

Abstract
Purpose
The electroretinogram c-wave is generated by the summation of the positive polarity hyperpolarization of the apical RPE membrane and a negative polarity slow PIII response of Müller glia cells. Therefore, the c-wave reduction noted in prior studies of mouse models of diabetes could reflect a reduction in the RPE component or an increase in slow PIII. The present study used a genetic approach to distinguish between these two alternatives.
Methods
Nyxnob mice lack the ERG b-wave, revealing the early phase of slow PIII. To visualize changes in slow PIII due to diabetes, Nyxnob mice were given streptozotocin (STZ) injections to induce diabetes or received vehicle as a control. After 1, 2, and 4 weeks of sustained hyperglycemia (>250 mg/dL), standard strobe flash ERG and dc-ERG testing were conducted. Histological analysis of the retina was performed.
Results
A reduced c-wave was noted at the 1 week time point, and persisted at later time points. In comparison, slow PIII amplitudes were unaffected after 1 week of hyperglycemia, but were significantly reduced in STZ mice at the 2-week time point. The decrease in amplitude occurred before any identifiable decrease to the a-wave. At the later time point, the a-wave became involved, although the slow PIII reductions were more pronounced. Morphological abnormalities in the RPE, including increased thickness and altered melanosome distribution, were identified in diabetic animals.
Conclusions
Because the c-wave and slow PIII were both reduced, these results demonstrated that diabetes-induced reductions to the c-wave cannot be attributed to an early increase in the Müller glia-derived potassium conductance. Furthermore, because the a-wave, slow PIII and c-wave reductions were not equivalent, and varied in their onset, the reductions cannot reflect the same mechanism, such as a change in membrane resistance. The presence of small changes to RPE architecture indicate that the c-wave reductions present in diabetic mice likely represents a primary change in the RPE induced by hyperglycemia.
Keywords: retinal pigment epithelium, Müller glia, diabetic retinopathy, electroretinogram
Müller glia cells and the retinal pigment epithelium (RPE) are essential supporting cells within the neuroretina, providing architectural as well as functional support to retinal neurons. In rodents, Müller glia cells become reactive in response to diabetes,1–4 displaying elevated levels of GFAP, dedifferentiation as identified by mislocalization of Kir4.1 voltage-dependent potassium channels, and a decreased potassium conductance after prolonged periods (4+ months) of disease.5–7 Müller glia cells further contribute to the progression of diabetic retinopathy and neovascularization by secretion of angiogenic factors and cytokines and their own apoptosis.6,8–10
The RPE also displays significant functional defects as a result of diabetes, exhibiting perturbations in movement of water out of the subretinal space (SRS), ion and nutrient transport, secretion of cytokines and growth factors, and transcellular permeability, each of which occurs prior to the development of vision-threatening diabetic macular edema.11–20 It has further been suggested that RPE damage contributes to and may be predictive of progression to proliferative retinopathy.21,22
The functional properties of the RPE can be evaluated using the direct current-coupled electroretinogram (dc-ERG).23,24 The amplitude of the dc-ERG is reduced in diabetes.23,25–28 In evaluating the changes, it is important to recognize that the components of the dc-ERG waveform reflect the activity of multiple retinal cell types.24,29 For example, the c-wave is generated mainly by the summation of two opposite polarity signals. One is a positive polarity waveform that stems from the hyperpolarization of the apical membrane of the RPE, generated in response to a light-driven decrease in subretinal [K+] induced by rod photoreceptor activity.30,31 The other is a negative polarity waveform (termed “slow PIII”) that also reflects the hyperpolarization of Müller glial cells due to the decrease in subretinal [K+].32,33 These two waveforms add to define the c-wave recorded at the corneal surface.24,34 Both components are initiated by a common stimulus, the change in K+ due to light stimuli. Although the positive apical RPE response and slow PIII overlap substantially, slow PIII has a faster onset and a faster time-to-peak before being interrupted by the positive polarity RPE response. This was demonstrated by Lurie and Marmor,35,36 who used aspartate to remove the b-wave, and is also revealed in the ERG waveform of mouse b-wave mutants, where slow PIII follows the a-wave and reaches a steady state that is then interrupted by the onset of the positive-polarity RPE response. This is inconsistent with a model in which these components have identical kinetics. The faster kinetics of slow PIII has been attributed to its proximity to the rod photoreceptor inner segments.33,37 The net result is that the response properties of slow PIII can be measured in relative isolation by removing the b-wave and focusing on early time points.24,34,38
Since the c-wave reflects two underlying components, changes in either slow PIII or the apical RPE response will impact c-wave amplitude. In the absence of any other changes, an increase in slow PIII will result in a smaller c-wave, while a reduction in slow PIII leads to an increase in c-wave amplitude.24 Recently, we demonstrated that after 2 weeks of streptozotocin (STZ)-induced hyperglycemia, C57BL/6J mice exhibited lower amplitudes in three components of the dc-ERG: the c-wave, fast oscillation, and off response.23 Because the ERG a-wave was unaffected, these changes could not be attributed to altered photoreceptor function, which provides the signal that drives these components. Müller glia cells become activated and display reductions in slow PIII after 4 weeks of hyperglycemia.39 However, this is generally thought to occur as a result of oxidative stress or injury which is not common to mouse models of diabetes at these early time points following onset of hyperglycemia.5,6,40 Therefore, a different mechanism may underlie the reductions in the dc-ERG components immediately following onset of hyperglycemia. We hypothesized that hyperglycemia could result in an increased slow PIII response that would in turn reduce the c-wave amplitude at early time points.
Historically, slow PIII has been isolated pharmacologically, using glutamate analogs to block synaptic transmission between the photoreceptor and depolarizing bipolar cell.41,42 Since this approach is not suitable for a long-term study, we used a genetic approach which allows for the study of multiple time points without the requirement for intraocular injection. Several mouse mutants harbor a mutation in a component of the depolarizing bipolar cell signal transduction cascade.43 These models allow slow PIII to be measured in vivo without any other experimental manipulation.34,38,44 In the present study we used the Nyxnob mouse,38,45,46 which carries a mutation in the nyctalopin gene, to examine the response of the slow PIII to STZ-induced diabetes. By extension, these studies allowed us to determine the extent to which the c-wave abnormalities noted in earlier work23 reflect a primary effect on the RPE generator of the c-wave, or a secondary effect of a change in generation of slow PIII by Müller cells.
Our results demonstrate that electrophysiological changes of the RPE in response to diabetes occur prior to reductions in the Müller glia and photoreceptor responses, revealing a direct functional challenge to the RPE as a result of hyperglycemia. We further detail subtle early morphological changes to the RPE identified in the absence of alterations to the inner neuroretina.
Methods
Ethical Approval
All animals were treated in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and all animal procedures were approved by the Institutional Animal Care and Use Committees at the Louis Stokes Cleveland VA Medical Center and the Cleveland Clinic.
Mouse Model of Type 1 Diabetes.
We obtained Nyxnob mice (backcrossed to C57BL/6J for 10 generations) from a local breeding colony. At ages 4 to 8 weeks, littermates were randomly assigned to diabetic (STZ) or nondiabetic control (CNTL) groups. Diabetes was induced by three sequential daily intraperitoneal injections of a freshly prepared solution of STZ in 0.1 M citrate buffer (pH 4.4) at 30 mg/kg body weight. Mice in the control group received citrate buffer in the same intervals as the STZ. Insulin was given to diabetic mice by intraperitoneal injection every other day, as needed, to prevent ketosis without preventing hyperglycemia and glucosuria (0–0.2 units of neutral protamine Hagedorn [NPH] Humulin N; Eli Lilly and Co., Indianapolis, IN, USA). Sterile saline was also administered as needed for dehydration. Body weight and nonfasting blood glucose were measured using a glucometer (One-Touch Ultra; Bio-Rad Laboratories, Hercules, CA, USA). All control mice exhibited blood glucose levels ≤250 mg/dL for the duration of the study. All mice injected with STZ became diabetic within 5 days of the final STZ injection and maintained blood glucose levels ≥250 mg/dL.
Western Blot Analysis
Following euthanasia, eyes were enucleated and placed in ice-cold PBS (pH 7.4). Retinas were dissected from eyecups and lysed in radioimmunoprecipitation assay buffer (Cell Signaling, Danvers, MD, USA) with complete protease inhibitors and phosphatase inhibitors (Roche, Indianapolis, IN, USA). Protein lysates (20 μg) were run on a 4% to 15% gradient gel by SDS-PAGE and transferred to a polyvinylidene fluoride membrane overnight. Membranes were blocked in 5% milk in Tris-buffered saline with 0.1% Tween 20 (TBS-T, pH 7.4) for 1 hour at room temperature (RT) and incubated with primary antibodies (rabbit anti-Kir4.1, 1:400 in 1% BSA; Millipore Corp., Billerica, MA, USA, and mouse anti-GAPDH, 1:1000 in 2% BSA; Abcam, Cambridge, UK) overnight at 4°C. Following 3 × 15 minute wash with TBS-T, horseradish peroxidase–conjugated secondary antibodies (1:20,000 in 5% milk TBS-T) were incubated for 1 hour at RT. Membranes were washed 4 × 15 minutes with TBS-T then rinsed once in TBS. Proteins were detected by chemiluminescene (Western Lightning ECL Pro; Perkin Elmer, Waltham, MA, USA). Densitometry was performed using ImageJ analysis of scanned films (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA).
Electroretinography
After overnight dark adaptation, mice were anesthetized (65 mg/kg sodium pentobarbital); the cornea was anesthetized (1% proparacaine hydrochloride); and the pupils were dilated (1% tropicamide, 2.5% phenylephrine hydrochloride, and 1% cyclopentolate). Mice were placed on a temperature-regulated heating pad throughout each recording session. The protocols used to record ERG components generated by the outer neural retina and the RPE have been described elsewhere.23 In brief, responses of the outer retina were recorded on a full-field Ganzfeld stimulator (Espion E3 ColorDome; Diagnosys, LLC, Lowell, MA, USA) with a Ag/AgCl electrode referenced to a Ag/AgCl pellet electrode placed in the mouth of the mouse. Ten steps of strobe-flash stimuli were presented in the dark in order of increasing stimulus strength (−3.6 to 2.1 log scot cd/m2) and the number of successive trials averaged together decreased from 20 for low-level flashes to 2 for the highest flash stimuli. The duration of the interstimulus interval increased from 4 seconds for low stimulus flashes to 90 seconds for the highest stimuli. The amplitude of the a-wave was measured 6.6 ms after flash onset from the pre-stimulus baseline. The amplitude of the slow PIII was measured from the baseline to the minimum value of the negative curve, approximately 100 ms. Immediately following the dark-adapted strobe-flash stimuli, components of the dc-ERG were recorded in response to a 5 cd/m2 stimulus presented for 7 minutes. The amplitude of the c-wave was measured from the prestimulus baseline to the peak of the c-wave. The amplitude of the fast oscillation was measured from the c-wave peak to the trough of the fast oscillation. The amplitude of the light peak was measured from the fast oscillation trough to the asymptotic value. The amplitude of the off-response was measured from the light peak asymptote to the peak of the off-response. The time span in which we evaluate slow PIII and the c-wave are greatly different and demonstrate that in response to a strobe flash stimulus, each waveform component has a different time-to-peak and can be evaluated separately. Slow PIII is evaluated in a 250 ms recording in response to a strobe flash stimulus. In this recording, slow PIII peaks at ∼100 ms and the c-wave is not apparent. Yoshimura et al.47 demonstrated in rabbits that in response to stimulus of less than 2 seconds, the slow PIII peak occurs well prior to the c-wave and further notes that at stimuli of 500 lux intensity, the peak time of the c-wave is longer than that of the slow PIII for stimulus durations of less than 3.5 seconds and almost the same at more than 3.5 seconds.47 In our study, we utilized the 7-minute stimulus to observe the response of the RPE, which remains confounded by the slow PIII. The strobe flash was used evaluate slow PIII in the same mice at the same time points of diabetes.
Retina and RPE Flat-Mounts
Eyes were enucleated and fixed in 4% paraformaldehyde (PFA) for 1 hour prior to dissection. Retinas were separated from RPE/sclera and fixed for an additional 2 hours in 4% PFA. Tissues were blocked in 1% BSA + 5% goat serum in 0.05% Triton X-100 in PBS (PBS-T) overnight. Retinas were incubated in peanut agglutinin (PNA)-conjugated 594 (1:100 in PBS-T, Invitrogen, Carlsbad, CA, USA) for 1 hour at RT in the dark or overnight at 4°C. We incubated RPE/scleras in phalloidin-conjugated 488 (1:10 in PBS, Sigma-Aldrich Corp., St. Louis, MO, USA) for 1 hour at RT in the dark or overnight at 4°C. Tissues were washed 4 × 5 minutes in PBS, and incubated in secondary antibody (1:500, AlexaFluor 488 or 594 in PBS-T; Life Technologies, Carlsbad, CA, USA) for 1 hour at RT. Tissues were washed 3 × 10 minutes in PBS-T and counterstained in DAPI (1:10,000 in PBS; Thermo Fisher Scientific, Waltham, MA, USA) prior to flat-mounting on slides. We imaged RPE flat-mounts with a fluorescence/differential interference contrast microscope (BX-61; Olympus Corp., Tokyo, Japan), equipped with a charge-coupled device monochrome camera (Hamamatsu Photonics, Bridgewater, NJ, USA). Medial and lateral micrographs of phalloidin were captured (SlideBook software, version 4.2; Intelligent Imaging Innovations, Denver, CO, USA) for each of the four petals of each flat-mount from at least three animals per group per time point. At least 25 cells per micrograph were analyzed using ImageJ software to determine RPE cell size. Staining of retinal flat-mounts with PNA was also captured and measured from low magnification micrographs of each flat-mount from at least three animals per group per time point.
Light Microscopy
Enucleated eyes were fixed in 0.1 M sodium cacodylate buffer (pH 7.4) containing 2% formaldehyde and 2.5% glutaraldehyde. The tissues were then osmicated, dehydrated though a graded ethanol series, and embedded in epoxy resin (Embed-812/DER73 Epon kit; Electron Microscope Services, Hatfield, PA, USA). Semi-thin sections (0.8 μm) were cut along the horizontal meridian through the optic nerve and stained with 1% toluidine blue O for evaluation. Low magnification photomicrographs were taken of sections traversing the optic nerve. The distance from the outer limiting membrane (OLM) to the inner limiting membrane (ILM) was measured in three sections per animal and averaged for a minimum of three animals per group using ImageJ software. Higher magnification photomicrographs were taken 250 μm from the optic nerve, and the length of the outer segment (OS), inner segment (IS), and outer nuclear layer (ONL) were measured in three equidistant areas per section per animal. Thickness of RPE was also measured at ×100 magnification from three sections of each mouse. To assess melanosome density, RPE cells from the inferior retina were bisected into apical and basolateral areas and total melanosomes were counted within each space and throughout the apical processes that extended into the outer segments. Melanosome density is presented as melanosomes within each area, and does not include melanosomes found outside of the apical RPE cell membrane. Nearest neighbor analysis was performed on ×40 images of the ONL using ImageJ software.
Statistical Analysis
For all analyses (except relative amplitude comparisons), data were compiled as average ±SEM or SD as indicated, and statistics were performed using nonrepeated measures, 2-way ANOVA with Tukey post-hoc analysis on graphing software (GraphPad Prism 6; GraphPad, Inc., La Jolla, CA, USA). Statistical significance was determined by achieving a P value for both the ANOVA and multiple comparisons test below 0.05. To normalize the amplitude of the slow PIII, c-wave, and fast oscillation to that of the mean control response, the amplitude of each individual animal was compared to the average a-wave response and calculated as the relative amplitude. Relative amplitudes were then averaged and compared to the control average. Statistical significance for the relative amplitude was measured by unpaired Student's t-test and values of P ≤ 0.05 were considered significant. At least three animals per condition (CNTL and STZ) per time point were used for all experiments.
Results
To determine if Müller glia exhibit an increased slow PIII response that contributed to the reduction in the c-wave at early time points following the onset of hyperglycemia, we employed Nyxnob mice that reveal the slow PIII component in the ERG waveform due to a null mutation in the nyctalopin gene.45 Type 1 diabetes was induced in Nyxnob mice by injection with STZ and we assessed retinal histology and the ERG at 1, 2, and 4 weeks following the onset of diabetes (Fig. 1A). At 2 and 4 weeks, STZ mice displayed slightly lower body weight compared with controls (n ≥ 5 for each group; ANOVA, P = 0.012; Tukey, *P ≤ 0.05; Fig. 1B). Significantly elevated levels of blood glucose (≥250 mg/dL) were observed in STZ mice throughout the testing period (ANOVA, P < 0.0001; Tukey, ***P < 0.0001; n ≥ 5 for each group; Fig. 1C). Because slow PIII is severely abrogated in Kir4.1−/− mice48 and changes in localization and expression of Kir4.1 have been documented in rodent models of diabetes,6,7,49 we assessed retinal Kir4.1 levels by Western blot (Fig. 1D). No significant difference was identified at the time points evaluated.
Figure 1.
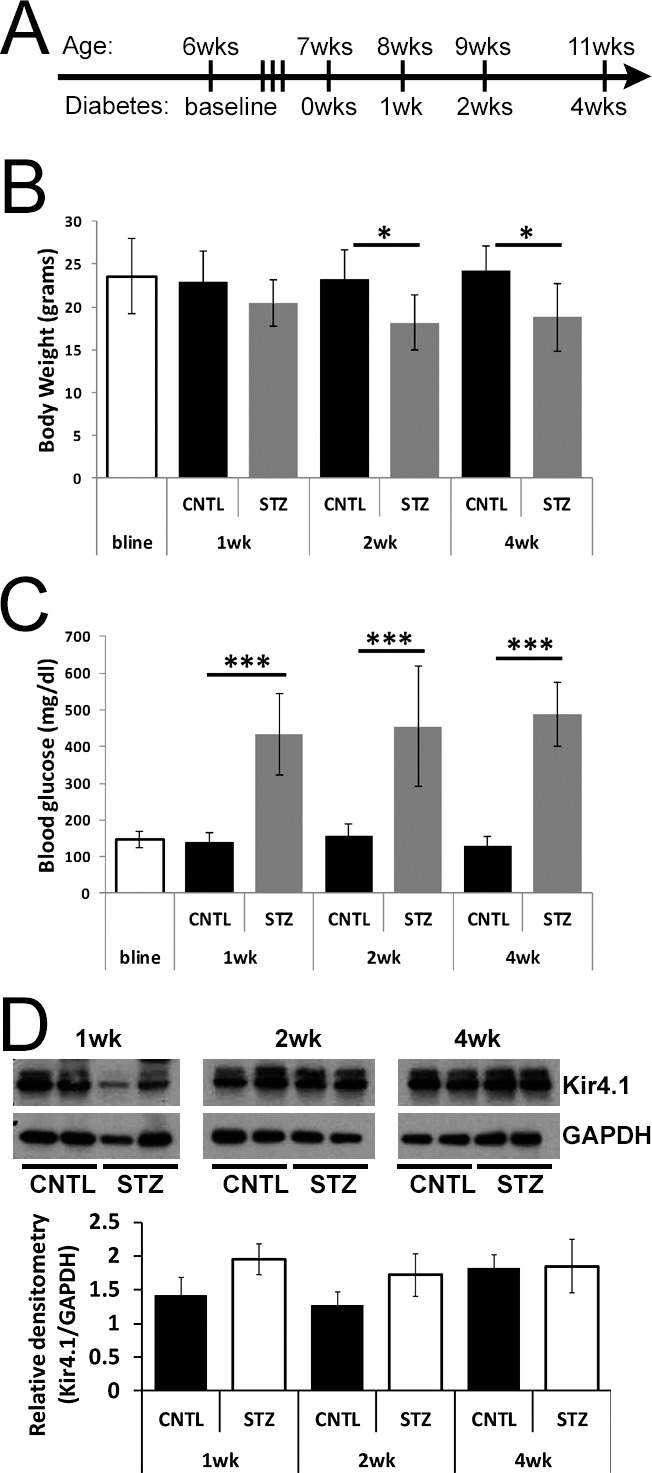
Streptozotocin-injected Nyxnob mice are hyperglycemic. (A) Schematic illustrating the time course of STZ injections and ERG testing paradigms for Nyxnob mice. Both males and females were utilized. (B) Body weight was measured at baseline and at the time of each testing period (n ≥ 5). (C) Systemic nonfasting blood glucose levels were measured at each time point. Values ≥250 mg/dL were considered diabetic (n ≥ 3). (D) Top: representative Western blot analysis of retinal lysates from STZ and CNTL mice at each time point. Bottom: quantification of Kir4.1 levels relative to GAPDH (n ≥ 2 for each group). For all panels, data points indicate the average ±SEM. *P ≤ 0.05. ***P ≤ 0.0001 between CNTL and STZ by 2-way ANOVA with Tukey's post-hoc analysis.
Electroretinography
Strobe flash ERGs were recorded from CNTL and STZ-Nyxnob mice at 1, 2, and 4 weeks following onset of hyperglycemia. Representative waveforms elicited in response to 3 of the 10 stimuli are depicted in Figure 2A. The main components of the strobe flash ERG of Nyxnob mice include the a-wave and slow PIII (labeled in Fig. 2A). Statistically significant reductions to the a-wave were not observed over the time course evaluated (Fig. 2B). Significant reductions to the slow PIII amplitude of STZ mice were identified at 2 and 4 weeks following onset of diabetes (Figs. 2A, 2C; 2-way ANOVA, **P ≤ 0.0004; Tukey, *P ≤ 0.05, **P ≤ 0.001, n ≥ 5). To normalize slow PIII to the mean control a-wave response, relative values of each were plotted (Fig. 2D). The diagonal line indicates a proportional change in the slow PIII and a-wave amplitude. We found that all points fall below the diagonal line, indicating that the reductions to slow PIII were not proportional to the a-wave amplitude. One week after diabetes onset, the relative slow PIII is reduced to a greater extent than the relative a-wave (Fig. 2D, the black diamond [1-week post-diabetes onset] falls below the diagonal line). At 2 and 4 weeks, the relative slow PIII reduction was significantly greater than the relative a-wave reduction (Fig. 2D, Student's t-test, *P ≤ 0.05, **P ≤ 0.001, gray and white diamonds). These data demonstrate that slow PIII did not exhibit increased amplitude at any time point following onset of hyperglycemia. Moreover, it was consistently affected to a greater degree than the a-wave throughout the testing period.
Figure 2.

Diabetic Nyxnob mice exhibit reduced slow PIII and a-wave amplitudes. (A) Averaged strobe flash ERG responses at 1, 2, and 4 weeks following onset of diabetes in response to −2.4, −0.6, and 1.4 log scot cd/m2 flashes. Control waveforms are in black, STZ-injected waveforms in gray. Scale bar: 200 μV/50 ms. (B) Amplitudes of a-waves obtained in response to increasing stimulus strength (−0.6 to 2.1 log scot cd/m2). (C) Average slow PIII amplitudes obtained in response to −2.4, −0.6, and 1.4 log scot cd/m2 flashes. *P ≤ 0.05. **P ≤ 0.005 between CNTL and STZ by 2-way ANOVA with Tukey's post-hoc analysis. (D) Relative change in a-wave and slow PIII amplitudes elicited by the 1.4 log scot cd/m2 stimulus observed in STZ mice plotted relative to the CNTL average response to the same stimulus. The diagonal line indicates an equivalent reduction in a-wave and slow PIII amplitudes. Data points indicate the average ±SEM. *P ≤ 0.05. **P ≤ 0.001 by Student's t-test.
The direct-current coupled ERG is composed of four main components generated secondary to rod photoreceptor activity24,50 (labeled on Fig. 3A). The initial positive polarity c-wave is the most reproducible and robust component and reflects the sum of slow PIII and a hyperpolarization of the apical RPE membrane. In our earlier study,23 STZ-injected C57BL/6J mice displayed reductions in c-wave amplitude in the absence of reductions to the rod photoreceptor derived a-wave. This is the same pattern we found in the Nyxnob mouse. Significant reductions to the c-wave of the STZ-Nyxnob mice were likewise identified at 1 week post onset of hyperglycemia and continued throughout the testing period (Figs. 3A, 3B; ANOVA, P ≤ 0.0001; Tukey, *P ≤ 0.05, **P ≤ 0.001, ***P ≤ 0.0001; n ≥ 5). We previously determined that the relative c-wave amplitude was correlated with degree of hyperglycemia.23 When we analyzed the c-wave amplitude as a function of blood glucose concentration in Nyxnob mice, we found that there was no significant correlation as blood glucose concentrations were consistent throughout the testing period (Fig. 3C). We next sought to determine if the c-wave was affected to a greater extent than the a-wave or slow PIII by plotting the relative mean c-wave as a function of each. As observed with the slow PIII response, the relative c-wave was affected prior to and to a greater extent than the a-wave as each point fell below the diagonal line (Fig. 3D, Student's t-test, *P ≤ 0.05, **P ≤ 0.001). The c-wave was also initially affected prior to and to a greater extent than slow PIII at 1 week as the black diamond fell below the diagonal line (Fig. 3E, Student's t-test, *P ≤ 0.05). However, the c-wave and slow PIII were equivalently reduced at 2 and 4 weeks as the gray and white diamonds fell along the line (Fig. 3E). These data demonstrate that the initial reduction to the c-wave as a result of diabetes is: (1) not caused by an increase in absolute value of the negative slow PIII amplitude; (2) occurs prior to significant reductions in slow PIII amplitude; and (3) is initially affected to a greater extent than slow PIII.
Figure 3.
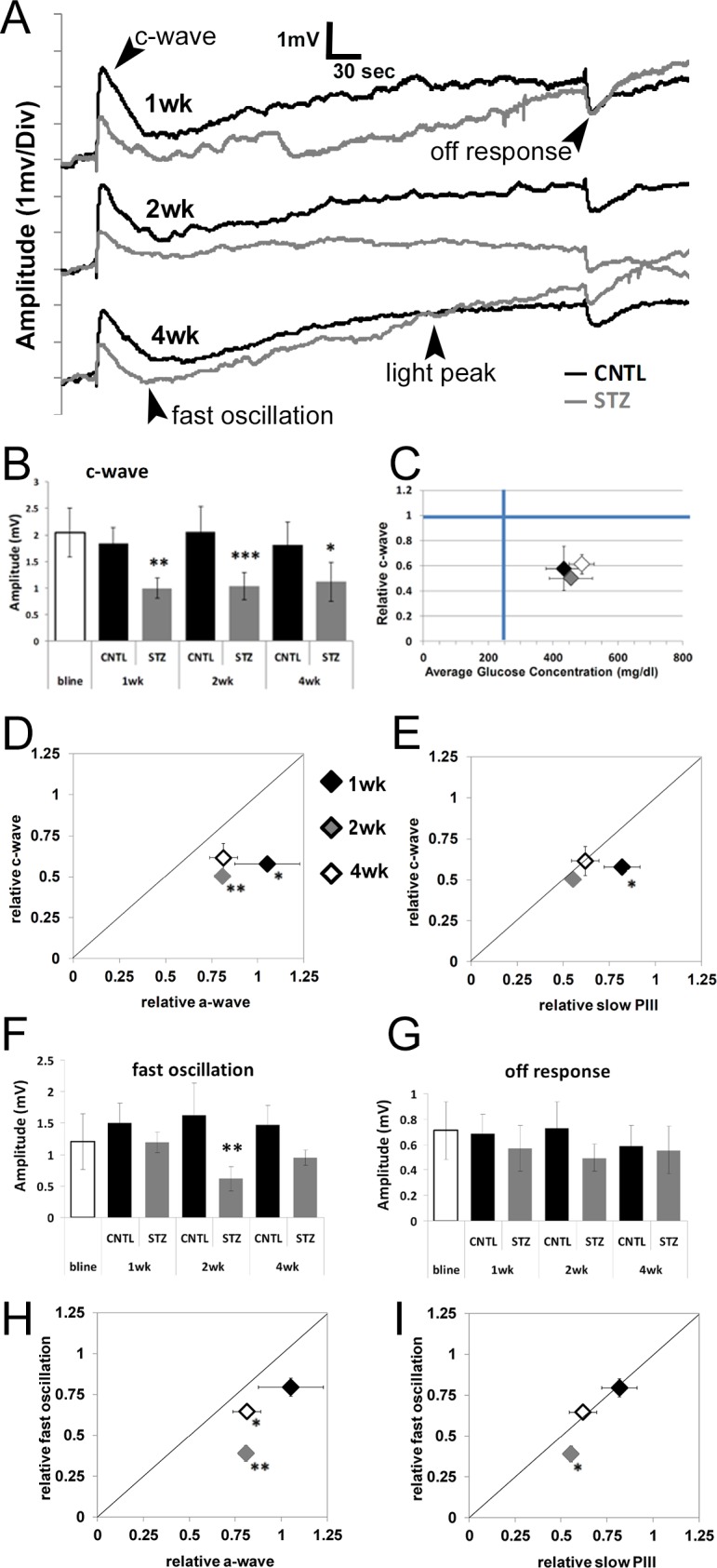
Diabetic Nyxnob mice display reduced c-wave amplitudes prior to and of a greater extent than a-wave and slow PIII reductions. (A) Representative dc-ERG waveforms from Nyxnob mice at 1, 2, and 4 weeks of diabetes. (B) Average c-wave amplitude at each time point. (C) Relative c-wave amplitude plotted as a function of systemic blood glucose level. (D) Relative c-wave amplitude plotted as a function of the relative a-wave elicited by the 1.4 log scot cd/m2 stimulus at each time point. (E) Relative c-wave amplitude plotted as a function of the relative slow PIII amplitude in response to a 1.4 log scot cd/m2 light stimulus. (F) Average fast oscillation amplitude at each time point. (G) Average off-response amplitude at each time point. (H) Relative fast oscillation amplitude plotted as a function of the relative a-wave elicited in response to the 1.4 log scot cd/m2 light stimulus. (I) Relative fast oscillation amplitude plotted as a function of the relative slow PIII amplitude in response to a 1.4 log scot cd/m2 light stimulus. (B, F, G) *P ≤ 0.05. **P ≤ 0.005. ***P ≤ 0.0001 by 2-way ANOVA with Tukey's post-hoc analysis. (D, E, H, I) *P ≤ 0.05. **P ≤ 0.001 by Student's t-test.
We additionally observed a significant reduction in the fast oscillation of Nyxnob-STZ mice at 2 weeks following onset of diabetes (Figs. 3A, 3F; ANOVA, P ≤ 0.0001; Tukey, **P < 0.001; n ≥ 5), which was consistent with our previous findings from STZ-injected C57BL/6J mice.23 This parameter is also more significantly affected than the a-wave at 2 and 4 weeks as the gray and white diamonds fell below the diagonal line (Fig. 3H, Student's t-test, *P ≤ 0.05, **P ≤ 0.001) and more significantly affected than the slow PIII at 2 weeks (Fig. 3I; Student's t-test, *P ≤ 0.05). The light peak was not significantly reduced. The off-response had a trend for reduction (Figs. 3A, 3G; ANOVA, P ≤ 0.05); however, this measure did not reach statistical significance by Tukey's post-hoc analysis). Because the fast oscillation, light peak, and off-response were reduced, but in a different time frame and to different degrees than the reduction to the c-wave, our data demonstrate that an overall reduction in membrane potential is not the cause of the altered dc-ERG response.
Histology
It has been noted that cone photoreceptors, the inner retina,51 and the RPE are particularly susceptible to hyperglycemia.52,53 Moreover, diabetic rats exhibit increased retinal thickness and water content, representative of diffuse central edema.54 Therefore, we assessed morphology of the outer retina and RPE of CNTL and STZ-Nyxnob mice by analyzing histology of semi-thin sections (Fig. 4A). No difference in overall length of the retina from the outer limiting membrane to the inner limiting membrane or in the IS length was observed (Figs. 4B, 4C, respectively). We observed a trend for a slightly smaller OS length in STZ retinas compared with CNTLs, which was significant at 1 week (Fig. 4D; ANOVA, P ≤ 0.0001; Tukey ***P < 0.0001; n ≥ 3 for each group). We observed a complementary trend for a slightly larger ONL length in STZ retinas (Fig. 4E; ANOVA, P = 0.0062; Tukey, not significant). Despite the change in ONL length, no difference was found in the number of rod photoreceptor nuclei per column unit (Fig. 4F). The inverse relationship between the change to the OS and ONL are thought to account for lack of an overall difference in OLM-ILM length. Further visual observation of the semi-thin sections led to a general assessment that the photoreceptor nuclei in STZ-injected animals were spaced farther apart than in the CNTLs. We performed a nearest neighbor analysis and found a trend for an increase in distance at 4 weeks which did not meet statistical significance by 2-way ANOVA (Fig. 4G).
Figure 4.

Diabetic Nyxnob mice exhibit normal retinal morphology. (A) Representative micrographs from semi-thin sections of CNTL and STZ retinas spanning the optic nerve. Scale bar: 20 μm, (B) Distance from the OLM to the ILM was measured 250, 500, and 1000 μm on the inferior and superior sides of the optic nerve. (C–E) Length of the IS, OS, and ONL was measured at each time point. ***P ≤ 0.0001 by 2-way ANOVA with Tukey's post-hoc analysis. (F) Rod nuclei per column were measured and averaged at each time point. (G) Nearest neighbor analysis of rod photoreceptor nuclei within the ONL.
Cone photoreceptor damage has been observed in other rodent models of diabetes and in zebrafish.51–53,55–59 To determine if a change in cone number could account for the increased spacing in the ONL, we measured cone density by PNA staining of retinal flat-mounts (Figs. 5A, 5B) and by counting cone photoreceptors/field of view as identified by nuclear morphology in toluidine blue stained semi-thin sections (Fig. 5E, red arrows; Fig. 5F). No differences in either parameter were identified. To measure connectivity in the inner retina we also counted cone pedicles in PNA-stained cryosections (Figs. 5C, 5D). No significant difference was found between groups.
Figure 5.
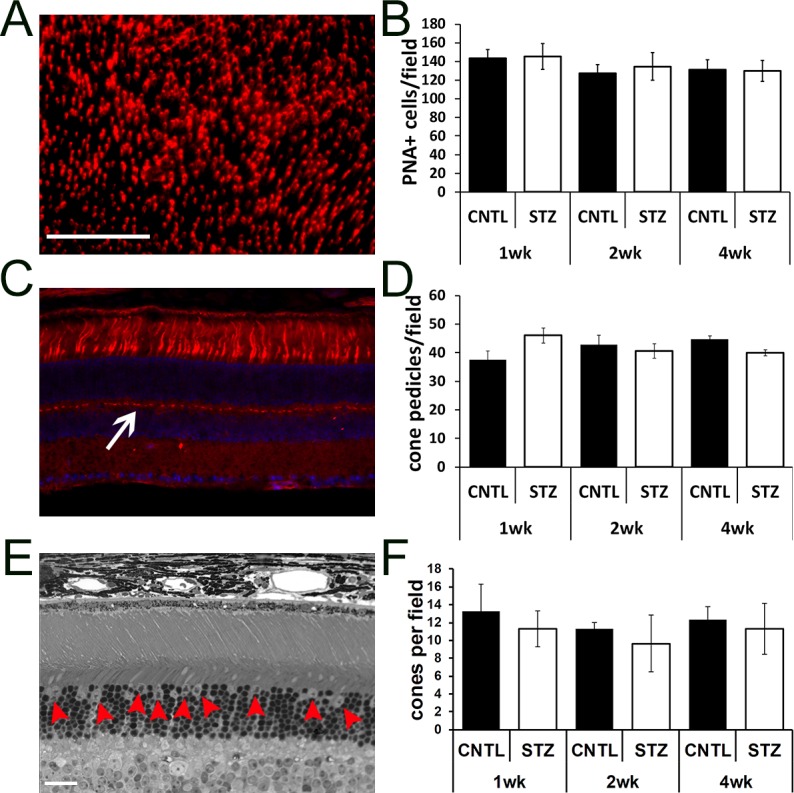
Diabetic Nyxnob mice have normal cone density. (A) Representative retinal flat-mount prepared and stained with PNA. Scale bar: 100 μm. (B) Peanut agglutinin–positive cells/field in each of four petals per flat-mount were counted using ImageJ analysis. (C) Representative cryosection stained with PNA (red) and counterstained with DAPI (blue). (D) Cone pedicles (white arrow) were counted using ImageJ analysis. (E) Representative micrograph from a semi-thin section spanning the optic nerve used for counting cone nuclei (red arrowheads). Scale bar (C, E): 20 μm. (F) Cone nuclei per field were counted and averaged.
These data demonstrate that the overt structure of the retina was normal in CNTL and STZ-Nyxnob mice through 4 weeks of hyperglycemia. However, at each time point analyzed, we noted prominent differences in the RPE of CNTL and STZ mice. First, from the toluidine blue stained semi-thin sections, we observed an increase in RPE thickness (measured from the basolateral to apical membrane) in STZ mice compared with CNTLs (Figs. 6A, 6B, red double-headed arrows; ANOVA, P = 0.0019; Tukey, not significant; n ≥ 3). To determine if RPE cells were larger in area, flat-mounts of isolated RPE sheets were prepared and stained with phalloidin (representative image, Fig. 6C). However, no statistical differences in RPE cell area were identified in medial (Fig. 6D) or peripheral (Fig. 6E) areas. In addition to changes in RPE thickness, the distribution of melanosomes within the RPE appeared different between CNTL and STZ mice (Fig. 6A, red arrowheads, Fig. 6F). At each time point, we observed a trend for a reduction in melanosome density in the basolateral region of the RPE (Fig. 6F; ANOVA, P = 0.0096; Tukey, not significant; n ≥ 3) and an increase in density of melanosomes within the apical half of the RPE cells (Fig. 6F; ANOVA, P = 0.003; Tukey, *P < 0.05; n ≥ 3). The total density of melanosomes was not significantly different. In addition to a general increase in density within the apical region of the RPE cell, we found a trend for an increase in the number of melanosomes located in the apical processes and/or outside of the RPE cells themselves (Fig. 6G; ANOVA, P = 0.043; Tukey, not significant, n ≥ 3).
Figure 6.

Diabetic Nyxnob mice have enlarged RPE thickness and altered melanosome distribution. (A) Representative micrographs from semi-thin sections used to analyze the RPE. Scale bar: 10 μm. Red, double-headed arrows denote RPE thickness and red arrowheads indicate melanosomes located outside the RPE. (B) Thickness of RPE was measured from semi-thin sections *P ≤ 0.05. **P ≤ 0.005. ***P ≤ 0.0001 by 2-way ANOVA with Tukey's post-hoc analysis. (C) Representative RPE flat-mount prepared and stained with phalloidin. Scale bar: 10 μm. (D–E) Area of RPE was measured from medial and peripheral regions (relative to the optic nerve) in each of four petals from each RPE flat-mount. (F) Melanosomes were counted within bisected apical and basolateral areas identified from micrographs of semi-thin sections of the inferior RPE. Melanosome density was determined from the total area and each bisected region. Bottom: apical (ap). Top: basolateral (bl). (G) Melanosomes found outside of the RPE, in the subretinal space and between outer segments, were counted and averaged for each field.
Discussion
We previously reported that the c-wave was smaller in STZ-injected C57BL/6J mice compared with controls at 2 and 4 weeks.23 As noted, the c-wave reflects the summed hyperpolarization of the apical RPE membrane and the negative slow PIII. Wu et al.34 demonstrated that Kir4.1+/−/Nyxnob mice have elevated c-wave amplitudes compared with wild-type Kir4.1+/+/Nyxnob mice, thus showing that a selective reduction of slow PIII will lead to an increase in the corneal c-wave. In Sprague Dawley rats injected with STZ, decreased levels of Kir4.1 have been found at 4 weeks post-hyperglycemia onset.49 However, we found by Western blot that retinal Kir4.1 levels were unchanged in STZ-Nyxnob mice through the 4-week time point. We therefore hypothesized that increased slow PIII amplitudes could underlie the reduction to the amplitude of the c-wave. By exploiting the characteristic ERG waveform of the Nyxnob mouse, we demonstrated that Müller glia exhibit smaller slow PIII responses as early as 1 week following the onset of hyperglycemia. These responses occur before photoreceptor activity is decreased and are of a greater extent than a-wave reductions at later times (Fig. 2). This is also when no apparent insult to bipolar cell function can be identified.23 Although it is possible that fast PIII can contribute to the waveform at 100 ms, we previously demonstrated that slow PIII and c-wave responses are resilient to the progressive degeneration and loss of photoreceptors as observed in NyxnobPrphRd2/+ mice.38
Importantly, the c-wave is reduced in the diabetic mice as early as 1 week following onset of hyperglycemia (Fig. 3). As we have shown here that the slow PIII is also smaller in STZ mice compared with CNTLs (Fig. 2), we can conclude that changes in slow PIII do not account for the reduction in the c-wave, in timing, or direction. Moreover, the rod photoreceptor response, reflected in the a-wave amplitude, also does not account for the reduction to the c-wave (Fig. 2A, 2C). Because a-wave amplitudes are not reduced, it is unlikely that the reduced RPE response is caused by an overall reduction in [K+] within the subretinal space. Therefore, the changes in the light-evoked responses of the RPE as a result of diabetes are most likely due to early changes in the RPE itself. This notion is supported by early histological changes observed in the RPE.
In addition to reductions to the c-wave, the other components of the dc-ERG, including the negative polarity fast oscillation, the light peak, and the off-response, also displayed a trend for decreased amplitudes (Fig. 3), correlating with our previous studies of mouse models of type 1 and 2 diabetes.23 Each of the dc-ERG components is derived from changes in ion conductance and membrane potential across the RPE.24,60–68 Importantly, although each dc-ERG component is generated secondarily to rod photoreceptor activation24 the dc-ERG alterations were observed at the earliest periods in diabetes, before statistically significant changes to the a-wave were identified.23 Therefore, it is likely that alterations in ion channel expression and/or conductance, or the mechanisms by which these channels are inserted into or maintained within the RPE membrane, underlie the defects in the dc-ERG.
Our physiological data is coupled with new detailed findings to outer retina histology. Notably, we documented altered melanosome distribution in the RPE of STZ mice (Fig. 6). These data are intriguing as they point toward an alteration in the cellular transport processes with the RPE. Although melanosomes within the RPE of mammals do not undergo a significant redistribution with light onset as observed with fish and amphibians, they have been shown to move into and out of apical processes.69 Within the inferior retina of C57BL/6J mice, melanosomes maintain a slightly more basal localization,70 regardless of light adaptation. Here we observe significantly more melanosomes in the apical region of the inferior RPE and within the apical processes that interdigitate with the OS. Melanosome transport is regulated by Rab27a in a Na-K-ATPase dependent manner.71 Importantly, a marked decrease in Na-K-ATPase activity in the diabetic RPE has been documented.26,72 Furthermore, melanosome location is also regulated by Myo7a, which plays an important role in the visual cycle by regulating translocation of RPE65.73 Of significant interest in this regard is new data suggesting that inhibition of the visual cycle by retinylamine prevents vascular and neuroretinal damage associated with diabetic retinopathy.21 Each of these findings suggests that general cell biology in the diabetic RPE is perturbed.
Our study also documents trends for increased RPE thickness (Fig. 6) and ONL length, and in spatial distribution of ONL nuclei by nearest neighbor analysis (Fig. 4). Alterations in RPE histology in diabetic rodents have been previously identified.52,72,74 However, our findings come on the heels of recent studies that help to shed some light on the etiology of our observed pathology. A primary role of both cell types is to regulate subretinal (RPE) and inner retina (Müller glia) fluid transport. Numerous experimental models of diabetes display defects in fluid transport across the RPE membranes,26,75–78 which has been suggested to play a role in pathogenesis of retinopathy. Indeed, isolated human RPE cells grown in high glucose media exhibit a 50% reduction in the regulatory volume increase compared with controls. This is hypothesized to be due to dysfunction of Na+/H+ and Cl−/HCO−3 antiporters.75 It has recently been suggested that insufficient fluid transport associated with diabetes is responsible for diabetic retinal edema, as diabetic mice do not display light-dependent increases in the SRS-apparent diffusion coefficient.19 Moreover, diabetic rats displayed increased retinal thickness and retinal water volume.54 Although we find no overt evidence of edema at 4 weeks of diabetes, we did observe an increase in outer segment length at 1 week and an increase in RPE thickness at each time point evaluated, displaying the possibility for insufficient or faulty fluid transport out of the retina by RPE cells.
Other recent reports point toward alternate mechanisms for disruptions in fluid/ion transport. New evidence revealed that the transient receptor potential isoform 4 (TRPV4) works in conjunction with aquaporin 4 (AQP4) to regulate cell volume in retinal Müller glia.79 Intriguingly, AQP4 has also been shown to be expressed in human fetal RPE cells80 and recently, so has TRPV4.81 Given the changes to ONL length, and an obvious increase in space between rod nuclei columnar units in the STZ-Nyxnob mice, it is possible that AQP4 and TRPV4 are not functioning at complete capacity to induce subretinal and RPE swelling. Of note is the recent observation that TRPV4 is also downregulated on retinal microvascular endothelial cells in response to hyperglycemia/diabetes82 and may play a significant role in the development of diabetic macular edema, irrespective of its expression on the RPE or Müller glia. These hypotheses warrant further investigations.
Herein we have identified novel changes to outer retina of STZ-treated Nyxnob mice by electrophysiology and histology. These data enhance our knowledge of the defects to the outer retina at very early time points following the onset of hyperglycemia. Hyperglycemia is known to induce the dysfunction of multiple retinal cell types prior to the development of microvascular damage.14,83 Photoreceptors, the photosensitive cells of the retina, have recently been implicated in the development of vascular disease within the retina as a result of diabetes.53 However, it is not entirely clear if the progression from nonproliferative diabetic retinopathy to the downstream blinding pathologies of neovascularization and diabetic retinal edema is due to hyperglycemia itself or instead, from the maladaptive responses of inflammation and oxidative stress that occur after chronic hyperglycemia. Nevertheless, outer retinal cells display functional (ERG) defects almost immediately following onset of hyperglycemia and those consequences are long-lasting.23,56,84–86 Here and previously, we find that alterations in physiology of the Müller glia and RPE occur prior to the earliest detectable signs of altered physiology in photoreceptors. Thus, dysfunction of the cells which support photoreceptors may be the primary target for therapeutic intervention.
Acknowledgments
We thank Neal Peachey, PhD, for providing Nyxnob mice and critical evaluation of the manuscript.
Supported by a VA Merit Award (I01BX002754); a Career Development award (ISS) from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Service; and an unrestricted grant from the Research to Prevent Blindness to the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University. The authors alone are responsible for the content and writing of the paper.
Disclosure: M.J. Tarchick, None; P. Bassiri, None; R.M. Rohwer, None; I.S. Samuels, None
References
- 1. Bringmann A,, Iandiev I,, Pannicke T,, et al. Cellular signaling and factors involved in Müller cell gliosis: neuroprotective and detrimental effects. Prog Retin Eye Res. 2009; 28: 423–451. [DOI] [PubMed] [Google Scholar]
- 2. Reichenbach A,, Bringmann A. New functions of Müller cells. Glia. 2013; 61: 651–678. [DOI] [PubMed] [Google Scholar]
- 3. Rungger-Brändle E,, Dosso AA,, Leuenberger PM. Glial reactivity an early feature of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2000; 41: 1971–1980. [PubMed] [Google Scholar]
- 4. Zong H,, Ward M,, Madden A,, et al. Hyperglycaemia-induced pro-inflammatory responses by retinal Müller glia are regulated by the receptor for advanced glycation end-products (RAGE). Diabetologia. 2010. ; 53: 2656–2666. [DOI] [PubMed] [Google Scholar]
- 5. Barber AJ,, Antonetti DA,, Gardner TW;, and The Penn State Retina Research Group Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. Invest Ophthalmol Vis Sci. 2000; 41: 3561–3568. [PubMed] [Google Scholar]
- 6. Bringmann A,, Pannicke T,, Grosche J,, et al. Müller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006; 25: 397–424. [DOI] [PubMed] [Google Scholar]
- 7. Curtis TM,, Hamilton R,, Yong PH,, et al. Müller glial dysfunction during diabetic retinopathy in rats is linked to accumulation of advanced glycation end-products and advanced lipoxidation end-products. Diabetologia. 2011. ; 54: 690–698. [DOI] [PubMed] [Google Scholar]
- 8. Giebel SJ,, Menicucci G,, McGuire PG,, Das A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest. 2005; 85: 597–607. [DOI] [PubMed] [Google Scholar]
- 9. Matteucci A,, Gaddini L,, Villa M,, et al. Neuroprotection by rat Müller glia against high glucose-induced neurodegeneration through a mechanism involving ERK1/2 activation. Exp Eye Res. 2014; 125: 20–29. [DOI] [PubMed] [Google Scholar]
- 10. Muto T,, Tien T,, Kim D,, Sarthy VP,, Roy S. High glucose alters Cx43 expression and gap junction intercellular communication in retinal Müller cells: promotes Müller cell and pericyte apoptosis. Invest Ophthalmol Vis Sci. 2014; 55: 4327–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crider JY,, Yorio T,, Sharif NA,, Griffin BW. The effects of elevated glucose on Na+/K(+)-ATPase of cultured bovine retinal pigment epithelial cells measured by a new nonradioactive rubidium uptake assay. J Ocul Pharmacol Ther. 1997; 13: 337–352. [DOI] [PubMed] [Google Scholar]
- 12. Minamizono A,, Tomi M,, Hosoya K. Inhibition of dehydroascorbic acid transport across the rat blood-retinal and -brain barriers in experimental diabetes. Biol Pharm Bull. 2006; 29: 2148–2150. [DOI] [PubMed] [Google Scholar]
- 13. Salceda R,, Contreras-Cubas C. Ascorbate uptake in normal and diabetic rat retina and retinal pigment epithelium. Comp Biochem Physiol C Toxicol Pharmacol. 2007; 146: 175–179. [DOI] [PubMed] [Google Scholar]
- 14. Simó R,, Hernández C. Neurodegeneration in the diabetic eye: new insights and therapeutic perspectives. Trends Endocrinol Metab. 2014; 25: 23–33. [DOI] [PubMed] [Google Scholar]
- 15. Simó R,, Villarroel M,, Corraliza L,, Hernández C,, García-Ramírez M. The retinal pigment epithelium: something more than a constituent of the blood-retinal barrier--implications for the pathogenesis of diabetic retinopathy. J Biomed Biotechnol. 2010; 2010: 190724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sparrow JR,, Hicks D,, Hamel CP. The retinal pigment epithelium in health and disease. Curr Mol Med. 2010; 10: 802–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005; 85: 845–881. [DOI] [PubMed] [Google Scholar]
- 18. Villarroel M,, García-Ramírez M,, Corraliza L,, Hernández C,, Simó R. Effects of high glucose concentration on the barrier function and the expression of tight junction proteins in human retinal pigment epithelial cells. Exp Eye Res. 2009; 89: 913–920. [DOI] [PubMed] [Google Scholar]
- 19. Berkowitz BA,, Kern TS,, Bissig D,, et al. Systemic retinaldehyde treatment corrects retinal oxidative stress, rod dysfunction, and impaired visual performance in diabetic mice. Invest Ophthalmol Vis Sci. 2015; 56: 6294–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berkowitz BA,, Grady EM,, Khetarpal N,, Patel A,, Roberts R. Oxidative stress and light-evoked responses of the posterior segment in a mouse model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2015; 56: 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu H,, Tang J,, Du Y,, et al. Retinylamine benefits early diabetic retinopathy in mice. J Biol Chem. 2015; 290: 21568–21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu HZ,, Song Z,, Fu S,, Zhu M,, Le YZ. RPE barrier breakdown in diabetic retinopathy: seeing is believing. J Ocul Biol Dis Infor. 2011; 4: 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Samuels IS,, Bell BA,, Pereira A,, Saxon J,, Peachey NS. Early retinal pigment epithelium dysfunction is concomitant with hyperglycemia in mouse models of type 1 and type 2 diabetes. J Neurophysiol. 2015; 113: 1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu J,, Peachey NS,, Marmorstein AD. Light-evoked responses of the mouse retinal pigment epithelium. J Neurophysiol. 2004; 91: 1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacGregor LC,, Matschinsky FM. Treatment with aldose reductase inhibitor or with myo-inositol arrests deterioration of the electroretinogram of diabetic rats. J Clin Invest. 1985; 76: 887–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. MacGregor LC,, Matschinsky FM. Experimental diabetes mellitus impairs the function of the retinal pigmented epithelium. Metabolism. 1986; 35: 28–34. [DOI] [PubMed] [Google Scholar]
- 27. Pautler EL,, Ennis SR. The effect of induced diabetes on the electroretinogram components of the pigmented rat. Invest Ophthalmol Vis Sci. 1980; 19: 702–705. [PubMed] [Google Scholar]
- 28. Samuels IS,, Lee CA,, Petrash JM,, Peachey NS,, Kern TS. Exclusion of aldose reductase as a mediator of ERG deficits in a mouse model of diabetic eye disease. Vis Neurosci. 2012; 29: 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Granit R. The components of the retinal action potential in mammals and their relation to the discharge in the optic nerve. J Physiol. 1933; 77: 207–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmidt R,, Steinberg RH. Rod-dependent intracellular responses to light recorded from the pigment epithelium of the cat retina. J Physiol. 1971; 217: 71–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steinberg RH,, Schmidt R,, Brown KT. Intracellular responses to light from cat pigment epithelium: origin of the electroretinogram c-wave. Nature. 1970; 227: 728–730. [DOI] [PubMed] [Google Scholar]
- 32. Fujimoto M,, Tomita T. Reconstruction of the slow PIII from the rod potential. Invest Ophthalmol Vis Sci. 1979; 18: 1091–1093. [PubMed] [Google Scholar]
- 33. Oakley B,, II, Flaming DG,, Brown KT. Effects of the rod receptor potential upon retinal extracellular potassium concentration. J Gen Physiol. 1979; 74: 713–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu J,, Marmorstein AD,, Kofuji P,, Peachey NS. Contribution of Kir4.1 to the mouse electroretinogram. Mol Vis. 2004; 10: 650–654. [PMC free article] [PubMed] [Google Scholar]
- 35. Lurie M,, Marmor MF. Analysis of the response properties and light-integrating characteristics of the c-wave in the rabbit eye. Exp Eye Res. 1980; 31: 335–349. [DOI] [PubMed] [Google Scholar]
- 36. Lurie M,, Marmor MF. Similarities between the c-wave and slow PIII in the rabbit eye. Invest Ophthalmol Vis Sci. 1980; 19: 1113–1117. [PubMed] [Google Scholar]
- 37. Oakley B,, II, Green DG. Correlation of light-induced changes in retinal extracellular potassium concentration with c-wave of the electroretinogram. J Neurophysiol. 1976; 39: 1117–1133. [DOI] [PubMed] [Google Scholar]
- 38. Samuels IS,, Sturgill GM,, Grossman GH,, Rayborn ME,, Hollyfield JG,, Peachey NS. Light-evoked responses of the retinal pigment epithelium: changes accompanying photoreceptor loss in the mouse. J Neurophysiol. 2010; 104: 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wong VH,, Vingrys AJ,, Bui BV. Glial and neuronal dysfunction in streptozotocin-induced diabetic rats. J Ocul Biol Dis Infor. 2011; 4: 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Q,, Zemel E,, Miller B,, Perlman I. Early retinal damage in experimental diabetes: electroretinographical and morphological observations. Exp Eye Res. 2002; 74: 615–625. [DOI] [PubMed] [Google Scholar]
- 41. Slaughter MM,, Miller RF. An excitatory amino acid antagonist blocks cone input to sign-conserving second-order retinal neurons. Science. 1983; 219: 1230–1232. [DOI] [PubMed] [Google Scholar]
- 42. Malchow RP,, Yazulla S. Separation and light adaptation of rod and cone signals in the retina of the goldfish. Vision Res. 1986; 26: 1655–1666. [DOI] [PubMed] [Google Scholar]
- 43. Pardue MT,, Peachey NS. Mouse b-wave mutants. Doc Ophthalmol. 2014; 128: 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peachey NS,, Sturgill-Short GM. Response properties of slow PIII in the Large (vls) mutant. Doc Ophthalmol. 2012; 125: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gregg RG,, Mukhopadhyay S,, Candille SI,, et al. Identification of the gene and the mutation responsible for the mouse nob phenotype. Invest Ophthalmol Vis Sci. 2003; 44: 378–384. [DOI] [PubMed] [Google Scholar]
- 46. Pardue MT,, McCall MA,, LaVail MM,, Gregg RG,, Peachey NS. A naturally occurring mouse model of X-linked congenital stationary night blindness. Invest Ophthalmol Vis Sci. 1998; 39: 2443–2449. [PubMed] [Google Scholar]
- 47. Yoshimura Y,, Onoe S,, Takahashi Y,, Mori T,, Sato T,, Tazawa Y. Electroretinogram c-wave and slow PIII of the rabbit: changes in peak time and amplitude under various stimulus durations. Doc Ophthalmol. 1988; 69: 187–193. [DOI] [PubMed] [Google Scholar]
- 48. Kofuji P,, Ceelen P,, Zahs KR,, Surbeck LW,, Lester HA,, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000; 20: 5733–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Y,, Xu G,, Ling Q,, Da C. Expression of aquaporin 4 and Kir4.1 in diabetic rat retina: treatment with minocycline. J Int Med Res. 2011; 39: 464–479. [DOI] [PubMed] [Google Scholar]
- 50. Steinberg RH. Interactions between the retinal pigment epithelium and the neural retina. Doc Ophthalmol. 1985; 60: 327–346. [DOI] [PubMed] [Google Scholar]
- 51. Hombrebueno JR,, Chen M,, Penalva RG,, Xu H. Loss of synaptic connectivity, particularly in second order neurons is a key feature of diabetic retinal neuropathy in the Ins2Akita mouse. PLoS One. 2014; 9: e97970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Énzsöly A,, Szabó A,, Kántor O,, et al. Pathologic alterations of the outer retina in streptozotocin-induced diabetes. Invest Ophthalmol Vis Sci. 2014; 55: 3686–3699. [DOI] [PubMed] [Google Scholar]
- 53. Kern TS,, Berkowitz BA. Photoreceptors in diabetic retinopathy. J Diabetes Investig. 2015; 6: 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berkowitz BA,, Bissig D,, Ye Y,, Valsadia P,, Kern TS,, Roberts R. Evidence for diffuse central retinal edema in vivo in diabetic male Sprague Dawley rats. PLoS One. 2012; 7: e29619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alvarez Y,, Chen K,, Reynolds AL,, Waghorne N,, O'Connor JJ,, Kennedy BN. Predominant cone photoreceptor dysfunction in a hyperglycaemic model of non-proliferative diabetic retinopathy. Dis Model Mech. 2010; 3: 236–245. [DOI] [PubMed] [Google Scholar]
- 56. Phipps JA,, Fletcher EL,, Vingrys AJ. Paired-flash identification of rod and cone dysfunction in the diabetic rat. Invest Ophthalmol Vis Sci. 2004; 45: 4592–4600. [DOI] [PubMed] [Google Scholar]
- 57. Zarebska A,, Czerny K,, Bakiera K,, et al. Histological changes in the retina in experimental alloxan-induced diabetes in rabbits. Ann Univ Mariae Curie Sklodowska Med. 2001; 56: 81–84. [PubMed] [Google Scholar]
- 58. Zarebska A,, Łańcut M,, Bakiera K,, et al. Ultrastructural changes in the receptor parts of retinal rods in experimental alloxan-induced diabetes in rabbits. Ann Univ Mariae Curie Sklodowska Med. 2001; 56: 77–80. [PubMed] [Google Scholar]
- 59. Zhang Y,, Wang Q,, Zhang J,, Lei X,, Xu GT,, Ye W. Protection of exendin-4 analogue in early experimental diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2009; 247: 699–706. [DOI] [PubMed] [Google Scholar]
- 60. Edwards MM,, Marin de Evsikova C,, Collin GB,, et al. Photoreceptor degeneration, azoospermia, leukoencephalopathy, and abnormal RPE cell function in mice expressing an early stop mutation in CLCN2. Invest Ophthalmol Vis Sci. 2010; 51: 3264–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fujii S,, Gallemore RP,, Hughes BA,, Steinberg RH. Direct evidence for a basolateral membrane Cl- conductance in toad retinal pigment epithelium. Am J Physiol. 1992; 262: C374–C383. [DOI] [PubMed] [Google Scholar]
- 62. Gallemore RP,, Steinberg RH. Effects of DIDS on the chick retinal pigment epithelium. I. Membrane potentials, apparent resistances, and mechanisms. J Neurosci. 1989; 9: 1968–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gallemore RP,, Steinberg RH. Light-evoked modulation of basolateral membrane Cl- conductance in chick retinal pigment epithelium: the light peak and fast oscillation. J Neurophysiol. 1993; 70: 1669–1680. [DOI] [PubMed] [Google Scholar]
- 64. Griff ER,, Steinberg RH. Changes in apical [K+] produce delayed basal membrane responses of the retinal pigment epithelium in the gecko. J Gen Physiol. 1984; 83: 193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Linsenmeier RA,, Steinberg RH. Origin and sensitivity of the light peak in the intact cat eye. J Physiol. 1982; 331: 653–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Marmorstein LY,, Wu J,, McLaughlin P,, et al. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (best-1). J Gen Physiol. 2006; 127: 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wu J,, Marmorstein AD,, Peachey NS. Functional abnormalities in the retinal pigment epithelium of CFTR mutant mice. Exp Eye Res. 2006; 83: 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wu J,, Marmorstein AD,, Striessnig J,, Peachey NS. Voltage-dependent calcium channel CaV1.3 subunits regulate the light peak of the electroretinogram. J Neurophysiol. 2007; 97: 3731–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Futter CE,, Ramalho JS,, Jaissle GB,, Seeliger MW,, Seabra MC. The role of Rab27a in the regulation of melanosome distribution within retinal pigment epithelial cells. Mol Biol Cell. 2004; 15: 2264–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Williams MA,, Pinto LH,, Gherson J. The retinal pigment epithelium of wild type (C57BL/6J +/+) and pearl mutant (C57BL/6J pe/pe) mice. Invest Ophthalmol Vis Sci. 1985; 26: 657–669. [PubMed] [Google Scholar]
- 71. Booth AE,, Tarafder AK,, Hume AN,, Recchi C,, Seabra MC. A role for Na+ K+-ATPase α1 in regulating Rab27a localisation on melanosomes. PLoS One. 2014; 9: e102851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bensaoula T,, Ottlecz A. Biochemical and ultrastructural studies in the neural retina and retinal pigment epithelium of STZ-diabetic rats: effect of captopril. J Ocul Pharmacol Ther. 2001; 17: 573–586. [DOI] [PubMed] [Google Scholar]
- 73. Williams DS,, Lopes VS. The many different cellular functions of MYO7A in the retina. Biochem Soc Trans. 2011; 39: 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Murakami T,, Iwanaga T,, Ogawa Y,, et al. Development of sensory neuropathy in streptozotocin-induced diabetic mice. Brain Behav. 2013; 3: 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Civan MM,, Marano CW,, Matschinsky FW,, Peterson-Yantorno K. Prolonged incubation with elevated glucose inhibits the regulatory response to shrinkage of cultured human retinal pigment epithelial cells. J Membr Biol. 1994; 139: 1–13. [DOI] [PubMed] [Google Scholar]
- 76. Greene DA,, Lattimer SA,, Sima AA. Sorbitol phosphoinositides, and sodium-potassium-ATPase in the pathogenesis of diabetic complications. N Engl J Med. 1987; 316: 599–606. [DOI] [PubMed] [Google Scholar]
- 77. Pierce GN,, Ramjiawan B,, Dhalla NS,, Ferrari R. Na(+)-H+ exchange in cardiac sarcolemmal vesicles isolated from diabetic rats. Am J Physiol. 1990; 258: H255–H261. [DOI] [PubMed] [Google Scholar]
- 78. Winegrad AI. Banting lecture 1986. Does a common mechanism induce the diverse complications of diabetes? Diabetes. 1987; 36: 396–406. [DOI] [PubMed] [Google Scholar]
- 79. Jo AO,, Ryskamp DA,, Phuong TT,, et al. TRPV4 and AQP4 channels synergistically regulate cell volume and calcium homeostasis in retinal Müller glia. J Neurosci. 2015; 35: 13525–13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Juuti-Uusitalo K,, Delporte C,, Gregoire F,, et al. Aquaporin expression and function in human pluripotent stem cell-derived retinal pigmented epithelial cells. Invest Ophthalmol Vis Sci. 2013; 54: 3510–3519. [DOI] [PubMed] [Google Scholar]
- 81. Zhao PY,, Gan G,, Peng S,, et al. TRP channels localize to subdomains of the apical plasma membrane in human fetal retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2015; 56: 1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Monaghan K,, McNaughten J,, McGahon MK,, et al. Hyperglycemia and diabetes downregulate the functional expression of TRPV4 channels in retinal microvascular endothelium. PLoS One. 2015; 10: e0128359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Antonetti DA,, Klein R,, Gardner TW. Diabetic retinopathy. N Engl J Med. 2012; 366: 1227–1239. [DOI] [PubMed] [Google Scholar]
- 84. Barber AJ. A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry. 2003; 27: 283–290. [DOI] [PubMed] [Google Scholar]
- 85. Barber AJ,, Lieth E,, Khin SA,, Antonetti DA,, Buchanan AG,, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998; 102: 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Han Y,, Bearse MA,, Jr,, Schneck ME,, Barez S,, Jacobsen CH,, Adams AJ. Multifocal electroretinogram delays predict sites of subsequent diabetic retinopathy. Invest Ophthalmol Vis Sci. 2004; 45: 948–954. [DOI] [PubMed] [Google Scholar]