

Abstract
We are currently in an exciting time where our understanding of genetic underpinnings of inflammatory bowel disease (IBD) has undergone a revolution, based in large part by novel genotyping and sequencing technologies. With >160 susceptible loci identified for IBD, the goals now are to understand at a fundamental level, the function of these susceptibility alleles. Clinical relevance of how these susceptible genes shape the development of IBD is also a high priority. The main challenge is to understand how the environment and microbiome play a role in triggering disease in genetically susceptible individual, as the interactions may be complex. To advance the field, novel in vitro and mouse models that are designed to interrogate complex genetics and be able to functionally test hypotheses are needed. Ultimately, the goal of genetics studies will be to translate genetics to the patients with IBD and improve their care.
Keywords: Crohn’s disease, ulcerative colitis, genome-wide association study, causative variants
Introduction
Two major subtypes comprise most cases of inflammatory bowel disease (IBD) and include Crohn’s disease (CD) and ulcerative colitis (UC). Though classification into either CD or UC is usually possible due to distinguishing features, it is important to note that CD and UC have substantial overlapping clinical and pathologic features which appear to be reflected in our emerging understanding of IBD genetics. Clinically, both CD and UC are associated with intermittent, abdominal pain and diarrhea that occurs over a wide spectrum of severity. Pathologically, both forms show acute and chronic inflammation within the mucosa of the intestine that includes cryptitis and crypt abscesses. (1). The major pathologic hallmarks that distinguish CD from UC are ‘skip lesions’ (inflamed areas admixed with uninflamed areas) that can occur anywhere within the gastrointestinal (GI) tract, non-caseating granulomas, as well as fistulas and deep penetrating ulcers. Perianal disease is a manifestation of an aggressive form of CD that is observed in a subset of patients (1). Conversely, UC classically affects the distal rectum and has the potential to extend proximally (2); the upper GI tract and ileum are usually not involved. Pathologically, UC lesions are homogeneous without skip areas and the inflammation is limited to the mucosa. Typically these ulcers do not penetrate into the muscle wall. A small subset of patients initially present with symptoms and pathology that doesn’t easily distinguish between CD and UC. These patients are classified as IBD-undetermined type, and can evolve into a more UC or CD-like clinical pictures in time. In all cases, cure is rare but clinical remission is often achievable by current medical therapies. The major clinical issues include the lack of durability of medical therapy, the high risk for eventual interventions (~50% of IBD cases) and the difficulty in managing patients post-resection.
The study of IBD genetics has flourished over the past decade and has contributed significantly to our understanding of the pathogenesis of IBD (Figure 1). To date, the majority of these studies have focused on patients in areas that historically have had high prevalence, though additional exciting studies are ongoing throughout the world including areas where IBD is rapidly increasing in incidence. In predominately Caucasian populations (North America, Europe and Israel), >160 susceptible loci been identified predominately through large-scale (>75,000 patients and controls) genome wide association studies (GWAS). Interestingly, there are a significant proportion of shared genes between UC and CD (3; 4). Through the application of state-of-the-art sequencing technology, susceptibility loci have been associated with specific genes or regulatory regions (5; 6). In addition, IBD geneticists have begun to identify rare variants and some of the causative genes that are transmitted through Mendelian inheritance (7). Taken together, this work has suggested that alteration of complex networks of host genetics, immune dysregulation, and environmental factors can trigger or exacerbate IBD (8; 9).
Figure 1.
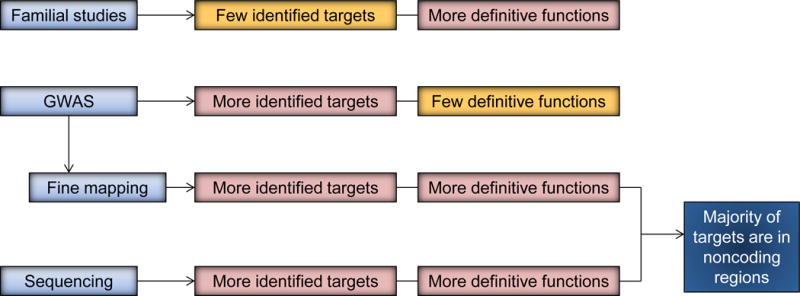
The characteristics of different study designs and methodology of IBD genetic studies. Familial studies represent a past approach that identified a few specific targets (e.g. NOD2). Currently GWAS with fine mapping are able to identify greater number of loci; however, not all of these loci have defined gene targets. Currently whole exome and whole genome sequencing have the potential to specifically defined more genetic targets.
Common vs. rare genetic variants
The biological basis and impact on disease differs when comparing rare and common polymorphisms (10–12). Common human genetic polymorphisms, as the term implies, occur in a given population with a minimum frequency of 5% (13). In IBD, many identified susceptibility loci are found at a much higher frequency (typically 20–50%). In part, because of this high frequency in target populations, several susceptibility alleles were identified in early, smaller cohort GWAS studies (4; 14) because of the greater power to detect associations of IBD with such variants (10–12). It is believed that such common variants that are associated with increased IBD susceptibility are retained in a given population because of some advantage that is conferred by the polymorphism. However, discussed later, common genetic variants identified through these studies typically have modest effects on IBD susceptibility, with most odds ratios < 1.1 for a given locus. In addition, the majority of these common variants are located in the non-coding regions (10–12). In contrast, rare variants, defined as occurring with < 0.5% frequency, are often specific to certain populations, and are highly deleterious. Rare variants are often missense alleles, and with greater effect sizes (odds ratio > 1.5). There is, however, less power with these variants to uncover associations with IBD (10–12). For example, GWAS on the order of 10,000 patients will miss these variants. Such variants have been associated with very early onset IBD (VEO-IBD), specific polymorphisms in adult IBD (i.e. NOD2)(5) and other mutations associated with immunodeficiency (15; 16).
Genetics of VEO-IBD
In addition to providing insights into pathogenesis, genetic studies in specified cohorts for IBD (eg. VEO-IBD) are clinically useful, as these cases are not easily diagnosed with standard histological and immunological features (17). The results from genetic analysis can also be used for genetic counseling for family members of patients (17). Thus, it has tremendous clinical implications beyond pathogenesis research (17). The key geneticalterations in VEO-IBD have been uncovered through a combination of genetic mapping, association studies and ultimately exome sequencing of specific patients (either global or targeted). As mentioned above, rare variants are often monogenic (17). Most identified genetic causes of VEO-IBD are associated with defects in the epithelial barrier, neutrophils, T-, and/or B-cells, regulatory T cells, and IL-10 signaling, or association with other hyperinflammatory and autoinflammatory disorders (17), Because of the well-defined genetics in VEO-IBD patients, translational studies identifying involved pathways and potential environmental triggers can lead to identification of therapeutic targets (Figure 2).
Figure 2.
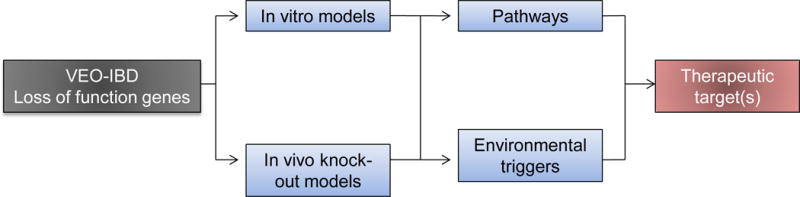
Translational functional studies based on the genetic studies of loss of function genes (i.e., VEO-IBD) can lead to identification of potential therapeutic targets. Due to the severity of the mutations, approaches using knock-out models, both in mice and cell culture, are well poised to develop systems to discover pathways targeted by these mutations as well as environmental triggers. This approach can potentially lead to novel therapeutic targets.
NADPH oxidase
Multiple SNPs associated with increased risk of IBD are located in coding regions of multiple NADPH oxidase complex genes. These mutations reduce reactive oxygen species (ROS) formation, but do not lead to chronic granulomatous disease (CGD). Specifically, SNPs associated with coding variants of the NCF2 gene (a.k.a. p67phox) were found in 4% of VEO-IBD patients. These mutations all affect binding with the downstream signaling molecule RAC2 and result in reduced ROS production (18; 19). Subsequently, several investigations focused on the genetic landscape of VEO-IBD and associated immunodeficiency disorders, including CGD, and primary immunodeficiency X-linked lymphoproliferative disease type 2 (XLP2). The rationale was that such cases are relatively rare and the hypothesis was that there were common genetic mechanisms. This was the case, as they found additional SNPs associated with genes that are part of the core NADPH oxidase complex genes (20). The majority of the VEO-IBD patients with the NADPH oxidase-associated SNPs were heterozygous for these mutations as compared to patients with CGD that are homozygous for such mutations. Other genes associated with the NADPH oxidase pathway included Annexin A1, which in mouse models has been shown to promote epithelial wound repair through a complex formed with Rac1, p120, Vav2, and Nox1 which in turn leads to activation of Src (21). In a follow up study, Leoni and colleagues showed that endogenous Annexin A1 can be released by intestinal epithelial cells as a component of extracellular vesicles, which activate wound repair process (22). Interestingly, patients with IBD have higher serum levels of Annexin A1-containing extracellular vesicles (22).
Tetratricopeptide repeat domain 7
In a separate study, patients with variants of the tetratricopeptide repeat domain 7 (TTC7A) gene were identified within a small cohort (n=41) of severe VEO-IBD cases. Whole exome and targeted sequencing showed truncation of the TTC7A gene. In vitro studies with primary intestinal epithelial cells grown as organoids showed that the TTC7A mutations reversed the apical and baso-lateral surfaces leading to organoid inversion. In addition, the epithelial cells in these organoids showed decreased adhesion, increased apoptosis, and decreased phosphatidylinositol 4-phosphate production (23). In a separate study, intestinal organoids isolated from patients with combined immunodeficiency and TTC7A mutation (determined by whole exome sequencing) also showed inversion of apicobasal polarity of the epithelial cells (24). Interestingly, the epithelial abnormality could be reversed pharmacologically by Rho kinase inhibitor (24).
XIAP
Rare variants in XIAP were identified in 4% of male VEO-CD patients in a study from Germany. Interestingly, the in vitro mechanistic studies suggested the lack of functional XIAP in immune cells from these patients may be related to NOD1/2 signaling rather than other alterations in innate and adaptive immunity (25). Pediatric IBD with these CD-unique private mutations tend to have ileocolonic disease location and respond to traditional therapy. Moreover, while the sample size is small, XIAP-deficiency does appear to carry a higher penetrance than NOD2-deficiency (25).
IL-10 receptors
IL-10 receptor genes (IL-10RA and IL-10RB) were among the first genes shown to be associated with early onset IBD (26; 27). The major function of this receptor is to suppress inflammation and tumor necrosis factor (TNF) secretion as validated in human monocytes from patients with deficient IL-10R. These patients are characterized by refractory colitis and perianal disease, but lack significant small intestinal involvement (26; 27). Interestingly, allogeneic stem cell transplantation appears to be a promising therapeutic approach (26; 27). Importantly, deficient IL-10R has not yet been observed in adult onset IBD. These studies highlight how genetic findings associate with clinical phenotype and can be translated to patient management (26).
Animal studies with targeted knockout of IL-10R genes have revealed its role in specific cell types. Within intestinal epithelial cells, CD1d protects inflammation in a IL-10/STAT3/HSP110-dependent manner (28). In both resident intestinal and bone marrow-derived macrophages, IL-10R deletion resulted in enhanced proinflammatory cytokine and reduced anti-inflammatory cytokine secretions (29). Interestingly, in mouse models, the IL-10 produced by macrophages did not affect gut homeostasis, supporting the clinical finding of IL-10R deficiency in VEO-IBD patients (30). Variants of other immunodeficiency-associated genes with somewhat variable age of onset of intestinal inflammation includes Wiscott-Aldrich syndrome, immune dysregulation, polyendocrinology, enteropathy, X-linked (IPEX; associated with FOXP3), and common variable immunodeficiency (LRBA) (17). Importantly, in a mouse model of immunodeficiency (HOIL-1 knockout), a hyperinflammatory phenotype can be induced by chronic Mycobacteriaum tuberculosis or murine γ-herpesvirus 68 infections, thus highlighting the importance of environmental factors in phenotypic studies of immunodeficiency (31).
IBD genetics in defined populations
Genetics studies for IBD (and other diseases) have benefited by the investigation of specific population subsets. For example, the modern Ashkenazi Jewish population resulted from recent narrow population bottleneck (~350 individuals) followed by rapid expansion (32). This population is known to have a distinct genetic landscape and harbors a high prevalence of autosomal recessive diseases as well as a higher risk of other common diseases. A recent study involving 128 complete genomes of Ashkenazi Jewish individuals showed 47% more novel variants per genome than control populations (32). Further studies confirmed the complementary value of studying this population. Novel IBD susceptible genes included RPL7, CPAMD8, PRG2, PRG3, and HEATR3 (33; 34). These genes are of unclear function and are targets for further study. This study supports the notion that variants that are rare in the general population are relatively not rare if studied in a defined population with enhanced susceptibility to IBD.
Another population where genetic associations have begun to shed light on IBD pathogenesis is Asian populations. Previous studies showed that Japanese and Korean IBD patients do not harbor NOD2 variants (which is specific to patients from European ancestry), and the contribution of ATG16L1 T300A to pathogenesis is likely minor (35; 36). GWAS studies from CD patients in Japan and Korea showed that while there is a fraction of susceptibility alleles that overlap with Caucasians with CD, there is clearly a panel of distinct alleles that show significant association in these Asian cohorts (37; 38). For example, TNFsf15 and MHC II are both highlighted by studies from Japan and Korea to be highly associated with CD (35; 36). Further studies with larger cohorts will clarify whether susceptibility alleles associated with ATG16L2 and ELF1 are significant in Asian CD patients (35; 36). In contrast, there is greater overlap with Asian and Caucasian populations when comparing UC susceptible loci (39). Thus, further study of these ethnically distinct populations will broaden our understanding of the genetic contribution of specific alleles and can help identify novel environmental triggers. Of note, many of the published genotyping work from these ethnically distinct populations were conducted using SNP chips based on Caucasian genetics. One future direction is to compile such chips that are based on the genetics specific to these populations.
The concept of genetic burden
The previous discussed rare genetic variants are associated with a nearly 100% likelihood of developing IBD. However, most susceptibility alleles for IBD are based on common variants and are therefore far less penetrant. Some studies have suggested that the cumulative genetic burden of these common genetic variants is associated with an early age of diagnosis. This has been reported for CD, but not UC (40) and the overall susceptible loci only account for ~30% of all cases (3; 40). These types of genetic studies are very challenging and require substantial populations of IBD patients that are well-phenotyped and genotyped.
Fine-mapping of the susceptible genes
GWAS studies identify markers that map to blocks of linkage disequilibrium and may extend from tens to hundreds of kilobases away from the target locus and may contain many genes (41). Functional involvement of any given gene is therefore not defined simply by the proximity of the gene to the most associated variant in the block (41; 42). One economical method to identify gene associations with candidate polymorphisms is through a process of fine mapping to SNPs that are concentrated to areas of susceptibility loci. Algorithms such as conditional regression can be used to determine whether the effect of each variant explains linkage disequilibrium (41). Designed in 2009 by researchers of 11 distinct autoimmune and inflammatory disorders and further developed by Illumina, Immunochip is a SNP microarray that interrogates 195,806 SNPs and 718 small insertion-deletions (43; 45). The use of Immunochip allowed deep replication of previous GWAS data, including the top 2,000 independent association signals for each disease (45; 46). In addition, it also allowed fine mapping of the loci identified by other previous GWAS (3; 35; 41–44). Another recent study using this high-density genotyping approach including 7,406 SNPs within the MHC region on >30,000 IBD patients and >30,000 control subjects showed a primary role for HLA-DB1*01:03 in both CD and UC (47). Thus, targeted high density mapping could potentially reveal additional susceptible loci.
By analyzing the 200,000 known SNPs associated with autoimmune disease-associated loci, Farh et al. found that approximately 90% of the causal variants are in non-coding regions, whereas 60% mapped to enhancers of immune cell types (42). This landmark study represents an important next step to advance GWAS studies. One recent study provided clues to the functional implications of the IBD causal variants in non-coding regions. Up to half of the tested SNPs (91/216) were associated with super enhancers of T cells, and these super enhancers were selectively targeted by tofacitinib, a JAK inhibitor (48). Super enhancers are a group of transcriptional factors important for genes associated with cell identity and genetic risk of disease (49). Thus, such a comprehensive super enhancer landscape of relevant cell types can be used in IBD genetics to facilitate discovery of druggable targets. In the future, whole genome sequencing will likely be the standard method to pinpoint IBD susceptibility loci.
Overlap with other diseases
One important consequence from the IBD Immunochip project was the discovery of overlapping susceptible loci of IBD with other diseases (3). A significant proportion (70%; 113/163) of the loci identified is shared with other complex diseases or traits (3). The spectrum of diseases with shared susceptible loci includes immune mediated diseases (e.g. rheumatoid arthritis), primary immunodeficiency, Mendelian susceptibility to mycobacterial disease, and leprosy (3). Susceptibility loci associated with IL12B, STAT1, IRF8, TYK2, STAT3, IFNGR2, and IFNGR1 are found in both IBD and mycobacterial infection, whereas susceptibility loci associated with NOD2, RIPK2, TNFsf15, LRRK2, IL23R, and C13orf31 are shared between IBD and leprosy (3). Interestingly, anti-TNF, one of the most effective therapies for IBD, can be associated with re-activation of latent mycobacterial disease (50). Thus, these findings may provide additional mechanistic insights into therapy-associated adverse effects.
Protective loci
The various GWAS, fine-mapping, and sequencing studies identified several protective alleles for IBD (3; 5; 14). These alleles are associated with IL23R, CARD9, NOD2, and PTPN22. Among the IL23R SNPs associated with CD, the less common glutamine allele of R381Q confers approximately 3-fold protection against developing CD (14). Further deep sequencing studies identified two additional rare, protective alleles in the IL23R gene, namely G149R and V362I (5). Given the critical role of IL23R in the differentiation of CD4+ Th17 cells, these findings suggest that modulating the IL23/Th17 pathway may be an important therapeutic approach. Indeed, ustekinumab, a monoclonal antibody targeting IL12 and IL23, shows great promise in CD (51).
A splice-site variant in CARD9 was also associated with a protection effect for IBD (5). The hypothetical transcript was found in cDNA from many cell types, including spleen, lymph node, and peripheral blood mononuclear cells (5). CARD9 functions as an adaptor protein that is downstream of cell surface receptors that include Dectin-1 which binds components of fungi. Functional studies with knockout mice for CARD9 show alterations in the intestinal microbiome and defects in wound repair and clearance of pathogenic bacterial infections (52).
One interesting finding from the GWAS-Immunochip study was that two CD susceptibility loci, PTPN22 and NOD2, showed significant protection effects for UC (3). PTPN22, a protein kinase phosphatase expressed mainly in hematopoietic cells, is associated with other immune diseases, including rheumatoid arthritis, systemic lupus erythematosus. type I diabetes, multiple sclerosis, and Grave’s disease (53). The risk allele associated with these diseases is associated with diminished IL-2 production of T cells upon TCR stimulation, and is a more potent negative regulator of T lymphocyte activation (53). A separate study showed that the R620W gain of function variant was significantly associated with CD, whereas the R263Q loss of function variant was associated with UC (54). Mechanistically, however, while it is likely that R263Q involves regulating lymphocyte activation through modulating Src family kinase, the functional consequence of the polymorphisms in human and mice are not well studied (55). Likewise, while the roles of NOD2 in inducing inflammation is known (56), the mechanisms of which a susceptible allele for CD may be a protective allele for UC is still unclear.
Expression quantitative trait loci (eQTL) mapping
Expression quantitative trait loci (eQTL) is a method to correlate specific polymorphisms to the abundance of certain gene transcript(s) and identify potential causal variants (57). This method is performed by correlating genotyping data with transcript abundances (either by RNA microarray or RNA seq analysis) within a tissue of interest. Other factors that alter the transcription status (e.g. epigenetic changes) can also be integrated (57–59). The resultant gene expression profile and that of eQTL can be further used to study potential gene-gene interactions and networks (59).
The complexity of the eQTL approach can be illustrated by the choice of a relevant cell type and the optimal physiologic context. Using 56 cell types and 21 immune-mediated diseases for the analysis of cis-regulatory elements, Farh et. al. found that transcripts for immune cells were largely enriched in IBD patients, compared to intestinal mucosa (42). In addition, they also found that the SNPs identified through GWAS and eQTLs from peripheral blood comprise a different landscape – noteworthy is the involvement of enhancer region in up to 60% in the GWAS SNPs vs. 14% in the eQTL SNPs (42). A separate study by Fairfax et. al. took the approach further and investigated in a ‘context-specific’ manner, in which peripheral monocytes from healthy individuals were stimulated with lipopolysaccharide (LPS) or interferon-gamma (IFN-ɤ) (60). Stimulation with LPS or IFN-ɤ resulted in local changes in eQTL (cis-eQTL) in immediate short term but also distant changes (trans-eQTL) on separate gene sets (e.g. MHC II), which collectively support an IFN-signaling cascade (60). Notably, induced eQTL were significantly enriched for GWAS loci, thus providing an independent signal for disease association (60). Therefore, it is plausible that the currently defined eQTLs may likely represent only a subset of the genuine eQTLs, and further studies taking the context-specific approach may yield further insight into the regulation of eQTL and the functions of identified polymorphisms.
Gene-gene interaction
Gene-gene interaction in IBD has been investigated using various statistical modeling approaches, including logic regression, and predictability of disease was assessed by area under the curve (AUC). For instance, a recent study by analyzing the UC GWAS datasets showed potential interactions between HLA-DQA1, RIT1/UBQLN4, and IFNG/IL26/IL22, highlighting known pathways of host response to microbial organisms (61). Likewise, a similar study performed in pediatric CD patients found additive gene-gene interaction involving TLR4, PSMG1, TNFRsf6B, and IRGM (62). However, while this is clearly of interest, there are very limited validation studies.
Causal pathways and networks
The abundant data generated from GWAS and transcriptional profiling studies identified signaling pathways that regulate IBD pathogenesis. The complexity of the affected pathways revealed potential signaling networks composed of various pathways that are critical for the pathogenesis of IBD. TNF, for example, can be secreted by a variety of cell types, including macrophages, dendritic cells, T cells, adipocytes, and fibroblasts. In turn, TNF acts on targeted cell types including effector T cells, endothelial cells, intestinal epithelial cells, Paneth cells, and myofibroblasts. NF-kB, a central player in TNF signaling, is also critical in the signaling of many other important pathways (i.e. TLRs) (63). Indeed, the link between NF-kB activation and intestinal inflammation and subsequent intestinal epithelial cell death is well documented (34; 64). Thus, by connecting the identified signaling pathways and networks, one is able to determine the key modulators which are the most connected ‘hot spots’ (3). Further superimposed on these networks are host-microbial interactions which will be discussed later.
Identifying environmental factors
Clinical clues are abundant that environmental factors play a prominent role in the IBD pathogenesis. The incidence rate of IBD is rapidly increasing worldwide and most notably in areas where IBD was rare prior to the 1990s (65). More detailed examinations of specific European and North American countries further support the role of the environment. In Spain, a north-south disease gradient occurs where the incidence is highest in the northern region of Spain and gradually decreases toward the southern region (66). Likewise, in Canada, an east-west disease gradient occurs with the highest incidence in the eastern region (66). Though interesting, the genesis of these population gradients is unclear. In addition, while patients with mutations in IL-10R gene will develop spontaneous colitis with 100% penetrance, in vivo studies using mouse model with deficiency in IL-10 signaling highlight the importance of the environmental triggers, in the development of colitis, and in this case, certain commensal bacteria that trigger colitis (67). Thus, it is possible that the required “dosage” of environmental exposure that will trigger IBD correlates with the genetics of the patients (Figure 3). For example, patients who harbor one of the Mendelian gene mutations or rare variants may require less of a dose (e.g. specific commensal microbe) to trigger disease, whereas patients with common variant(s) may require a greater exposure dosage (e.g. complex environmental trigger).
Figure 3.
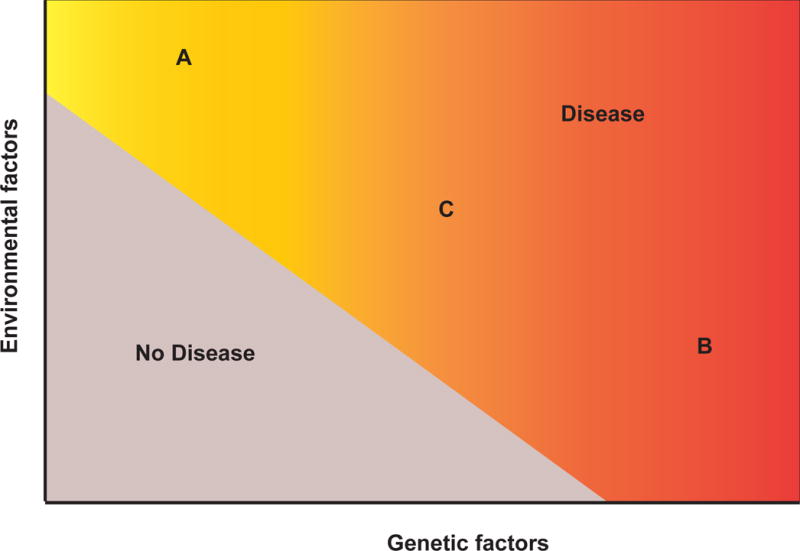
Hypothetical thresholds for genetic and environmental factors required to trigger IBD. The three scenarios include (A) environmental factors are dominant and requires less genetic risk factors (similar to an infectious disease model); (B) genetic factors are dominant (e.g. VEO-IBD), which likely requires less environmental dosage; and (C) ‘medium’ dosage of genetic and environmental factors, similar to other autoimmune disorders.
Several potential environmental factors have been evaluated in detail and include smoking, diet, use of antibiotics, and overall sanitization status. In Western countries, smoking consistently is shown to be detrimental for CD but protective for UC (66). In contrast, a recent Asia-Pacific study failed to identify any link between smoking status and the incidence of CD (68). Interestingly, in Asia, ex-smoking appears to be a risk factor for developing UC (68). The polymorphisms of various nicotine metabolizing enzymes have been postulated to be associated with an effect on the incidence of IBD in patients who are exposed to smoking. Indeed, a recent study showed that SNP of one such gene, CYP2A6, may increase the incidence of CD among smokers (69). Similarly, the SNPs in GSTP1 gene is associated with increased risk of UC in ex-smoker (69).
The mechanism by which smoking modulates intestinal inflammation in IBD patients is still unclear, but animal studies provide some insights into its diverse effects in both the small and large intestine (70). However, one major limitation of these animal studies is the lack of standardization for smoking exposure. The type of exposure ranged from nicotine patches, cigarette extracts, to actual cigarettes, and amongst each type of exposure, the dosages used are variable among studies (70). Added to the complexity is the use of various injury models (i.e. chemical- vs. pathogen-induced colitis) to trigger intestinal damage, as smoking by itself does not result in spontaneous enterocolitis in most animal models (70). Nonetheless, in vitro application of cigarette extract on patient-derived dendritic cells showed that it increased the prevalence of Foxp3+ CD4 T cells in UC whereas it skewed toward Th1 subset in CD (71), suggesting a possible differential effect in immune cells in CD vs. UC. One possible scenario is that to trigger/aggravate IBD requires combinations of specific sets of genes and smoking exposure, thereby explaining the seemingly different effects in patients from different ethnic groups. Future studies investigating the potential interplay between susceptible genes in Asian vs. Caucasian IBD patients in this regard may yield new insights.
Given the increased incidence and prevalence of obesity and associated metabolic symptoms over the last decades, which coincide with the increased incidence of that of IBD, the role of diet in the pathogenesis of IBD must be considered (65; 66; 72). In humans, genetics only appears to account for a small part of the increase in body mass index (~5%) (73; 74). Preclinical models have highlighted the diverse impact of nutrients in regulating intestinal mucosal immunity and shaping microbiota composite, both of which have significant implications to the pathogenesis of IBD (72; 75). In addition to human data, animal studies illustrate how certain diets (e.g. “Western” diet) can induce dysbiosis (76), presumably through affecting the metabolites (77; 78), which in turn is associated with host mucosal barrier dysfunction. Studies investigating the impact of diet on microbiota and metabolomics are ongoing and will be of particular interest in the future.
Challenges in studying gene-environment interactions in IBD
The notion that a combination of genetic and environmental factors is required in causing a disease phenotype is not a novel concept in medicine. This is illustrated by studies from phenylketonuria, an autosomal recessive disease (79). Individuals with mutations in phenylalanine hydroxylase require exposure to phenylalanine in the diet to develop the disease (80). Likewise, in intestinal tract, the pathogenesis of Celiac disease involves susceptible genes (HLA-DQ2 and DQ8) and environmental exposure (gluten-containing diet) (81). It is therefore perceivable that certain gene-environment interactions are important in the pathogenesis of IBD. However, unlike phenylketonuria or Celiac disease, both of which involves relatively limited numbers of susceptible genes and known triggers, IBD is a complex genetic disease with as yet unclear triggers. Therefore it is very difficult to study the gene-environment interactions in these patients. Added to the complexity is that the ‘dosage’ of the environmental exposure is difficult to quantify. Therefore, the traditional 2 × 2 approach of dichotomous genotype (dominant and recessive) and dichotomous exposure (plus and minus) used in traditional genetic epidemiology studies do not necessarily address these questions (79).
The study design is another challenge when trying to incorporate the massive information obtained from the various GWAS and fine mapping studies to identify gene-environment interactions. The GWAS and fine mapping studies are performed mostly by a cross-section and/or case-control (retrospective) study design (82). While this is the most cost-efficient approach for genetics studies, for identifying environmental exposure, there is a high potential for selection bias and recall bias (79; 82). Nonetheless, the potential environmental exposure identified from these studies (if available), can be used as a screening for further validation (79). The use of various pre-existing databases for nested case-control studies may be used as additional sources to identify environmental exposure. We envision environmental factors will be clustered based on their effects on specific genotype of the IBD patients (Figure 4). For example, as seen in the genetic studies in Caucasian and Asian CD populations, while many susceptible loci are shared between the two populations, there are clear subsets of susceptible alleles that are predominantly identified in each population. Likewise, it is possible that environmental triggers will follow the same classifications.
Figure 4.
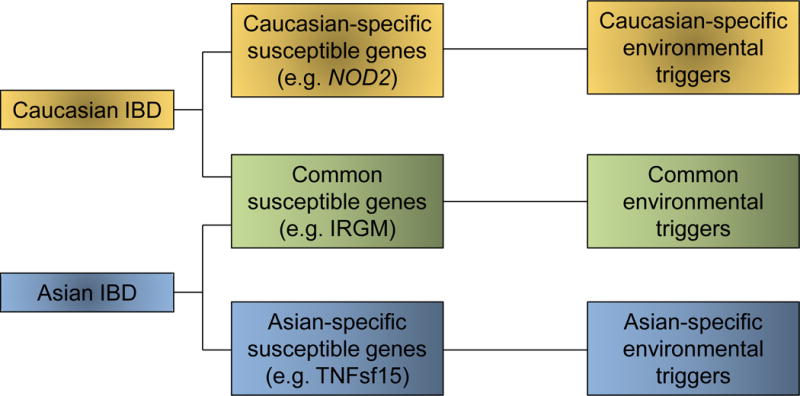
Different combinations of genetic and environmental factors may contribute to development of IBD in different ethnic populations. Caucasian and Asian populations appear to have some shared genetic risk factors, while others are distinct. What remains to be seen is whether the distinct genetic factors would affect common pathways. The other unknown issue is the role of environmental factors for each ethnic group.
In addition to study design, sample size possesses another challenge. For instance, using a simplified model of dichotomous genetics and dichotomous environmental exposure, the minimum sample size required to determine the joint effect of gene-environment is at least 4 times the sample size that is needed to evaluate the effect of each of the component (83). Therefore, assuming this very simplistic model, the currently available database designed to study IBD genetics is not powered to study gene-environment interactions. Further statistical models and new studies are necessary and must take into account more complex models.
Even with sufficient sample size, additional questions remain. As in genetics one has to differentiate linkage disequilibrium from true causative variants, many of the environmental factors are part of complex mixtures and are highly correlated with each other, thus separating the effects of each individual environmental factor may not be easy.
Role of microbiota in IBD patients
The importance of gut microbiota in induction, training, and function of the immune system is well recognized (84). In addition, elements of the environment may act on host indirectly through the microbiome. Thus, understanding the details of host-microbial interactions is highly relevant to IBD.
Recent studies of the microbiome in IBD patients, including improvements in the standardization of methodology and study design, have advanced our understanding of IBD pathogenesis. However, while these data are intriguing, most studies provide associations and not cause-effect relationships (85–87). For instance, studies examining the microbiome from fecal and mucosal biopsies from twins with IBD have confirmed the reduced diversity and changes in abundance in certain phyla in ileal CD but not colonic CD as compared to healthy controls (88). This study however, found that the effect of disease phenotype was greater than that of the host genotype (88). A similar study examining the fecal microbiota from CD patients showed that unaffected relatives of CD patients also have a different composition of microbiota compared to healthy controls, including less Collinsella aerofaciens, a member of the Escherichia coli-Shigella group, and more Ruminococcus torques, suggesting a potential subclinical mucin degradation in unaffected relatives of CD (89). It was, however, unclear whether any of the unaffected relatives developed CD later, which would be suggestive of early disease-causing mechanism. In a separate study, the microbiome of a large cohort of treatment-naive pediatric CD patients was analyzed. They found that the microbiome of CD patients contained an increased abundance in specific bacterial families including Enterobacteriaceae, Pasteurellacaea, Veillonellaceae, and Fusobacteriaceae, and decreased abundance in Erysipelotrichales, Bacteroidales, and Clostridiales (90). In the same study, it was found that the degree of mucosal dysbiosis was greater in patients who had previous exposure to antibiotics, suggesting that the dysbiosis status may be affected by antibiotics usage (90). No genetic correlations were performed in this study.
Further studies have examined correlations between the host genotype and mucosal microbiome by comparing areas with active and inactive disease in CD patients. The hypothesis was that certain host genotypes (i.e. ATG16L1 T300A) predispose to defects in microbial clearing by immune cells, and hence the geographic composition of microbiome in active areas may be different then in the uninvolved areas. Indeed, the microbiome of inflamed ileums in CD patients that contained the ATG16L1 T300A SNP was associated with greater abundance of Bacterioides, Fusobacteria and E. coli, and less Lachnospiraceae compared to patients with wild type ATG16L1. However, there was no difference in microbiota composition in uninvolved areas when considering genotypes (91). Another study that analyzed the fecal microbiome from 416 twin pairs without IBD showed that the abundance of several microbial taxa can be shaped by host genetics (92).
In UC patients, one study showed a lower abundance of Roseburia hominis and Faecalibacterium prausnitzii compared to controls, and both species were inversely correlated with disease activity (93). These bacteria are thought to have beneficial properties including the production of short chain fatty acids (94). Interestingly, it was noted in a separate study that there was a reduction in short chain fatty acid in UC patients (predominantly propionate and acetate but not butyrate) (93). As larger IBD cohorts are analyzed for the microbiome composition, the impact of host genetics will need to be addressed.
The ‘genetics’ of the microbiome
Many investigators are utilizing mouse models to functionally test components of the microbiome and determine how it interacts with the host immune system (95). The importance of homeostasis of the microbiome in reducing inflammatory diseases is demonstrated in a study by Olszak et. al., where germ-free animals are more susceptible to chemical-induced colitis compared to specific pathogen-free animals (96). Exposure of germ-free animals to specific pathogen-free conditions early in life, but not in adults, ameliorated the colitis. Furthermore, this was shown to be mediated by invariant natural killer (NK) cells through chemokine ligand CXCL16 (96).
Another emerging concept for role of the microbiome is metagenomics. Several recent studies have highlighted that microbial genes are ‘inherited’ by maternal transmission and strongly influence phenotypes much like the effects of host genetics. We have recently shown that maternal transmission of specific bacteria are important in modulating mucosal immunity and inflammation through immunoglobulin (Ig)A (97). In another study, mice with early life contact with commensal microbes (maternally transmitted) establish mucosal tolerance to later environmental exposures. There is experimental evidence that shows this is mediated through invariant NK T cells (96). The emerging understanding that microbial genes can influence inflammation will add a layer of complexity to our ongoing studies of IBD genetics in humans.
In addition to bacterial microbiome, recent advances include the identification of enteric virome associated with IBD. By analyzing different cohorts of IBD patients from geographically distinct locations, Norman and Handley et. al. found that both CD and UC patients have alterations in the enteric virome compared to control patients, with enriched presence of Caudoviralies bacteriophages (98). The virome appear to be CD and UC disease and cohort specific, and importantly, the changes were not associated with bacterial population changes (98). While functional studies are clearly needed to explain the mechanisms, future studies of IBD microbiome need to take into consideration of possible interplay between enteric virome and bacterial microbiome.
The role of IgA, which is secreted exclusively in the mucosa, and its interplay with gut microbiota have been recently investigated. A fraction of the microbiota in the fecal samples in CD patients, but not non-IBD controls, are coated with IgA, thus allowing sorting based on the IgA coating patterns (99). These bacteria coated with IgA, when transferred to germ-free mice, were able to aggravate the severity of intestinal damage upon treatment with Dextran-sodium sulfate (DSS) (99). Thus, commensal microbiota with high IgA coating may play a role in triggering colitis in IBD. More recently, we have found that fecal IgA can be used as a marker for microbial variability, as mice with low IgA concentration in the feces (not the level of IgA coating; to be differentiated from the abovementioned study) showed increased intestinal damage in DSS injury model (97). The microbes associated with the IgA-low mice (enriched for Sutterella species) can degrade at least the bound secretory component of IgA partially through proteolysis (97). Thus, identification of additional microbial species that modulate IgA status may be of interest in further understanding the role of microbiota in IBD.
Next generation animal models to evaluate host and microbial genetics
One important approach for understanding the functional consequences of causative genes is through genetic engineered animal models. Mouse models continue to be important due to similarities with humans in terms of epithelial cell biology, immunology, microbial ecology and physiology. One key CD susceptibility gene that has been highly studied in mouse models is Atg16L1, because of its significance in GWAS studies and that the major susceptibility allele is a coding variant (100; 101). Early in vivo studies were conducted using multiple hypomorphic mouse lines (Atg16L1HM) generated through the gene trap. We found that Atg16L1HM mice displayed abnormal Paneth cell granule morphology and secretion defects in antimicrobial peptides (102). Importantly, defects in Atg16L1HM Paneth cells were triggered by a chronic viral infection (102). In addition, we found similar abnormalities in Paneth cells in a subset of CD patients with susceptibility mutations in ATG16L1 and NOD2 (103; 104). Atg16L1HM mice were used to evaluate the effects on bacterial infection by the pathogen Citrobacter rodentium. These mice showed a resistance to infection by this pathogen suggesting that diminished function of Atg16L1 can be protective in this circumstance (105). Atg16L1 interacts with other pathways that affect gut inflammation. Atg16L1 function is required to counteract effects of loss of the unfolded protein response and inhibit inflammation (64).
More recently, mouse models with a knockin of the Atg16L1 T300A susceptibility variant have been generated (100; 101). Comparing the Atg16L1HM and Atg16L1 T300A mice, while Paneth cell defects were observed in both, the magnitude was largely reduced in the T300A mice, which more closely reflects what was seen in CD patients (103). This is important as it demonstrated how the phenotypes tend to be amplified in mouse models with complete or nearly complete loss of function. The subtle changes created by susceptibility mutations will require close attention. In addition, an unexpected finding with the ATG16L1 T300A mice is the central role of the ATG16L1 T300A SNP in apoptosis rather than autophagy as originally hypothesized (100; 101). Under conditions of cell stress, the T300A allele is more sensitive to degradation. In our opinion, future animal models focusing on IBD-specific polymorphisms will yield further insights into the functions of the genes and polymorphisms. In addition, while limited studies comparing knock-out and knock-in approaches that were done showed that the phenotypes seen with knock-in animals may be a subset of the spectrum of what can be seen with knock-out animals. However, there may be genes where the phenotypes between the two approaches may be distinct.
Another major challenge in IBD is a dearth of truly relevant mouse models of enteritis/colitis. Most of the studies require induction with chemical agents (e.g. DSS) on genetic susceptible hosts (102; 106). This is a suboptimal set up and does not recapitulate the development of enterocolitis in patients.
Focus on cell types in IBD genetics
One logical next question following identification of any given potential causative variant is whether the presence of certain polymorphisms elicits different effects in different cell types, and whether the impact is more important in certain cell types. Currently there are two major approaches to address this issue. The first is the development of mouse models using cell type-specific engineering (e.g. intestinal mucosa vs inflammatory cells) which offer great insight in delineating the how variants function within a given cell type. For instance, mice with deficient Atg16L1 in the intestinal epithelial cells, but not CD11c-expressing dendritic cells, showed defective clearance of Salmonella infection (107). This study provides mechanistic insights into how specific genes function in tissue. A second approach is through eQTL mapping (as discussed above). This approach, while currently limited, can be enhanced by combining a ‘context-specific’ stimulus to determine the eQTL under more disease-relevant physiologic conditions (60). The data generated herein can in turn inform and guide experimental designs for creating animal models.
Models to study complex genetics
As described above, the state of the art animal models for IBD studies have shifted from the traditional knock-in and knock-out approach to a fine mapping and targeted cell type engineering. The recently described CRISPR-Cas9 system will certainly facilitate these approaches (108). However, while technologies have improved for generating animal models, up until recently it was difficult to manipulate more than two genes simultaneously. For instance, Il-10 knockout mice that develop spontaneous colitis have been used widely as a model for IBD(29; 109–111). We believe this approach will continue to be important for understanding the functional consequences of single genes, in particular the Mendelian genes and rare variants. However, for the majority of the IBD cases that involve complex genetics, this approach may not provide directly biologically relevant information. Novel technology that allows manipulating multiple genes simultaneously in animals will allow further facilitation of the IBD genetics research. Alternatively, given the complexity of the network of genes and pathways involved, studying cells and tissue directly isolated from the patients may prove to be informative. Recent advances in streamlining the culture conditions have enabled a high throughput assay to create such a platform (112; 113). This will allow better understanding of the biology and have the potential to enable personalized medicine.
Genotype-phenotype correlation and clinical applications
In addition to providing insight into etiopathogenesis of IBD, another primary goal of IBD genetics is to provide new information that will translate into better diagnostic, prognostic, or therapeutic applications/decisions. Progress towards this goal has been best in VEO-IBD. Diagnostically, when clinicians encounter young (< 6 years old), male patients with enterocolitis, patients from consanguineous family (in particular those with family history of IBD), or patients with primary immunodeficiency that develop IBD-type intestinal disease, targeted exome sequencing is suggested to exclude the possibility of harboring known rare variants or genes transmitted through Mendelian inheritance. However, genetic analysis so far does not provide additional diagnostic/prognostic value to IBD patients outside of the abovementioned criteria. The use of selective genotyping, however, remains under active discussion. Commercial test kits are now available that incorporate genotyping of 4 genes with known susceptibility: ATG16L1, ECM1, NKX2-3, and STAT3. This test is performed in combination with a panel of serum markers and anti-bacterial serum antibodies (114), the latter of which appears to be the key element of the commercial diagnostic kit (115). On the other hand, the CD prognostic test kit from the same vendor contained only anti-bacterial serum antibodies but no genotyping, suggesting the limitation of our current knowledge in IBD genetics.
However, several studies have attempted to correlate genotype to phenotype and apply genotyping information to predict disease course. Apart from the rare variants and genes transmitted through Mendelian inheritance, both of which showed correlation with early onset and severe colitis, whether genotype correlates with phenotype has been the subject of multiple investigations. Study from the European IBDchip project found that susceptibility loci in the NOD2 gene were significantly associated with ileal location, stenosing and penetrating behavior, and need for surgery (116). Likewise, in CD, fistulizing disease was found to be associated with IL23R, LOC441108, PRDM1, and NOD2, whereas the need for surgery was associated with IRGM, TNFSF15, C13ORF31, and NOD2. Stenosing phenotype was found to be associated with NOD2, JAK2, and ATG16L1 (116). A separate recent study examining >10,000 European CD patients through GWAS of clinical phenotypes found that MAGI1 variant (which encodes a protein involved in intestinal epithelial tight junction) was associated with a complicated structuring phenotype, whereas variants in CLCA2, 2q24.1, and LY75 loci were associated with ileal involvement, mild disease course, and the presence of erythema nodosum, respectively (117). In UC, a SNP-based risk scoring system including 46 SNPs was able to differentiate medically refractory UC from non-medically refractory patients, thus predicting the need for surgery (118). In the same study, MHC was found to be a major genetic determinant for severe UC (118). However, other studies did not support the use of genetic testing to predict disease course (40; 119).
In addition, while no data has suggested how genetics may be able to predict response to certain therapies, genetics could be used to identify patients that may develop severe adverse events after certain medication. For instance, severe leucopenia can be a life threatening condition in IBD patients receiving thiopurine, and this was thought to be solely due to genetic variation in TPMT, which encodes a thiopurine S-methyltransferase (120). However, in patient population with low frequency of TPMT mutations (Asians), it was found that a SNP in NUDT15 was significantly associated with thiopurine-associated leucopenia (120). Thus, this highlights how genetics could be applied to predict toxicity of therapy. Also, as described earlier, early onset IBD patients with deficient IL-10R may benefit from allogeneic stem cell transplantation (26; 27).
One challenge is to develop readouts that link genetics with environmental factors that either trigger or exacerbate disease in genetically susceptible individuals. More recently, we have shown that morphologic defects of the small intestinal Paneth cells can be applied as an integrated readout that synthesize the effect of CD genetics and yet identified environmental factors and predicts prognosis in patients (104). There are multiple genetic loci associated with ATG16L1 and NOD2 that confer this effect. Further testing of this approach in clinical studies and identification of additional disease-relevant cell types that can allow the testing of this approach is critical in advancing the field forward.
Future directions
The panel of expanded susceptible loci and genomic sequencing technique has identified the genetic targets associated with IBD. Future functional and eQTL studies will advance our understanding in pathogenesis of these genes. The identification and mechanistic studies of environmental factors will be keys to explain the rising incidence of IBD, despite the stable genetic landscape over the years. We expect to identify several gene-environment multiple-hit modules. From human studies, using a ‘biological plausible’ approach to screen for possible environmental exposure will be key in translational studies. The mechanisms of potential environmental factors can be interrogated using next generation animal models to further clarify its impact, and more importantly, the ‘dosage’ and exposure duration for these environmental factors. Development of enteritis/colitis animal models other than using chemical-induced injury and be more clinically relevant will be of value. These can then inform next phase clinical studies for further validation. While the type and dosage of environmental factors are difficult to measure, an emerging concept is to that their ‘byproducts’ (e.g. metabolites) may offer indication of the exposure. This is exemplified by the recent enthusiasm in microbiome and metabolomics research; however, none of which has been able to establish causality. This will be a priority in IBD pathogenesis research. On clinical side, whether the field should move from phenotype-based IBD sub-classification for patient management to a “genotype-first” approach remains to be tested (121). Finally, with the advanced informatics technology, collecting and interpreting the growing big data will be able to provide an integrated analysis that links genetics, environmental exposure, disease phenotype, to prognosis.
Summary Points.
The majority of the identified common polymorphisms are non-coding and only account for a subset of IBD cases.
Most of the identified susceptible genes control responses to host-microbes interactions.
Different patient populations are affected by a different landscape of susceptible genes.
More precise mechanistic studies can be performed using next generation animal models engineered with identical polymorphism(s) found in human instead of complete knockout.
Changes in gut microbiota are associated with IBD. Whether this is a cause or effect remains unclear.
There is a combination of dosage effect in genetics and environmental factors in triggering IBD. Rare variants and Mendelian genes likely require less ‘dose’ of environmental trigger (i.e., commensal microbiota is sufficient), whereas common variants require additional environmental trigger (e.g. enteropathogens).
Footnotes
Disclosure statement:
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Literature cited
- 1.Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380:1590–605. doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 2.Ordas I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380:1606–19. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- 3.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nature genetics. 2011;43:246–52. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nature genetics. 2011;43:1066–73. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellinghaus D, Zhang H, Zeissig S, Lipinski S, Till A, et al. Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology. 2013;145:339–47. doi: 10.1053/j.gastro.2013.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christodoulou K, Wiskin AE, Gibson J, Tapper W, Willis C, et al. Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut. 2013;62:977–84. doi: 10.1136/gutjnl-2011-301833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–17. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham DB, Xavier RJ. From genetics of inflammatory bowel disease towards mechanistic insights. Trends Immunol. 2013;34:371–8. doi: 10.1016/j.it.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nature reviews. Genetics. 2008;9:356–69. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 11.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology. 2011;140:1704–12. doi: 10.1053/j.gastro.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. American journal of human genetics. 2014;95:5–23. doi: 10.1016/j.ajhg.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orange JS, Glessner JT, Resnick E, Sullivan KE, Lucas M, et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. The Journal of allergy and clinical immunology. 2011;127:1360–7 e6. doi: 10.1016/j.jaci.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stray-Pedersen A, Backe PH, Sorte HS, Morkrid L, Chokshi NY, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. American journal of human genetics. 2014;95:96–107. doi: 10.1016/j.ajhg.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, et al. The Diagnostic Approach to Monogenic Very Early Onset Inflammatory Bowel Disease. Gastroenterology. 2014;147:990–1007 e3. doi: 10.1053/j.gastro.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muise AM, Walters T, Xu W, Shen-Tu G, Guo CH, et al. Single nucleotide polymorphisms that increase expression of the guanosine triphosphatase RAC1 are associated with ulcerative colitis. Gastroenterology. 2011;141:633–41. doi: 10.1053/j.gastro.2011.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muise AM, Xu W, Guo CH, Walters TD, Wolters VM, et al. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut. 2012;61:1028–35. doi: 10.1136/gutjnl-2011-300078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhillon SS, Fattouh R, Elkadri A, Xu W, Murchie R, et al. Variants in nicotinamide adenine dinucleotide phosphate oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology. 2014;147:680–9 e2. doi: 10.1053/j.gastro.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Leoni G, Alam A, Neumann PA, Lambeth JD, Cheng G, et al. Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. The Journal of clinical investigation. 2013;123:443–54. doi: 10.1172/JCI65831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leoni G, Neumann PA, Kamaly N, Quiros M, Nishio H, et al. Annexin A1-containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. The Journal of clinical investigation. 2015 doi: 10.1172/JCI76693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avitzur Y, Guo C, Mastropaolo LA, Bahrami E, Chen H, et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology. 2014;146:1028–39. doi: 10.1053/j.gastro.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bigorgne AE, Farin HF, Lemoine R, Mahlaoui N, Lambert N, et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. The Journal of clinical investigation. 2014;124:328–37. doi: 10.1172/JCI71471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeissig Y, Petersen BS, Milutinovic S, Bosse E, Mayr G, et al. XIAP variants in male Crohn’s disease. Gut. 2015;64:66–76. doi: 10.1136/gutjnl-2013-306520. [DOI] [PubMed] [Google Scholar]
- 26.Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–45. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143:347–55. doi: 10.1053/j.gastro.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 28.Olszak T, Neves JF, Dowds CM, Baker K, Glickman J, et al. Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature. 2014;509:497–502. doi: 10.1038/nature13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–19. doi: 10.1016/j.immuni.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40:720–33. doi: 10.1016/j.immuni.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 31.MacDuff DA, Reese TA, Kimmey JM, Weiss LA, Song C, et al. Phenotypic complementation of genetic immunodeficiency by chronic herpesvirus infection. eLife. 2015;4 doi: 10.7554/eLife.04494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carmi S, Hui KY, Kochav E, Liu X, Xue J, et al. Sequencing an Ashkenazi reference panel supports population-targeted personal genomics and illuminates Jewish and European origins. Nat Commun. 2014;5:4835. doi: 10.1038/ncomms5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kenny EE, Pe’er I, Karban A, Ozelius L, Mitchell AA, et al. A genome-wide scan of Ashkenazi Jewish Crohn’s disease suggests novel susceptibility loci. PLoS Genet. 2012;8:e1002559. doi: 10.1371/journal.pgen.1002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang W, Hui KY, Gusev A, Warner N, Ng SM, et al. Extended haplotype association study in Crohn’s disease identifies a novel, Ashkenazi Jewish-specific missense mutation in the NF-kappaB pathway gene, HEATR3. Genes Immun. 2013;14:310–6. doi: 10.1038/gene.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang SK, Hong M, Zhao W, Jung Y, Baek J, et al. Genome-wide association study of Crohn’s disease in Koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut. 2014;63:80–7. doi: 10.1136/gutjnl-2013-305193. [DOI] [PubMed] [Google Scholar]
- 36.Yamazaki K, Umeno J, Takahashi A, Hirano A, Johnson TA, et al. A genome-wide association study identifies 2 susceptibility Loci for Crohn’s disease in a Japanese population. Gastroenterology. 2013;144:781–8. doi: 10.1053/j.gastro.2012.12.021. [DOI] [PubMed] [Google Scholar]
- 37.Hirano A, Yamazaki K, Umeno J, Ashikawa K, Aoki M, et al. Association study of 71 European Crohn’s disease susceptibility loci in a Japanese population. Inflamm Bowel Dis. 2013;19:526–33. doi: 10.1097/MIB.0b013e31828075e7. [DOI] [PubMed] [Google Scholar]
- 38.Hong SN, Park C, Park SJ, Lee CK, Ye BD, et al. Deep resequencing of 131 Crohn’s disease associated genes in pooled DNA confirmed three reported variants and identified eight novel variants. Gut. 2015 doi: 10.1136/gutjnl-2014-308617. [DOI] [PubMed] [Google Scholar]
- 39.Yang SK, Hong M, Zhao W, Jung Y, Tayebi N, et al. Genome-wide association study of ulcerative colitis in Koreans suggests extensive overlapping of genetic susceptibility with Caucasians. Inflamm Bowel Dis. 2013;19:954–66. doi: 10.1097/MIB.0b013e3182802ab6. [DOI] [PubMed] [Google Scholar]
- 40.Ananthakrishnan AN, Huang H, Nguyen DD, Sauk J, Yajnik V, Xavier RJ. Differential effect of genetic burden on disease phenotypes in Crohn’s disease and ulcerative colitis: analysis of a North American cohort. Am J Gastroenterol. 2014;109:395–400. doi: 10.1038/ajg.2013.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polychronakos C, Li Q. Understanding type 1 diabetes through genetics: advances and prospects. Nature reviews. Genetics. 2011;12:781–92. doi: 10.1038/nrg3069. [DOI] [PubMed] [Google Scholar]
- 42.Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2014 doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nature reviews. Genetics. 2013;14:661–73. doi: 10.1038/nrg3502. [DOI] [PubMed] [Google Scholar]
- 44.Yang SK, Hong M, Choi H, Zhao W, Jung Y, et al. Immunochip Analysis Identification of 6 Additional Susceptibility Loci for Crohn’s Disease in Koreans. Inflamm Bowel Dis. 2014 doi: 10.1097/MIB.0000000000000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis research & therapy. 2011;13:101. doi: 10.1186/ar3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nature genetics. 2011;43:1193–201. doi: 10.1038/ng.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goyette P, Boucher G, Mallon D, Ellinghaus E, Jostins L, et al. High-density mapping of the MHC identifies a shared role for HLA-DRB1*01:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nature genetics. 2015;47:172–9. doi: 10.1038/ng.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vahedi G, Kanno Y, Furumoto Y, Jiang K, Parker SC, et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature. 2015 doi: 10.1038/nature14154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–47. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 51.Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N Engl J Med. 2012;367:1519–28. doi: 10.1056/NEJMoa1203572. [DOI] [PubMed] [Google Scholar]
- 52.Sokol H, Conway KL, Zhang M, Choi M, Morin B, et al. Card9 mediates intestinal epithelial cell restitution, T-helper 17 responses, and control of bacterial infection in mice. Gastroenterology. 2013;145:591–601 e3. doi: 10.1053/j.gastro.2013.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vang T, Congia M, Macis MD, Musumeci L, Orru V, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nature genetics. 2005;37:1317–9. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 54.Diaz-Gallo LM, Espino-Paisan L, Fransen K, Gomez-Garcia M, van Sommeren S, et al. Differential association of two PTPN22 coding variants with Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2011;17:2287–94. doi: 10.1002/ibd.21630. [DOI] [PubMed] [Google Scholar]
- 55.Rhee I, Veillette A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol. 2012;13:439–47. doi: 10.1038/ni.2246. [DOI] [PubMed] [Google Scholar]
- 56.Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14:9–23. doi: 10.1038/nri3565. [DOI] [PubMed] [Google Scholar]
- 57.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nature reviews. Genetics. 2009;10:184–94. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, et al. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–7. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, et al. A genome-wide association study of global gene expression. Nature genetics. 2007;39:1202–7. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 60.Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science. 2014;343:1246949. doi: 10.1126/science.1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang MH, Fiocchi C, Zhu X, Ripke S, Kamboh MI, et al. Gene-gene and gene-environment interactions in ulcerative colitis. Hum Genet. 2014;133:547–58. doi: 10.1007/s00439-013-1395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wagner J, Sim WH, Ellis JA, Ong EK, Catto-Smith AG, et al. Interaction of Crohn’s disease susceptibility genes in an Australian paediatric cohort. PLoS One. 2010;5:e15376. doi: 10.1371/journal.pone.0015376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hedl M, Lahiri A, Ning K, Cho JH, Abraham C. Pattern recognition receptor signaling in human dendritic cells is enhanced by ICOS ligand and modulated by the Crohn’s disease ICOSLG risk allele. Immunity. 2014;40:734–46. doi: 10.1016/j.immuni.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272–6. doi: 10.1038/nature12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rogler G, Vavricka S. Exposome in IBD: Recent Insights in Environmental Factors that Influence the Onset and Course of IBD. Inflamm Bowel Dis. 2014 doi: 10.1097/MIB.0000000000000229. [DOI] [PubMed] [Google Scholar]
- 66.Ng SC, Bernstein CN, Vatn MH, Lakatos PL, Loftus EV, Jr, et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut. 2013;62:630–49. doi: 10.1136/gutjnl-2012-303661. [DOI] [PubMed] [Google Scholar]
- 67.Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, et al. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe. 2011;9:390–403. doi: 10.1016/j.chom.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ng SC, Tang W, Leong RW, Chen M, Ko Y, et al. Environmental risk factors in inflammatory bowel disease: a population-based case-control study in Asia-Pacific. Gut. 2014 doi: 10.1136/gutjnl-2014-307410. [DOI] [PubMed] [Google Scholar]
- 69.Ananthakrishnan AN, Nguyen DD, Sauk J, Yajnik V, Xavier RJ. Genetic polymorphisms in metabolizing enzymes modifying the association between smoking and inflammatory bowel diseases. Inflamm Bowel Dis. 2014;20:783–9. doi: 10.1097/MIB.0000000000000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Verschuere S, De Smet R, Allais L, Cuvelier CA. The effect of smoking on intestinal inflammation: what can be learned from animal models? J Crohns Colitis. 2012;6:1–12. doi: 10.1016/j.crohns.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 71.Ueno A, Jijon H, Traves S, Chan R, Ford K, et al. Opposing effects of smoking in ulcerative colitis and Crohn’s disease may be explained by differential effects on dendritic cells. Inflamm Bowel Dis. 2014;20:800–10. doi: 10.1097/MIB.0000000000000018. [DOI] [PubMed] [Google Scholar]
- 72.Lee D, Albenberg L, Compher C, Baldassano R, Piccoli D, et al. Diet in the Pathogenesis and Treatment of Inflammatory Bowel Diseases. Gastroenterology. 2015 doi: 10.1053/j.gastro.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–96. doi: 10.1038/nature14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. 2015;519:92–6. doi: 10.1038/nature14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martinez-Medina M, Denizot J, Dreux N, Robin F, Billard E, et al. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut. 2014;63:116–24. doi: 10.1136/gutjnl-2012-304119. [DOI] [PubMed] [Google Scholar]
- 77.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–8. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci U S A. 2012;109:594–9. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hunter DJ. Gene-environment interactions in human diseases. Nature reviews. Genetics. 2005;6:287–98. doi: 10.1038/nrg1578. [DOI] [PubMed] [Google Scholar]
- 80.Khoury MJ, Adams MJ, Jr, Flanders WD. An epidemiologic approach to ecogenetics. American journal of human genetics. 1988;42:89–95. [PMC free article] [PubMed] [Google Scholar]
- 81.Ludvigsson JF, Leffler DA, Bai JC, Biagi F, Fasano A, et al. The Oslo definitions for coeliac disease and related terms. Gut. 2013;62:43–52. doi: 10.1136/gutjnl-2011-301346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Khoury MJ, Flanders WD. Nontraditional epidemiologic approaches in the analysis of gene-environment interaction: case-control studies with no controls! American journal of epidemiology. 1996;144:207–13. doi: 10.1093/oxfordjournals.aje.a008915. [DOI] [PubMed] [Google Scholar]
- 83.Smith PG, Day NE. The design of case-control studies: the influence of confounding and interaction effects. International journal of epidemiology. 1984;13:356–65. doi: 10.1093/ije/13.3.356. [DOI] [PubMed] [Google Scholar]
- 84.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–41. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, et al. Conducting a microbiome study. Cell. 2014;158:250–62. doi: 10.1016/j.cell.2014.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–99. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62:1505–10. doi: 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–54 e1. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 89.Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60:631–7. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- 90.Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–92. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sadaghian Sadabad M, Regeling A, de Goffau MC, Blokzijl T, Weersma RK, et al. The ATG16L1-T300A allele impairs clearance of pathosymbionts in the inflamed ileal mucosa of Crohn’s disease patients. Gut. 2014 doi: 10.1136/gutjnl-2014-307289. [DOI] [PubMed] [Google Scholar]
- 92.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–99. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–83. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 94.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–73. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Virgin HW, Todd JA. Metagenomics and personalized medicine. Cell. 2011;147:44–56. doi: 10.1016/j.cell.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Olszak T, An D, Zeissig S, Vera MP, Richter J, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–93. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moon C, Baldridge MT, Wallace MA, Burnham CA, Virgin HW, Stappenbeck TS. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature. 2015 doi: 10.1038/nature14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160:447–60. doi: 10.1016/j.cell.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158:1000–10. doi: 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014;111:7741–6. doi: 10.1073/pnas.1407001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506:456–62. doi: 10.1038/nature13044. [DOI] [PubMed] [Google Scholar]
- 102.Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–45. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.VanDussen KL, Liu TC, Li D, Towfic F, Modiano N, et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology. 2014;146:200–9. doi: 10.1053/j.gastro.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marchiando AM, Ramanan D, Ding Y, Gomez LE, Hubbard-Lucey VM, et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe. 2013;14:216–24. doi: 10.1016/j.chom.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–57. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology. 2013;145:1347–57. doi: 10.1053/j.gastro.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pelletier S, Gingras S, Green DR. Mouse Genome Engineering via CRISPR-Cas9 for Study of Immune Function. Immunity. 2015;42:18–27. doi: 10.1016/j.immuni.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 110.Liu B, Tonkonogy SL, Sartor RB. Antigen-presenting cell production of IL-10 inhibits T-helper 1 and 17 cell responses and suppresses colitis in mice. Gastroenterology. 2011;141:653–62. doi: 10.1053/j.gastro.2011.04.053. 62 e1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Im E, Jung J, Pothoulakis C, Rhee SH. Disruption of Pten speeds onset and increases severity of spontaneous colitis in Il10(−/−) mice. Gastroenterology. 2014;147:667–79 e10. doi: 10.1053/j.gastro.2014.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.VanDussen KL, Marinshaw JM, Shaikh N, Miyoshi H, Moon C, et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut. 2014 doi: 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–45. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Plevy S, Silverberg MS, Lockton S, Stockfisch T, Croner L, et al. Combined serological, genetic, and inflammatory markers differentiate non-IBD, Crohn’s disease, and ulcerative colitis patients. Inflamm Bowel Dis. 2013;19:1139–48. doi: 10.1097/MIB.0b013e318280b19e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van Schaik FD, Oldenburg B, Hart AR, Siersema PD, Lindgren S, et al. Serological markers predict inflammatory bowel disease years before the diagnosis. Gut. 2013;62:683–8. doi: 10.1136/gutjnl-2012-302717. [DOI] [PubMed] [Google Scholar]
- 116.Cleynen I, Gonzalez JR, Figueroa C, Franke A, McGovern D, et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: results from the IBDchip European Project. Gut. 2013;62:1556–65. doi: 10.1136/gutjnl-2011-300777. [DOI] [PubMed] [Google Scholar]
- 117.Alonso A, Domenech E, Julia A, Panes J, Garcia-Sanchez V, et al. Identification of risk Loci for Crohn’s disease phenotypes using a genome-wide association study. Gastroenterology. 2015;148:794–805. doi: 10.1053/j.gastro.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 118.Haritunians T, Taylor KD, Targan SR, Dubinsky M, Ippoliti A, et al. Genetic predictors of medically refractory ulcerative colitis. Inflamm Bowel Dis. 2010;16:1830–40. doi: 10.1002/ibd.21293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ryan JD, Silverberg MS, Xu W, Graff LA, Targownik LE, et al. Predicting complicated Crohn’s disease and surgery: phenotypes, genetics, serology and psychological characteristics of a population-based cohort. Aliment Pharmacol Ther. 2013;38:274–83. doi: 10.1111/apt.12368. [DOI] [PubMed] [Google Scholar]
- 120.Yang SK, Hong M, Baek J, Choi H, Zhao W, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nature genetics. 2014;46:1017–20. doi: 10.1038/ng.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stessman HA, Bernier R, Eichler EE. A genotype-first approach to defining the subtypes of a complex disease. Cell. 2014;156:872–7. doi: 10.1016/j.cell.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]