

ABSTRACT
Multidrug resistance poses grand challenges to the effective treatment of infectious diseases and cancers. Integral membrane proteins from the multidrug and toxic compound extrusion (MATE) family contribute to multidrug resistance by exporting a wide variety of therapeutic drugs across cell membranes. MATE proteins are conserved from bacteria to humans and can be categorized into the NorM, DinF and eukaryotic subfamilies. MATE transporters hold great appeal as potential therapeutic targets for curbing multidrug resistance, yet their transport mechanism remains elusive. During the past 5 years, X-ray structures of 4 NorM and DinF transporters have been reported and guided biochemical studies to reveal how MATE transporters extrude different drugs. Such advances, although substantial, have yet to be discussed collectively. Herein I review these structures and the unprecedented mechanistic insights that have been garnered from those structure-inspired studies, as well as lay out the outstanding questions that present exciting opportunities for future work.
Keywords: cation binding, membrane transporter, multidrug efflux inhibitor, multidrug resistance, substrate recognition
Multidrug resistance has emerged as a major obstacle to the effective treatment of infectious diseases and cancers, and exerts devastating human and economic tolls.1,2 As the current pace of drug discovery is woefully inadequate to address the relentless rise in multidrug resistance, we will remain on the losing side of the war unless we fully understand the molecular mechanisms underlying multidrug resistance and how they can be tackled. A major mechanism underlying multidrug resistance is mediated by multidrug transporters, which are integral membrane proteins that can remove cytotoxic chemicals from cells.1 Among the known multidrug transporters are the multidrug and toxin extrusion (MATE) proteins, which can function as molecular pumps to flush various drugs out of cells and confer multidrug resistance.3-5 Over the past 5 years, major progress has been made toward illuminating how MATE transporters function and how they can be inhibited, but such progress has yet to be discussed in one setting. Here I present such research advances and focus on what we have learned about how MATE transporters extrude structurally and chemically distinct drugs, as well as the ramifications of these new discoveries in battling multidrug resistance.
MATE transporters
To date, at least 5 families of multidrug transporters have been identified: the ABC (ATP-binding cassette) family, the MATE family, the MFS (major facilitator superfamily), the RND (resistance-nodulation-division) family and the SMR (small multidrug resistance) family.1 Among them, the ABC multidrug transporters are primary membrane transport proteins which are powered by ATP hydrolysis. The remaining 4 families of multidrug transporters are all secondary transporters that utilize either the transmembrane H+ or Na+ electrochemical gradient to export drugs. In particular, the MFS, RND and SMR multidrug transporters typically employ the preexisting H+ gradient to extrude drugs across cell membrane, whereas the MATE transporters seem to be more functionally versatile and can use either the H+ or Na+ electrochemical gradient to export drugs.4,5
Based on their amino-acid sequence similarity, the ˜900 MATE transporters identified thus far can be separated into the NorM, DinF (DNA damage-inducible protein F) and eukaryotic subfamilies.3 Members of the NorM and DinF subfamilies can utilize either the H+ or Na+ electrochemical gradient, while the eukaryotic MATE proteins appear to be H+-dependent.6-13 Furthermore, both the NorM and DinF subfamilies contain eubacterial and archaeal members, although they typically share rather low amino-acid sequence similarity. Like the multidrug transporters from the other 4 families, MATE transporters can extrude polyaromatic drugs that carry positive charges at physiological pH, although the exported drugs often exhibit rather diverse chemical structures and properties.4,5
Notably, many characterized MATE transporters seem incapable of exporting compounds that carry negative charges. As such, MATE transporters are “poly-specific,” rather than non-specific. Significantly, since bacterial and human MATE transporters can export various antibiotics, anti-cancer and anti-diabetic drugs, they are promising therapeutic targets for tackling multidrug resistance in pathogens as well as managing drug-drug interactions in humans.4,5 Furthermore, elucidating how MATE transporters can recognize and extrude chemically and structurally dissimilar drugs will substantially advance our understanding of the molecular mechanisms underlying multidrug resistance in general.1
Cation-bound NorM-VC structure
To understand how MATE transporters work, their molecular structures are essential. The first MATE transporter structure, that of a NorM transporter from Vibrio cholera (NorM-VC), was reported in 2010.14 The 3.65-Å resolution X-ray structure of NorM-VC captures the transporter in an extracellular-facing, substrate-free state, and reveals the arrangement of 12 transmembrane helices (TM1-TM12) (Fig. 1). As viewed from the membrane, the NorM-VC structure displays a “V” shape, with each arm of the “V” being composed of 6 TMs, separating the transporter into 2 similarly-folded domains: the N (TM1-TM6) and C domain (TM7-TM12).
Figure 1.

Structure of cation-bound NorM-VC. (A) NorM-VC structure as viewed from the membrane plane. (B) The periplasmic view of the NorM-VC structure. The N and C domains of NorM-VC are colored cyan and yellow, respectively. Rb+ is drawn as a green sphere. Relevant transmembrane helices are numbered.
The N and C domains in NorM-VC exhibit a pseudo-twofold symmetry along an axis that is perpendicular to the membrane bilayer, and the structures of the 2 domains can be overlaid onto each other to yield a root mean squared deviation of <3 Å. Furthermore, the N and C domains have the same membrane topology, with both of their amino and carboxyl termini projecting into the cytoplasm. This type of “parallel” arrangement of 2 symmetry-related domains can also be found in membrane transporters belonging to the MSF family.15,16 Similar to the MATE transporters, MSF proteins typically consist of 12 TMs as well. However, this similarity is the point at which the architectural resemblance between the 2 transporter families ends, as the structural fold of NorM-VC is different from that of a MSF transporter.14
NorM-VC couples the efflux of various cytotoxic compounds to the influx of cation (i.e., Na+), and the cation-binding site was located within the C domain of the transporter, as revealed by the 4.2-Å resolution structure of NorM-VC bound to Rb+, a Na+ analog.14 Based on the Rb+-bound NorM-VC structure, a number of amino acids were suggested to coordinate Na+. Furthermore, the alanine or asparagine substitution of NorM-VCD371, abolished the binding of NorM-VC to Rb+ or Cs+, yet another Na+ analog.
However, likely owing to the low resolution limits of the Rb+-bound structure, the cation-coordination arrangement in NorM-VC is poorly established. Among all the amino acids that were implicated in cation binding, only NorM-VCY367, which is semi-conserved in the NorM subfamily, is positioned close enough to make plausible coordination to Rb+. As such, the NorM-VC structure yields little insight into how a MATE transporter binds Na+, or more importantly, how it catalyzes drug-Na+ exchange. It should also be noted that, although Rb+ and Cs+ are similar to Na+, they cannot support the drug efflux mediated by characterized MATE transporters. Nevertheless, Rb+ or Cs+ has often been used to localize the Na+-binding sites in membrane transporters since Na+ ions are not visible at moderate resolutions (3.5 Å or worse).
Substrate-bound NorM-NG structure
Mechanistic insights into MATE-mediated multidrug transport can only come from the structure of a MATE transporter captured in its substrate-bound state. In 2013, the structures of a substrate-bound NorM transporter, that from Neisseria gonorrhea (NorM-NG), were published.17 The NorM-NG structures, determined up to 3.5 Å resolutions, all portray the transporter in its extracellular-facing, substrate-bound state and reveal, for the first time, how a MATE transporter interacts with its polyaromatic and cationic substrates.
The overall structure of NorM-NG, which is composed of 12 TMs, bears remarkable resemblance to that of NorM-VC.17 Near the center of the membrane bilayer, the N and C domains of NorM-NG diverge and point away from one another toward the extracellular space, giving rise to an overall “V”-shaped structure as viewed from the membrane (Fig. 2). Similar to their counterparts in NorM-VC, the N and C domains in NorM-NG are related by a pseudo-twofold symmetry around the membrane normal. Furthermore, a central cavity is found at the interface between the symmetry-related N and C domains.17 In the extracellular-facing NorM-NG structure, the bottom of the central cavity is well-sealed and shielded from the cytoplasmic side of the membrane by highly ordered protein structure, ˜20 Å thick. By contrast, the cavity is only partly closed toward the extracellular space by 2 flexible, extracellular loops, which may enable ions and/or solvent molecules to diffuse freely into the central cavity.17
Figure 2.

Structure of substrate-bound NorM-NG. The structure is viewed from the membrane plane, and the views in (A) and (B) are related by 180° rotation around the membrane normal. The N and C domains of NorM-NG are colored cyan and yellow respectively. Monobody, a crystallization chaperone, is drawn as a magenta ribbon. Bound substrate is shown as magenta sticks.
The substrate-binding site of NorM-NG was established based on the Na+-free structures of the transporter in complexes with 3 different substrates.17 These structures suggest that the central cavity between the N and C domains constitutes the multidrug- or substrate-binding site in NorM-NG and shed light on how a MATE transporter binds and recognizes chemically and structurally dissimilar substrates (Fig. 3). In the structures of NorM-NG, ˜30% surface area of the bound substrates remains accessible to the solvent from the extracellular side of the membrane.17 Therefore, these structures capture the transporter in an outward-open, substrate-bound state, likely portraying a state in which NorM-NG is poised to release the drug into the periplasm.
Figure 3.

Structure of the multidrug-binding site in NorM-NG. (A), Structure of NorM-NG (in ribbon rendition) as viewed from the membrane plane and colored as in Figure 2. (B, C and D) Close-up views of the binding site for TPP, ethidium and R6G, respectively. Relevant amino acids are drawn as stick models and labeled.
In the NorM-NG structures, the 3 different substrates occupy similar locations in the central cavity and make numerous close-range interactions with the amino acids from both the N and C domains (Fig. 3). The functional importance of these drug-binding amino acids is further supported by site-directed mutagenesis and biochemical studies.17 Strikingly, NorM-NG makes substantially more ionic and H-bonding contacts than van der Waals interactions with the cationic and lipophilic substrates. This observation is unexpected since it has been generally accepted that hydrophobic interactions, mediated by aromatic amino acids, dominate the contacts between multidrug transporters and their drug substrates.18,19 The unusual multidrug-binding site thus implies that NorM-NG has evolved a different transport mechanism from other known multidrug transporters.
Cation-bound NorM-NG structure
Besides the Na+-free, substrate-bound structures of NorM-NG, the structure of a Cs+-bound NorM-NG was also determined to 3.7 Å resolutions.17 The structure of Cs+-bound NorM-NG is very similar to that of substrate-bound NorM-NG, apparently because a yet-unidentified ligand served as a surrogate substrate to stabilize the protein in its substrate-bound form, and/or the crystallization packing interactions favored the substrate-bound conformation.17 Furthermore, the cation-bound NorM-NG structure provides new insights into the Na+-coordination arrangement in this Na+-coupled MATE transporter and suggests a molecular mechanism that explains how Na+ triggers the release of drugs into the periplasm.17
Specifically, the cation-bound NorM-NG structure implicates one aromatic (NorM-NGY294) and 2 carboxylate amino acids (NorM-NGE261 and NorM-NGD377) in Na+-coordination (Fig. 4).17 These 3 amino acids are highly conserved in the NorM and eukaryotic subfamilies (Fig. 5), implying that the transport mechanism may be somewhat conserved between these 2 subfamilies of MATE transporters. Furthermore, those 3 amino acids and their counterparts in another bacterial NorM transporter were found to be functionally indispensable for multidrug efflux, further attesting to the biological relevance of the cation-bound structure of NorM-NG.17,20
Figure 4.

Cation-induced conformational changes in NorM-NG. (A) Structural overlay of substrate-bound NorM-NG (cyan and yellow) and substrate-free NorM-VC (gray). Bound substrate in NorM-NG is drawn as magenta sticks, relevant transmembrane helices are numbered. Red arrows high light the movement of TM7 and TM8 relative to TM10. (B) Close-up view of the cation-binding site in NorM-NG. Cs+ is drawn as a green sphere and overlaid with a difference Fourier map (magenta mesh). A substrate taken from the drug-bound structure of NorM-NG is shown in magenta sticks to indicate the location of the multidrug-binding site. Relevant amino acids are drawn as stick models and labeled. (C) The suggested rearrangement of Na+-coordination in NorM-NG during transport. Na+ is shown as a gray sphere and relevant amino acids are drawn as stick models and labeled.
Figure 5.

Sequence alignment of representative MATE transporters. Residues that are conserved among the 5 MATE proteins are colored magenta. Regions of secondary structural elements are outlined, with every 10th residue marked. Red and blue dots highlight amino acids that likely bind cations in DinF and NorM transporters, respectively. Residues 1 in PfMATE and NorM-NG, residues 1-20 in eukaryotic hMATE1 are omitted for clarity. Notably, the H+-coupled hMATE1 bears the cation-binding amino acids as found in NorM-NG and NorM-VC (blue dots), while lacking the 2 aspartates (red dots) as seen in DinF-BH and PfMATE.
Notably, NorM-NGY294 seems to coordinate Na+ through a cation-π interaction, offering the first piece of structural evidence that a membrane protein can make a Na+-π interaction. It also seems likely that the Na+-π interaction enables NorM-NG to selectively as well as dynamically bind Na+ during transport.17 Moreover, Na+-π interaction has also been suggested for yet another secondary transporter, BetP, a Na+-coupled symporter.21 As such, Na+-π interaction may prove to be an important element in Na+-binding by membrane transporters, including both antiporters (NorM-NG) and symporters.
Perhaps surprisingly, the cation-bound NorM-NG structure strongly suggests that the transporter utilizes distinct subsets of amino acids to interact with cation and drugs, and that it can bind them simultaneously.17 Therefore, the coupling between Na+ and drug is likely indirect and mediated by protein conformational changes in NorM-NG, likely involving the rearrangement of cation-bound transmembrane helices (Fig. 4). As such, the cation-bound NorM-NG structure corresponds to a fully-loaded state of the MATE transporter and supports an unconventional allosteric coupling mechanism (Fig. 6). Under this scenario, Na+ elicits the release of drugs from NorM-NG by shifting 2 Na+-binding transmembrane helices (TM7 and TM8), which also form part of the multidrug-binding site (Fig. 3). This finding sets a new conceptual framework in the transporter field, as it starkly contrasts the mechanism proposed for other antiporters including EmrE and NhaA, which states that an antiporter can bind either the counter-transported cation or substrate, but not to both at the same time.22,23
Figure 6.

Proposed antiport mechanism for NorM-NG. Briefly, Na+ (green circle) binds to a cation-free, drug-bound, extracellular-facing NorM-NG (pink) and triggers the movement of TM7 and TM8 (red arrow) in the cation-bound, drug-bound state, causing the drug (red) to be dissociated from NorM-NG. The cation-bound, drug-free NorM-NG then switches to an intracellular-facing conformation (gray) to intercept another drug from the cytoplasm. Drug binding subsequently induces the release of Na+ into the cytoplasm, and the protein returns to the drug-bound, extracellular-facing state. The known structures (specified by their PDB codes) correspond to the 3 extracellular-facing states. TM1 and TM2 are simplified as a thick cyan stick, TM7 and TM8 as a thick yellow stick, and TM10 as a thin yellow stick, respectively.
PfMATE structures
After the NorM-NG structures were reported, several crystal structures of the H+-coupled PfMATE were determined at 2.4 to 3.0 Å resolutions and published.24 PfMATE belongs to the DinF subfamily of MATE transporters and lacks the amino acids that constitute the cation-binding site in NorM-NG (Fig. 5). Despite this difference, the overall structure of PfMATE is similar to that of NorM-NG or NorM-VC. Among the published PfMATE structures are those determined at low (6.0-6.5) and high (7.0-8.0) pH, with the most significant difference between the 2 residing in TM1 (Fig. 7).
Figure 7.

Structures of PfMATE obtained at high and low pH. The high (A) and low (B) pH structures are viewed from the periplasm, respectively. N and C domains of PfMATE are colored cyan and yellow, respectively, except in the low pH structure, in which the extracellular half of TM1 is colored red to highlight the kinked helix (B).
Specifically, in the low pH structure, the TM1 in PfMATE is bent near the center of the helix, whereas TM1 remains unbent, or straight, in the high pH structure (Fig. 7). Since the TM1 is also unbent in the structure of PfMATE in complex with a substrate analog, a mechanism was put forward to suggest that the protonation of an aspartate in TM1 (PfMATED41) triggers the bending of TM1 and gives rise to the release of substrate from the transporter.24 Apparently, this mechanism is based on the untested assumption that PfMATED41 is protonated in the low pH structure but deprotonated in the high pH form.
However, in both the low and high pH structures, the calculated pKa of PfMATED41 is lower than 4, suggesting that in these 2 structures, PfMATED41 is always deprotonated.25 Furthermore, the TM1-bending-based mechanism is unlikely applicable to H+-dependent eukaryotic MATE transporters, since their TM1 generally lacks a protonatable amino acid (Fig. 5). Furthermore, the TM1 is unbent in the cation-bound structure of NorM-VC or NorM-NG, arguing that the TM1-bending is unlikely a shared feature for cation-bound NorM transporters either.14,17
More recently, the structure of a protonated transporter from the DinF subfamily was reported, in which the TM1 is also unbent (see below).26 In light of the accumulated experimental data, it seems that the TM1-bending-based transport mechanism needs to be critically re-evaluated. Indeed, it is likely that the conformation of TM1 in PfMATE, bent vs. straight, is affected by the pH-dependent interactions between the transporter and exogenous lipids, which were used to aid crystallization, rather than by the protonation or deprotonation of PfMATED41.26
Substrate-bound DinF-BH structure
Shortly after the PfMATE structures were published, the crystal structures of yet another H+-coupled DinF transporter, that from Bacillus halodurans (DinF-BH), were reported.25 Perhaps unexpectedly, the 3.7-Å resolution substrate-bound and 3.2-Å resolution substrate-free DinF-BH structures both reveal an asymmetric arrangement of the 12 transmembrane helices (Fig. 8). The pseudo-twofold symmetry is broken in DinF-BH by the kinking of 2 helices (TM7 and TM8) and is likely induced by proline residues.25 By contrast, the structure of NorM-VC, NorM-NG and PfMATE all preserve a pseudo-2-fold symmetry between their N and C domains.17,24
Figure 8.

Structure of DinF-BH. The structure as viewed from the membrane (A) and from the periplasm (B). The N and C domains of DinF-BH are colored cyan and yellow, respectively, except for the extracellular halves of TM7 and TM8, which are colored red to highlight the asymmetric arrangement of the transmembrane helices. Relevant helices are numbered.
The structural asymmetry in DinF-BH creates a substrate-binding chamber as well as shields TM1 from the solvent, the latter of which may explain the upward shift in pKa for DinF-BHD40 (equivalent of PfMATED41).25 Within the substrate-binding chamber, DinF-BHD40 makes a single charge-charge interaction with a cationic substrate (Fig. 9). Furthermore, in the 3.0-Å resolution structure of a protonation-mimetic mutant of DinF-BH, DinF-BHD40N forms an H-bond with DinF-BHD184 (Fig. 10).26 Both DinF-BHD40 and DinF-BHD184 are essential for the transport function, and neutralization of DinF-BHD40 substantially reduced the substrate binding to DinF-BH.25,26 Taken together, these data support a direct-competition-based mechanism in which the protonation of DinF-BHD40 breaks the charge-charge interaction between DinF-BHD40N and substrate, thereby triggering drug release from the transporter (Fig. 11).25,26
Figure 9.

Structure of the substrate-binding site in DinF-BH. (A) The structure of drug-bound DinF-BH as viewed from the membrane plane, with the N and C domains colored cyan and yellow, respectively. The bound substrate is drawn as magenta sticks and overlaid with experimental electron density map (cyan wire). (B) Close-up view of the substrate-binding site. Relevant amino acids are drawn as stick models and labeled. Bound substrate is shown as magenta sticks.
Figure 10.

Structural basis for the direct competition in DinF-BH. (A and B) Close-up views of the H-bonding networks in DinF-BHD40N (A) and deprotonated DinF-BH (B), respectively. Relevant amino acids are drawn as stick models and H-bonding interactions are indicated by dotted-lines. Red arrow highlights the interaction between DinF-BHD40N and DinF-BHD184 (A).
Figure 11.
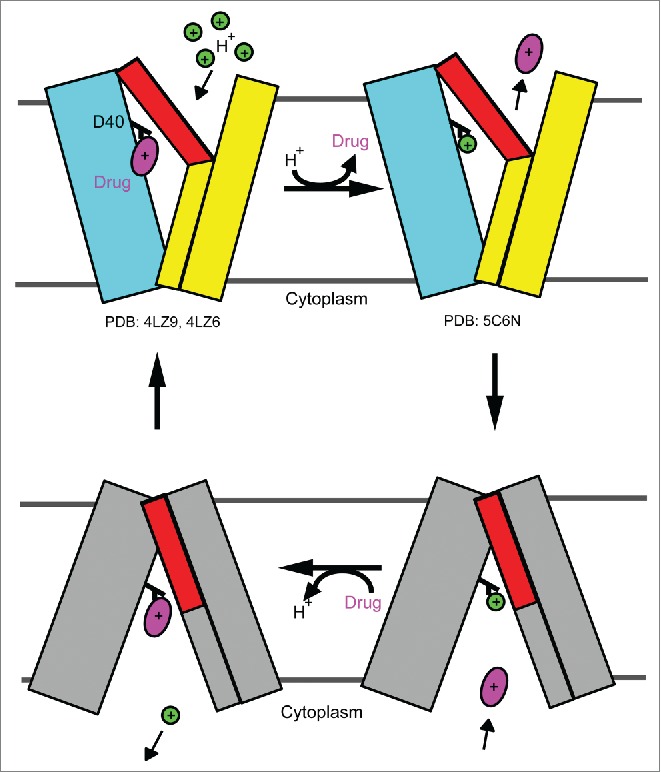
Proposed antiport mechanism for DinF-BH. In brief, protonation of DinF-BHD40 in the drug-bound, extracellular-facing transporter triggers the release of drug into the periplasm. The protonated, extracellular-facing DinF-BH then changes to the protonated intracellular-facing state. Drug binding to the transporter from the cytoplasm subsequently elicits the deprotonation of DinF-BHD40 and yields a drug-bound, intracellular-facing DinF-BH, which returns to the drug-bound, extracellular-facing state to complete the transport cycle. The N and C domains of the extracellular-facing DinF-BH are simplified as cyan and yellow rectangles, whereas those of the intracellular-facing transporter are colored gray, except for the extracellular portions of TM7 and TM8, which are in red. Drug and proton are drawn as a magenta oval and a green circle, respectively. DinF-BHD40 is colored in black and labeled. The known structures of DinF-BH (as specified by their PDB codes) portray the extracellular-facing states of the transporter.
This antiport mechanism markedly differs from that of NorM-NG and implies striking mechanistic divergence between the NorM and DinF transporters.25 Notably, DinF-BH lacks the cation-binding amino acids seen in NorM-NG and interacts with substrate differently from NorM-NG, which makes numerous charge-charge interactions with cationic drugs (Fig. 3). Furthermore, although eukaryotic MATE transporters are typically H+-dependent, they appear to be more similar to Na+-coupled NorM-NG than H+-dependent DinF-BH.25,26 Firstly, eukaryotic MATE transporters bear the cation-binding motif seen in NorM-NG, and hence may form a similar cation-binding site to that of NorM-NG (Fig. 5). Secondly, the counterparts for DinF-BHD184 in NorM-NG, NorM-VC and eukaryotic MATE transporters such as hMATE1 are asparagine residues, even although a DinF-BHD184N mutation abolished the transport activity of DinF-BH.26 As such, it is likely that NorM and eukaryotic MATE transporters have evolved transport mechanisms that are different from that of DinF proteins.26
Structures of verapamil-bound DinF-BH and NorM-NG
Most recently, the 3.0-Å resolution X-ray structures of DinF-BH and NorM-NG in complexes with verapamil were published.26 Verapamil is a currently marketed pharmaceutical, an ion-channel blocker as well as a broad-spectrum inhibitor of multidrug transporters.27-30 Verapamil can inhibit multidrug transporters from the ABC, MATE and MFS families, and given the structural differences among those multidrug transporters, it seems remarkable that verapamil can exert inhibitory effects on so many structurally and mechanistically different membrane transporters. The published structures of verapamil-bound MATE transporters thus provide the first glimpses into how this remarkable functional versatility can be accomplished by verapamil.
In the verapamil-bound DinF-BH and NorM-NG structures, verapamil occupies a location that is overlapping with the multidrug-binding sites.26 In vitro, verapamil can also inhibit the binding of radio-labeled substrate to either DinF-BH or NorM-NG.26 Therefore, verapamil appears to inhibit MATE-mediated multidrug efflux simply by preoccupying the multidrug-binding site, despite the structural and mechanistic differences between DinF-BH and NorM-NG.25 This inhibitory mechanism is reminiscent of that for the inhibitors of RND multidrug transporters.31 However, those RND inhibitors cannot be extruded by their cognate RND transporters, whereas both DinF-BH and NorM-NG can export verapamil.26,31
Despite a similar inhibitory mechanism, DinF-BH and NorM-NG make rather different molecular interactions with verapamil. Firstly, verapamil adopts different conformations within DinF-BH and NorM-NG. Specifically, verapamil displays an almost linear conformation in DinF-BH (Fig. 12), but assumes a rather folded, horse-shoe-shaped conformation in NorM-NG (Fig. 13), which may be important for maximizing the favorable interactions between the MATE transporter and verapamil. Secondly, the structure of DinF-BH remains largely unaltered upon binding verapamil, whereas the conformations of 2 extracellular loops in NorM-NG change significantly upon association with verapamil.26 These differences may reflect the mechanistic and structural difference between DinF-BH and NorM-NG, as well as the structural flexibility and functional versatility of verapamil. It is thus conceivable that verapamil also adopts different conformations upon binding other structurally and mechanistically distinct multidrug transporters.
Figure 12.

Structure of verapamil-bound DinF-BH. (A) Structure of verapamil-bound DinF-BH (ribbon) as viewed from the membrane plane. The N and C domains are colored cyan and yellow, respectively. Verapamil is drawn as magenta spheres. (B) Close-up view of the verapamil-binding site. Verapamil (magenta) and relevant amino acids are drawn as stick models and labeled.
Figure 13.

Structure of verapamil-bound NorM-NG. (A) Structure of NorM-NG in complex with verapamil as viewed from the membrane plane. NorM-NG is shown in ribbon rendition and its N and C domains are colored cyan and yellow, respectively. Verapamil is shown as magenta spheres. (B) Close-up view of the verapamil-binding site. Verapamil (magenta) and relevant amino acids are displayed as stick models and labeled.
Outlook
All the published structures depict the MATE transporters in their extracellular-facing states, in which the substrate- or multidrug-binding sites always open toward the periplasm. It is therefore unclear as to how the MATE transporters switch to their intracellular-facing conformations in order to acquire substrates from the cytoplasm. Although the intracellular-facing models of DinF-BH and NorM-NG have been suggested and are also consistent with available biochemical data, they both lack direct structural proof and are unlikely to be accurate in all the details.25 As such, the structure elucidation of an intracellular-facing MATE transporter would substantially advance our understanding of MATE-mediated multidrug extrusion.
Aside from the transport mechanism, little is known about the molecular basis for cation selectivity in a MATE transporter. Although the structures of cation-bound NorM-NG and DinF-BH are known, they give little insight into how a MATE transporter selects between Na+ and H+, as the cation-binding sites are located in entirely different domains within the NorM and DinF transporters.17,25 Therefore, to reveal the chemical basis of cation selectivity, it is essential to obtain the structures of an H+-coupled NorM and a Na+-coupled DinF, both trapped in their cation-bound states, in order to uncover the similarity and difference between the Na+- and H+-binding sites and to shed new light on cation selectivity.
Last but not least, based on amino-acid sequence analysis and available biochemical data, it is likely that the mechanism as well as structure of eukaryotic MATE transporters differ from those of DinF and NorM transporters.25,26 In order to understand how the structural similarity and divergence between eukaryotic and prokaryotic MATE transporters account for their unique functionalities, future efforts must also be made at investigating the structure and mechanism of eukaryotic MATE transporters.
In closing, although the past 5 years have witnessed tremendous progress made toward understanding how MATE transporters bind and extrude drugs, it remains largely unknown how a MATE transporter selects cations or captures drugs from the cytoplasm. These major gaps in our knowledge pose great challenges for unraveling the molecular underpinnings of multidrug transport, as well as spawn new and exciting opportunities for overcoming multidrug resistance.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
I thank my colleagues, particularly Jindrich Symersky, Martha Radchenko and Rongxin Nie, for making the research on MATE transporters possible in my laboratory. I also thank Kathy Aschheim (Nature Publishing Group) for discussions about this review.
Funding
The research in my laboratory is in part supported by the US National Institutes of Health (R01-GM094195), which is gratefully acknowledged.
References
- [1].Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007; 446:749-57; PMID:17429392; http://dx.doi.org/ 10.1038/nature05630 [DOI] [PubMed] [Google Scholar]
- [2].Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science 2009; 325:1089-93; PMID:19713519; http://dx.doi.org/ 10.1126/science.1176667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brown MH, Paulsen IT, Skurray RA. The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol Microbiol 1999; 31:394-5; PMID:9987140; http://dx.doi.org/ 10.1046/j.1365-2958.1999.01162.x [DOI] [PubMed] [Google Scholar]
- [4].Omote H, Miasa M, Matsumoto T, Otsuka M, Moroyama Y. The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol Sci 2006; 27:587-93; PMID:16996621; http://dx.doi.org/ 10.1016/j.tips.2006.09.001 [DOI] [PubMed] [Google Scholar]
- [5].Kuroda T, Tsuchiya T. Multidrug efflux transporters in the MATE family. Biochim. Biophys. Acta 2009; 1794:763-8 [DOI] [PubMed] [Google Scholar]
- [6].Morita Y, Kataoka A, Shiota S, Mizushima T, Tsuchiya T. NorM of Vibrio parahaemolyticus is an Na(+)-driven multidrug efflux pump. J Bacteriol 2000; 182:6694-7; PMID:11073914; http://dx.doi.org/ 10.1128/JB.182.23.6694-6697.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen J, Morita Y, Huda MN, Kuroda T, Mizushima T, Tsuchiya T. VmrA, a member of a novel class of Na(+)-coupled multidrug efflux pumps from Vibrio parahaemolyticus. J Bacteriol 2002; 184:572-6; PMID:11751837; http://dx.doi.org/ 10.1128/JB.184.2.572-576.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Long F, Rouquette-Loughlin C, Shafer WM, Yu EW. Functional cloning and characterization of the multidrug efflux pumps NorM from Neisseria gonorrhoeae and YdhE from Escherichia coli. Antimicrob Agents Chemother 2008; 52:3052-60; PMID:18591276; http://dx.doi.org/ 10.1128/AAC.00475-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].He GX, Kuroda T, Mima T, Morita Y, Mizushima T, Tsuchiya T. An H+-coupled multidrug efflux pump, PmpM, a member of the MATE family of transporters, from Pseudomonas aeruginosa. J Bacteriol 2004; 186:262-5; PMID:14679249; http://dx.doi.org/ 10.1128/JB.186.1.262-265.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Su XZ, Chen J, Mizushima T, Kuroda T, Tsuchiya T. AbeM, an H+-coupled Acinetobacter baumannii multidrug efflux pump belonging to the MATE family of transporters. Antimicrob Agents Chemother 2005; 49:4362-4; PMID:16189122; http://dx.doi.org/ 10.1128/AAC.49.10.4362-4364.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li L, He Z, Pandey GK, Tsuchiya T, Luan S. Functional cloning and characterization of a plant efflux carrier for multidrug and heavy metal detoxification. J Biol Chem 2002; 277:5360-8; PMID:11739388; http://dx.doi.org/ 10.1074/jbc.M108777200 [DOI] [PubMed] [Google Scholar]
- [12].Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, Moriyama Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci USA 2005; 102:17923-8; http://dx.doi.org/ 10.1073/pnas.0506483102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Masuda S, Terada T, Yonezawa A, Tanihara Y, Kishimoto K, Katsura T, Ogawa O, Inui K. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Sci Nephrol 2006; 17:2127-35; http://dx.doi.org/ 10.1681/ASN.2006030205 [DOI] [PubMed] [Google Scholar]
- [14].He X, Szewczyk P, Karyakin A, Evin M, Hong WX, Zhang Q, Chang G. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 2010; 467:991-4; PMID:20861838; http://dx.doi.org/ 10.1038/nature09408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science 2003; 301:610-5; PMID:12893935; http://dx.doi.org/ 10.1126/science.1088196 [DOI] [PubMed] [Google Scholar]
- [16].Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 2003; 301:616-20; PMID:12893936; http://dx.doi.org/ 10.1126/science.1087619 [DOI] [PubMed] [Google Scholar]
- [17].Lu M, Symersky J, Radchenko M, Koide A, Guo Y, Nie R, Koide S. Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc Natl Acad Sci USA 2013; 110:2099-104; http://dx.doi.org/ 10.1073/pnas.1219901110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen Y, Pornillos O, Lieu S, Ma C, Chen AP, Chang G. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci USA 2007; 104:18999-9004; http://dx.doi.org/ 10.1073/pnas.0709387104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, et al.. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009; 323: 1718-22; PMID:19325113; http://dx.doi.org/ 10.1126/science.1168750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Otsuka M, Yasuda M, Morita Y, Otsuka C, Tsuchiya T, Omote H, Moriyama Y. Identification of essential amino acid residues of the NorM Na+/multidrug antiporter in Vibrio parahaemolyticus. J Bacteriol 2005; 187:1552-8; PMID:15716425; http://dx.doi.org/ 10.1128/JB.187.5.1552-1558.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Khafizov K, Perez C, Koshy C, Quick M, Fendler K, Ziegler C, Forrest LR. Investigation of the sodium-binding sites in the sodium-coupled betaine transporter BetP. Proc Natl Acad Sci USA 2012; 109:E3035-44; http://dx.doi.org/ 10.1073/pnas.1209039109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Schuldiner S. Competition as a way of life for H(+)-coupled antiporters. J Mol Biol 2014; 426:2539-46; PMID:24862284; http://dx.doi.org/ 10.1016/j.jmb.2014.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Padan E. Functional and structural dynamics of NhaA, a prototype for Na(+) and H(+) antiporters, which are responsible for Na(+) and H(+) homeostasis in cells. Biochim Biophys Acta 2014; 1837:1047-62; PMID:24361841; http://dx.doi.org/ 10.1016/j.bbabio.2013.12.007 [DOI] [PubMed] [Google Scholar]
- [24].Tanaka Y, Hipolito CJ, Maturana AD, Ito K, Kuroda T, Higuchi T, Katoh T, Kato HE, Hattori M, Kumazaki K, et al.. Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature 2013; 496:247-51; PMID:23535598; http://dx.doi.org/ 10.1038/nature12014 [DOI] [PubMed] [Google Scholar]
- [25].Lu M, Radchenko M, Symersky J, Nie R, Guo Y. Structural insights into H+-coupled multidrug extrusion by a MATE transporter. Nat Struct Mol Biol 2013; 20:1310-7; PMID:24141706; http://dx.doi.org/ 10.1038/nsmb.2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Radchenko M, Symersky J, Nie R, Lu M. Structural basis for the blockade of MATE multidrug efflux pumps. Nat Commun 2015; 6:7995; PMID:26246409; http://dx.doi.org/ 10.1038/ncomms8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Miyamae S, Ueda O, Yoshimura F, Hwang J, Tanaka Y, Nikaido H. A MATE family multidrug efflux transporter pumps out fluoroquinolones in Bacteroides thetaiotaomicron. Antimicro Agents Chemother 2001; 45:3341-6; http://dx.doi.org/ 10.1128/AAC.45.12.3341-3346.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jonas BM, Murray BE, Weinstock GM. Characterization of emeA, a norA homolog and multidrug resistance efflux pump, in Enterococcus faecalis. Antimicro Agents Chemother 2001; 45:3574-9; http://dx.doi.org/ 10.1128/AAC.45.12.3574-3579.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Spoelstra EC, Westerhoff HV, Pinedo HM, Dekker H, Lankelma J. The multidrug-resistance-reverser verapamil interferes with cellular P-glycoprotein-mediated pumping of daunorrubicin as a non-competing substrate. Eur J Biochem 1994; 221:363-73; PMID:7909520; http://dx.doi.org/ 10.1111/j.1432-1033.1994.tb18748.x [DOI] [PubMed] [Google Scholar]
- [30].Ohta KY, Inoue K, Yasujima T, Ishimaru M, Yuasa H. Functional characteristics of two human MATE transporters: kinetics of cimetidine transport and profiles of inhibition by various compounds. J Pharm Pharm Sci 2009; 12:388-96; PMID:20067714 [DOI] [PubMed] [Google Scholar]
- [31].Nakashima R, Sakurai K, Yamasaki S, Hayashi K, Nagata C, Hoshino K, Onodera Y, Nishino K, Yamaguchi A. Structural basis for the inhibition of bacterial multidrug exporters. Nature 2013; 500:102-6 (2013); PMID:23812586; http://dx.doi.org/ 10.1038/nature12300 [DOI] [PubMed] [Google Scholar]