1.4 (−13 mV) and Nav1.6 (− 20 mV).16 In contrast with their effect on steady-state activation, neither OD1-KPH nor OD1-KPH-E55A have a statistically significant effect on the Nav1.7 voltage-dependence of fast inactivation (Fig. 3B).
Table 1.
Parameters of Nav1.7 activation and steady-state fast inactivation.
| G/Gmax |
I/Imax |
|||||
|---|---|---|---|---|---|---|
| V0.5 (mV) | n | p | V0.5 (mV) | n | p | |
| Control | −29.1 ± 0.2 | 14 | — | −72.2 ± 0.4 | 14 | — |
| OD1 (10 nM) | −35.2 ± 0.3 | 7 | 0.004 | −66.7 ± 0.4 | 5 | 0.1 |
| KPH (0.5 nM) | −35.3 ± 0.1 | 5 | 0.005 | −69.9 ± 0.2 | 5 | 0.3 |
| KPH-E55A (1 nM) | −39.1 ± 0.3 | 7 | 0.0001 | −69.5 ± 0.4 | 7 | 0.3 |
P values calculated with Student t test against control.
Recovery from fast inactivation
OD1 and its analogs KPH and KPH-E55A enhance Nav1.7 recovery from fast inactivation (Fig. 4). The recovery rate was estimated from the ratio of peak current evoked by a 40 ms test pulse to 0 mV, and peak current recorded during the preceding 40 ms pre-pulse to 0 mV, as a function of time interval between these 2 pulses. Data obtained in the absence of the toxin could be successfully fit by a single exponential function with a time constant of τ = 15 ± 2 ms (n = 14).
Figure 4.
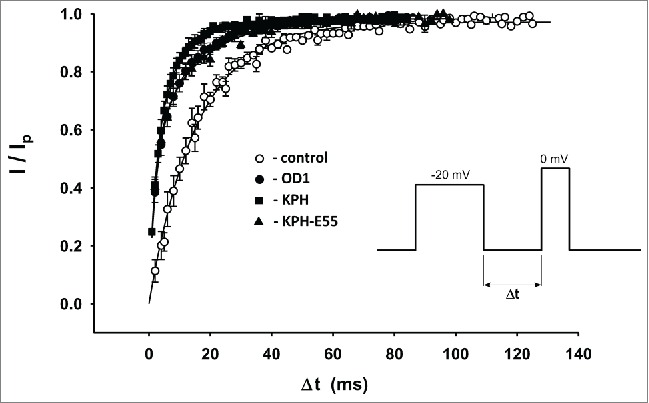
Recovery from fast inactivation in control conditions (open circles) and in the presence of 10 nM OD1, 500 pM KPH and 1 nM KPH-E55A.
In contrast, applying all toxins resulted in pronounced biphasic recovery from inactivation kinetics, with both kinetically distinct phases having approximately equal amplitudes. Experimental points were fit by a double exponential function. Parameters of the fits are given in Table 2.
Table 2.
Amplitudes (A) and time constants (τ) of recovery from fast inactivation state.
| As | τs (ms) | Af | τf (ms) | n | |
|---|---|---|---|---|---|
| Control | 0.94 ± 0.02 | 15 ± 2 | — | — | 14 |
| OD1 | 0.48 ± 0.03 | 15 ± 2 | 0.48 ± 0.04 | 2.8 ± 0.5 | 12 |
| KPH | 0.46 ± 0.04 | 13 ± 2 | 0.50 ± 0.05 | 2.8 ± 0.4 | 5 |
| KPH-E55A | 0.53 ± 0.05 | 15 ± 2 | 0.48 ± 0.09 | 2.5 ± 0.3 | 8 |
Single Na+ channel recordings
It is well established that the observed whole-cell current waveform may be realized by substantially different molecular mechanisms of gating.17 To get a deeper insight into these mechanisms, we investigated the action of OD1 at the microscopic level using single-channel patch clamp recording.18 Representative examples of single-channel Nav1.7 currents from outside-out patches are shown in Figure 5A. Applying wild-type OD1 (100 nM) did not affect the current amplitude, which was 1.4 ± 0.2 pA and 1.36 ± 0.09 pA (n = 3) in the absence and presence of OD1, respectively. Surprisingly, the mean open time of the channels also was not substantially changed by the toxin (Fig. 5B). In contrast, Na channels exhibited flickering behavior in the presence of the toxin, which in some sweeps lasted for the duration of the depolarizing voltage step (Fig. 5A).
Figure 5.
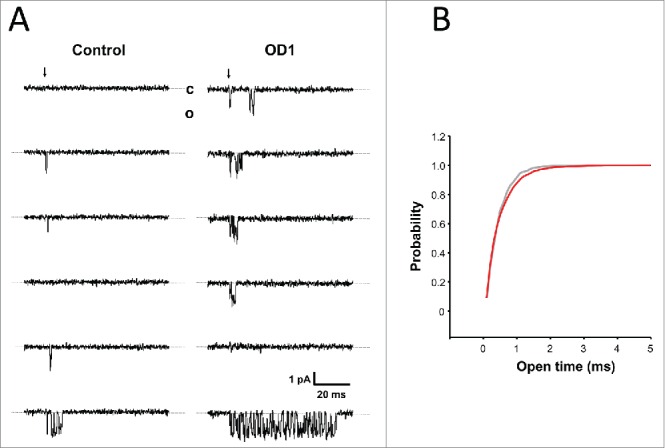
(A) Representative example of consecutive single-channel current traces recorded in the absence and presence of 100 nM OD1. Inward Na+ currents were evoked by step depolarization from a holding potential of −100 mV to −20 mV. The beginning of the step is indicated by the arrow and dashed lines correspond to the channel closed state (c). (B) Cumulative distribution of channel open times in control (gray) and the presence of OD1 (red).
Kinetic models
We attempted to find an appropriate kinetic model of channel conformational states in terms of Markov's formalism, and define the toxin's action through the transition rates changes. As a base model (i.e. without toxin), we chose a minimal model that contained one open state, 2 closed19,20 and 2 inactivated states (Fig. 6A). Rate constants were considered stationary and not dependent on membrane potential.
Figure 6.
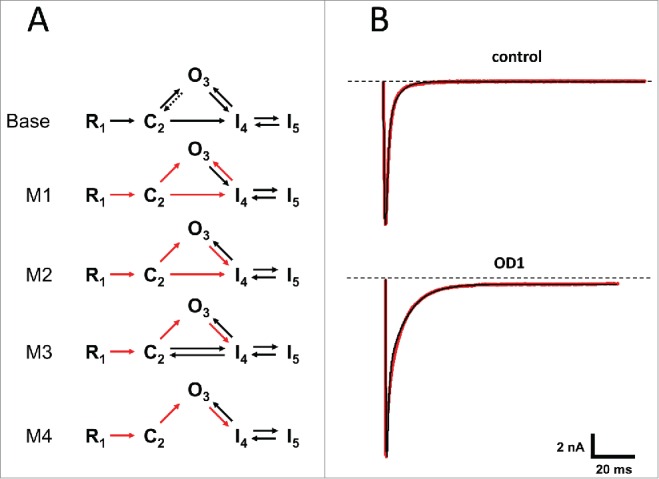
(A) Kinetic models of the Nav1.7 sodium channel. The top diagram represents a base model that was used to fit experimental traces in the absence of the toxins. The other models were used in fitting traces obtained in the presence of OD1. Transition rates derived from the base model were fixed (red arrows). (B) Examples of fit by base model (top traces) and model 4 (bottom). Experimental whole-cell Na+ currents evoked by step depolarization from −100 mV to −20 mV are in shown red and those calculated from the best fit model are in black.
To evaluate this approach, we used whole-cell experimental traces recorded in the absence of toxin under voltage-clamp conditions at a holding potential of −100 mV and a depolarizing step to −20 mV. This aimed to make sure that, at the beginning of depolarization, Na channels were mostly in a ‘resting’ state R, and depolarizations were sufficiently strong to activate most of the channel populations. Despite a substantial reduction in the number of free parameters compared with previous studies,21 our base model adequately described the experimental data (Fig. 6B, upper panel).
Given our previous observations on toxin effects on steady-state channel activation (see above), we further assumed that activation pathways were not significantly affected by the toxins22 and placed constraints on the pathways responsible for transitioning channels from closed to open states, while leaving the others that are associated with transitioning channels to inactivated states free floating (Table 3).
Table 3.
Transition rates (ms−1) and logarithm error ratios for different kinetic models. Rate constants for models 1–4 which were constrained to those of ‘base’ model are given in red.
| Model |
|||||
|---|---|---|---|---|---|
| Rate | Base | M1 | M2 | M3 | M4 |
| k12 | 4.76 | 4.76 | 4.764 | 4.75 | 4.75 |
| k23 | 4.54 | 4.54 | 4.54 | 4.54 | 4.54 |
| k24 | 0.243 | 0.243 | 0.243 | 3.51 | – |
| k32 | 0 | – | – | – | – |
| k34 | 4.42 | 1.17 | 4.42 | 4.30 | 4.30 |
| k42 | – | – | – | 0.563 | – |
| k43 | 0.471 | 0.472 | 1.22 | 0.784 | 1.24 |
| k45 | 0.716 | 0.161 | 0.173 | 0.194 | 0.174 |
| k54 | 0.0116 | 0.00663 | 0.00632 | 0.00625 | 0.00634 |
| LER | – | 0.205 | 0.0123 | 0 | 0.0255 |
The boundaries of constraints were chosen according to the rule: , where is matrix of rate constants derived from the base model, and is deviation that comes from differences between experimental traces used in base model simulations. By doing so, we kept constrained rate constants close to those derived from the base model (Table 3).
To compare the different models (M1–M4) that describe the toxins’ actions, we used a logarithm error ratio (LER) used previously.21,23 Based on the LER value, model M3 best fit the experimental traces. Models that allowed transition from state C2 to state I4 in one direction and/or unconstrained transitions from state O3 to state I4 did not yield realistic values of corresponding transition rates.
Discussion
Similar to our previous observations,16 OD1 and its analogs dramatically slowed the time course of the current decay phase. This apparent effect on channel inactivation is similar to that of α-scorpion toxins and has been convincingly linked to binding of the neurotoxin to site 3 on VGSCs, which comprises extracellular loops of the domain IV voltage sensor.5-7 According to the voltage-sensor trapping model, α-scorpion toxins trap the S4 transmembrane segment of the domain IV voltage sensor in the inward position and obstruct its outward movement, preventing fast inactivation.24 Single-channel recordings revealed that in the presence of toxins, channels rapidly fluctuate between open and closed states for relatively long intervals of time. On the other hand, the toxin did not affect the mean open time of the channel. This ‘flickering’ channel activity may explain the prolonged decay phase of the whole-cell Na+ current. The similar behavior of macroscopic currents in neuroblastoma cells affected by the α-scorpion toxin resulted from an increase in channel open time and channel reopenings.25
In addition to slowing down whole-cell current inactivation, OD1 and analogs affected the time course of the channels’ recovery from fast inactivation. In the absence of toxins, the time course of recovery followed a single exponential function, whereas an additional exponential component emerged in the presence of toxins. The time constant of the additional component was almost 5 times smaller than the recovery time constant in control conditions, and may be due to the number of channels that did not reach an inactivated state.
In terms of the Markov model, the trapping mechanism may be caused by an increase in the rate of transition from state C2 to state I4. On the other hand, the rate constant for transition from state I4 to state O3 doubles in the presence of the toxin, making transition back to the open state more favorable. The transition rate from an open to inactivated state I4 which determines channel open time was conserved compared to that in the absence of a toxin. All of the models that we tested yielded a lower rate for the transition from the intermediate inactivated state I4 to absorbing state I5. In agreement with this mechanism, strong depolarizing pre-pulses force S4 in the DIV to move into the activated position. This might also affect the binding site for the toxin on the S3−S4 linker and abolish the toxin's effect.8 While the importance of DIV in the channel inactivation process is well established, its involvement in the channel activation process is less clear.
The steady-state, current-voltage relationship in the presence of OD1 and its analogs showed a minor, but significant, shift to more hyperpolarized potentials. This effect is less pronounced for Nav1.7 than it is for Nav1.6 or Nav1.4, which are also affected by OD1 in a similar manner.16 It could also suggest that the DIV voltage sensors of the various isoforms are not functionally equivalent and involved in channel activation to differing degrees. However, at this stage, we cannot rule out the possibility that additional high-affinity binding (for example on DII) is responsible for the dual α/β toxin effect.
In terms of reversibility of the toxin's action, the effect of OD1 on Nav1.7 channel inactivation differs from that of previous studies involving α-scorpion site 3 toxin Ts3 (Tityus serulatus) on Nav1.4,9,22 but with a similar effect on macroscopic current inactivation. Whereas OD1s effects on Nav1.7 are fully reversible after washout, Ts3 remains tightly bound to Nav1.4. These findings for wild-type OD1 prompted us to analyze the mechanism of action of 2 OD1 analogs, OD1-KPH and OD1-KPH-E55A, which carry 3 or 4 non-conservative amino acid substitutions, respectively, in a structural region known as the ‘NC domain’. Both mutant toxins showed significantly improved potency in terms of their ability to inhibit fast inactivation (Fig. 1B), but from a mechanistic perspective, were otherwise indistinguishable from the wild-type toxin. This suggests that the observed increase in potency is primarily a result of an increase in binding affinity. The potency of the OD1 analogs is one order of magnitude higher than that of wild-type OD1 but there was no statistically significant difference between the V0.5 values obtained in the presence of OD1 and its analogs.
Materials and methods
Preparation of OD1 analogs
OD1 and its triple (D9K, D10P, K11H) and quadruple mutants (D9K, D10P, K11H, E55A) (hereafter referred to as KPH and KPH-E55A, respectively) were prepared by chemical synthesis via ligation of 3 unprotected peptide segments, as described recently with minor modifications.16 One-pot chemical ligation was used to join the peptide fragments, which yielded the fully reduced 65-mer peptides. OD-KPH observed mass was 7219.2 ± 1.0 Da, calculated mass (average isotope composition) 7218.3 Da. OD1-KPH-E55A observed mass was 7160.1 ± 1.0 Da, calculated mass (average isotope composition) 7160.2 Da.
For in-vitro folding and disulfide bond formation, 10 mg (∼1.4 μmol) of the purified and fully reduced peptides were dissolved in 6 M GdmHCl at a concentration of 4.0 mg/mL. Folding was initiated by rapid 1:20 dilution of the peptide solution with folding buffer (50 mM sodium phosphate, 8 mM reduced glutathione (GSH), 1mM oxidized glutathione (GSSG), pH 7.5). The reaction was left stirring at room temperature for 4 hours. The mixture was filtered and purified by HPLC on a Zorbax C3 column (10x250, 3 μm, 300 Å). OD1 KPH observed mass was 7210.5 ± 1.0 Da, calculated mass (average isotope composition) was 7210.3 Da. Yield 3.9 mg (0.6 μmol, 42%). OD1-KPH-E55A observed mass was 7153.2 ± 1.0 Da, calculated mass (average isotope composition) was 7152.2 Da. Yield: 3.2 mg (0.4 μmol, 29%).
Electrophysiology
CHO cells stably expressed with human Nav1.7 channels (Genionics AG, Zürich, Switzerland) were cultured as described previously16 and plated on round coverslips 1–4 d before experiments. Whole-cell Na+ currents were recorded and filtered at 10 kHz using Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) under voltage clamp conditions. Single-channel currents were filtered at 5kHz with a 24 dB/octave filter (Ithaco). The currents were digitized at 100 kHz using a Digidata 1400 interface and analyzed using pCLAMP 10.3 software package (Molecular Devices). Recording electrodes in whole-cell experiments were pulled from TG150TF borosilicate glass capillaries (Harvard Apparatus) and had resistance of 1.2–1.6 MΩ when filled with a solution containing (mM): 130 CsF, 10 CsCl, 10 EGTA, 1 MgCl2 and 10 HEPES-CsOH, pH 7.2. The extracellular solution contained (mM): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose and 10 HEPES-NaOH, pH 7.3. Membrane potential was corrected for a calculated liquid junction potential of 6.9 mV. A series resistance of 2.7 ± 0.6 MΩ was compensated by 85–90%. Capacitive and leakage currents were digitally subtracted using a P/8 protocol with hyperpolarizing conditioning pulses. Single-channel recordings were performed in a outside-out configuration. Recording electrodes were pulled from aluminosilicate glass capillaries and had a resistance of 6–10 MΩ when filled with an intracellular solution.
Data analysis
The inactivation rate was estimated by the ratio of the current measured at 1.5 ms after the peak (I1.5) to the peak current value (Ip). Toxin concentration–response relationships were defined as a function of I1.5/Ip against concentration, and were fitted with a logistic equation: , where C is toxin concentration; R0, R1 are asymptotic values for I1.5/Ip ratio. Conductance was calculated as: ,where Vr is the reversal potential estimated by fitting current–voltage relationships with the equation:
, where V1/2 is the potential at which current peak reaches its half-maximum value. Voltage-dependence of activation and inactivation data were fitted by a Boltzmann function:. Single-channel amplitude was estimated by fitting the amplitude histogram of elementary events with Gauss distribution. All non-linear regression analysis was performed using SigmaPlot 8.0 software. Results are shown as the mean ± standard error of the mean.
Kinetic modeling
To find the parameters of a kinetic model that give the best fit to whole-cell experimental current traces, we used a custom-developed application written in a MATLAB R2007b environment. In this approach, we resolved the optimization problem for the function , where is the matrix of transition rates, is a vector of n current samples derived from model equations and is vector of sampled experimental whole-cell current. The problem was resolved by using a genetic algorithm minimizer with constraints – ga, and/or gradient descent minimizer with constraints – fmincon (MATLAB). Differential equations associated with a kinetic model were numerically solved using ode45 solver (MATLAB).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by a National Health and Medical Research Council (NHMRC) Program grant (569927; D.J.A.). D.J.A. is an Australian Research Council (ARC) Australian Professorial Fellow.
References
- [1]. Goldin AL. Diversity of mammalian voltage-gated sodium channels. Ann N Y Acad Sci 1999; 868:38-50; PMID:10414280; http://dx.doi.org/ 10.1111/j.1749-6632.1999.tb11272.x [DOI] [PubMed] [Google Scholar]
- [2]. Catterall WA, Goldin AL, Waxman SG. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 2005; 57(4):397-409; PMID:16382098; http://dx.doi.org/ 10.1124/pr.57.4.4 [DOI] [PubMed] [Google Scholar]
- [3]. Lampert A, Dib-Hajj SD, Tyrrell L, Waxman SG. Size matters: erythromelalgia mutation S241T in Nav1.7 alters channel gating. J Biol Chem 2006; 281(47):36029-35; PMID:17008310; http://dx.doi.org/ 10.1074/jbc.M607637200 [DOI] [PubMed] [Google Scholar]
- [4]. Cregg R, Laguda B, Werdehausen R, Cox JJ, Linley JE, Ramirez JD, Bodi I, Markiewicz M, Howell KJ, Chen YC, et al. Novel mutations mapping to the fourth sodium channel domain of Nav1.7 result in variable clinical manifestations of primary erythromelalgia. Neuromolecular Med 2013; 15(2):265-78; PMID:23292638; http://dx.doi.org/ 10.1007/s12017-012-8216-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Cestèle S, Catterall WA. (2000) Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000;82:883-92; PMID:11086218; http://dx.doi.org/ 10.1016/S0300-9084(00)01174-3 [DOI] [PubMed] [Google Scholar]
- [6].Bosmans F, Tytgat J. Voltage-gated sodium channel modulation by scorpion a-toxins. Toxicon 2007; 49:142-58; PMID:17087986; http://dx.doi.org/ 10.1016/j.toxicon.2006.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]. Gurevitz M. Mapping of scorpion toxin receptor sites at voltage-gated sodium channels. Toxicon 2012; 60:502-11; PMID:22694883; http://dx.doi.org/ 10.1016/j.toxicon.2012.03.022 [DOI] [PubMed] [Google Scholar]
- [8]. Rogers JC, Qu YS, Tanada TN, Scheuer T, Catterall WA. Molecular determinants of high affinity binding of a-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel a subunit. J Biol Chem 1996; 271(27):15950-62; PMID:8663157; http://dx.doi.org/ 10.1074/jbc.271.27.15950 [DOI] [PubMed] [Google Scholar]
- [9]. Campos FV, Coronas FI, Beirão PS. Voltage-dependent displacement of the scorpion toxin Ts3 from sodium channels and its implication on the control of inactivation. Br J Pharmacol. 2004; 142(7):1115-22; PMID:15249424; http://dx.doi.org/ 10.1038/sj.bjp.0705793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. Leipold E, Hansel A, Olivera BM, Terlau H, Heinemann SH. Molecular interaction of d-conotoxins with voltage-gated sodium channels. FEBS Lett. 2005; 579(18):3881-4; PMID: 15990094; http://dx.doi.org/ 10.1016/j.febslet.2005.05.077 [DOI] [PubMed] [Google Scholar]
- [11]. Klint JK, Senff S, Rupasinghe DB, Er SY, Herzig V, Nicholson GM, King GF. Spider-venom peptides that target voltage-gated sodium channels: pharmacological tools and potential therapeutic leads. Toxicon 2012; 60(4):478-91; PMID:22543187; http://dx.doi.org/ 10.1016/j.toxicon.2012.04.337 [DOI] [PubMed] [Google Scholar]
- [12]. Xiao Y, Blumenthal K, Jackson JO, 2nd, Liang S, Cummins TR. The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol Pharmacol 2010; 78(6):1124-34; PMID:20855463; http://dx.doi.org/ 10.1124/mol.110.066332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Xiao Y, Blumenthal K, Cummins TR. Gating-pore currents demonstrate selective and specific modulation of individual sodium channel voltage-sensors by biological toxins. Mol Pharmacol 2014; 86(2):159-67; PMID:24898004; http://dx.doi.org/ 10.1124/mol.114.092338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. Jalali A, Bosmans F, Amininasab M, Clynen E, Cuypers E, Zaremirakabadi A, Sarbolouki MN, Schoofs L, Vatanpour H, Tytgat J. OD1, the first toxin isolated from the venom of the scorpion Odonthobuthus doriae active on voltage-gated Na+ channels. FEBS Lett 2005; 579(19):4181-6; PMID:16038905; http://dx.doi.org/ 10.1016/j.febslet.2005.06.052 [DOI] [PubMed] [Google Scholar]
- [15]. Maertens C, Cuypers E, Amininasab M, Jalali A, Vatanpour H, Tytgat J. Potent modulation of the voltage-gated sodium channel Nav1.7 by OD1, a toxin from the scorpion Odonthobuthus doriae. Mol Pharmacol 2006; 70(1):405-14; PMID:16641312 [DOI] [PubMed] [Google Scholar]
- [16]. Durek T, Vetter I, Wang CIA, Motin L, Knapp O, Adams DJ, Lewis RJ, Alewood PF. Chemical engineering and structural and pharmacological characterization of the a-scorpion toxin OD1. ACS Chem Biol 2013; 8:1215-22; PMID:23527544; http://dx.doi.org/ 10.1021/cb400012k [DOI] [PubMed] [Google Scholar]
- [17]. Aldrich RW. Voltage-dependent gating of sodium channels: towards an integrated approach. TINS 1986; 9:82-6 [Google Scholar]
- [18]. Hamill OP, Marty A, Neher E, Sakmann B, Sigworth F. Improved patch clamp techniques for high current resolution from cells and cell-free membrane patches. Pflugers Arch. 1981; 391:85-100; PMID:6270629; http://dx.doi.org/ 10.1007/BF00656997 [DOI] [PubMed] [Google Scholar]
- [19]. Horn R, Vandenberg CA, Lange K. Statistical analysis of single sodium channels. Effects of N-bromoacetamide. Biophys J 1984; 45(1):323-35; PMID:6324912; http://dx.doi.org/ 10.1016/S0006-3495(84)84158-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]. Zilberter YI, Motin LG. Existence of two fast inactivation states in cardiac Na channels confirmed by two-stage action of proteolytic enzymes. Biochim Biophys Acta 1991; 1068(1):77-80; PMID:1654106; http://dx.doi.org/ 10.1016/0005-2736(91)90063-E [DOI] [PubMed] [Google Scholar]
- [21]. Gurkiewicz M, Korngreen A, Waxman SG, Lampert A. Kinetic modeling of Nav1.7 provides insight into erythromelalgia-associated F1449V mutation. J Neurophysiol 2011; 105:1546-57; PMID:21289137; http://dx.doi.org/ 10.1152/jn.00703.2010 [DOI] [PubMed] [Google Scholar]
- [22]. Campos FV, Chanda B, Beirão PSL, Bezanilla F. α-Scorpion toxin impairs a conformational change that leads to fast inactivation of muscle sodium channels. J Gen Physiol 2008; 132(2):251-63; PMID:18663133; http://dx.doi.org/ 10.1085/jgp.200809995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23]. Horn R. Statistical methods for model discrimination . Applications to gating kinetics and permeation of the acetylcholine receptor channel. Biophys J 1987; 51(2):255-63; PMID:2435330; http://dx.doi.org/ 10.1016/S0006-3495(87)83331-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang J, Yarov-Yarovoy V, Kahn R, Gordon D, Gurevitz M, Scheuer T, Catterall WA. Mapping the receptor site for a-scorpion toxins on a Na+ channel voltage sensor. Proc Natl Acad Sci U S A 2011; 108(37):15426-31; PMID:21876146; http://dx.doi.org/ 10.1073/pnas.1112320108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Mozhaeva GN, Naumov AP, Kuryshev YA, Nosyreva ED. Some properties of sodium channels in neuroblastoma cells modified with scorpion toxin and chloramine-T. Single channel measurements. Gen Physiol Biophys 1990; 9:3-18; PMID:2155849 [PubMed] [Google Scholar]