

Summary
Upon heterologous overexpression, many proteins misfold or aggregate, thus resulting in low functional yields. Human acetylcholinesterase (hAChE), an enzyme mediating synaptic transmission, is a typical case of a human protein that necessitates mammalian systems to obtain functional expression. We developed a computational strategy and designed an AChE variant bearing 51 mutations that improved core packing, surface polarity, and backbone rigidity. This variant expressed at ∼2,000-fold higher levels in E. coli compared to wild-type hAChE and exhibited 20°C higher thermostability with no change in enzymatic properties or in the active-site configuration as determined by crystallography. To demonstrate broad utility, we similarly designed four other human and bacterial proteins. Testing at most three designs per protein, we obtained enhanced stability and/or higher yields of soluble and active protein in E. coli. Our algorithm requires only a 3D structure and several dozen sequences of naturally occurring homologs, and is available at http://pross.weizmann.ac.il.
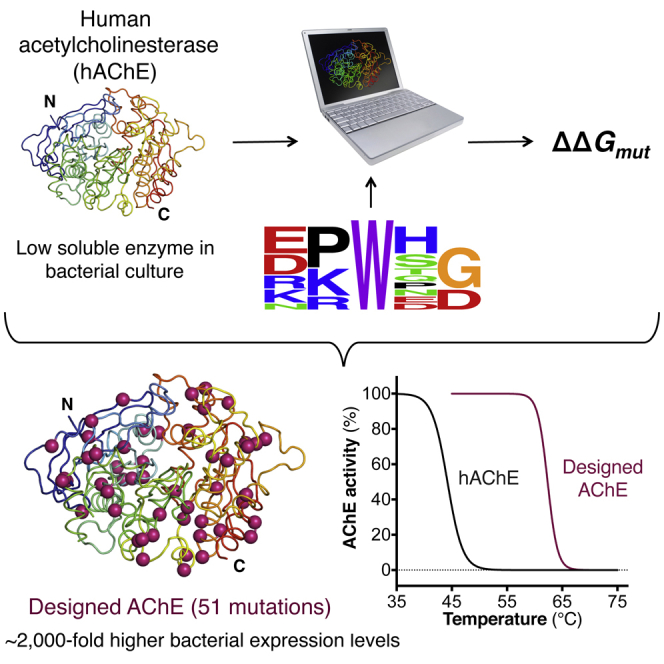
Graphical Abstract
Highlights
-
•
A new computational method is used to stabilize five recalcitrant proteins
-
•
Designed variants show higher expression and stability with unmodified function
-
•
A designed human acetylcholinesterase variant expresses solubly in bacteria
-
•
The method is fully automated and implemented on a webserver
Heterologous expression of proteins and their mutants often results in misfolding and aggregation. Goldenzweig et al. (2016) developed an automated algorithm for protein stabilization requiring minimal experimental testing; for instance, the five tested variants of human acetylcholinesterase showed ≥100-fold higher soluble bacterial expression and higher melting temperatures than wild-type.
Introduction
Most natural proteins are only marginally stable (Magliery, 2015). Thus, taken out of their natural context, through either overexpression, heterologous expression, or changes in environmental conditions, many proteins misfold and aggregate. The most general origin of overexpression challenges is low stability of the protein’s native, functional state relative to alternative nonfunctional or aggregation-prone states. By designing variants with more favorable native-state energy, yields of soluble and functional protein obtained by heterologous overexpression can be dramatically increased, alongside other merits such as longer storage and usage lifetimes and enhanced engineering potential.
Engineering stable protein variants is a widely pursued goal. Methods based on phylogenetic analysis (Lehmann et al., 2000, Steipe et al., 1994) and structure-based rational or computational design (Borgo and Havranek, 2012, Jacak et al., 2012, Korkegian et al., 2005, Lawrence et al., 2007) yielded proteins with improved stability and higher functional expression (Magliery, 2015). Individual mutations, however, contribute little to stability (typically ≤1 kcal/mol) (Zhao and Arnold, 1999), whereas stabilizing large and poorly expressed proteins typically requires many mutations. However, since even a single severely destabilizing mutation can undermine the benefit accruing from all others, high prediction accuracy is essential. Despite improvements in accuracy, existing approaches have a relatively high probability of inadvertently introducing disruptive mutations (false-positive predictions) (Borgo and Havranek, 2012, Magliery, 2015, Potapov et al., 2009). Published efforts to stabilize large proteins therefore either incorporate only a few predicted stabilizing mutations (typically ≤4) at each experimental step or use library approaches to identify optimal combinations of stabilizing mutations (Khersonsky et al., 2011, Sullivan et al., 2012, Trudeau et al., 2014, Whitehead et al., 2012, Wijma et al., 2014). Such approaches are laborious and impractical for proteins without established medium-to-high throughput screens, let alone for proteins of unknown function. To address the demand for stabilizing large, recalcitrant proteins by a wide range of researchers, who lack background in computational design, we developed an automated algorithm based on atomistic Rosetta modeling and phylogenetic sequence information. We specifically aimed to develop a general method that minimizes false-positive predictions to ensure that only a few variants need to be experimentally tested to achieve high functional yields (ideally, just one variant). We applied this algorithm to four different enzymes and one protein of unknown function. In each case, up to five variants were designed as the default output, encoding from 9 to 67 mutations relative to wild-type. These variants exhibited enhanced bacterial expression yields and stability, without sacrificing or altering activity.
Design
Computational Mutation Scanning Minimizes False-Positive Mutations
To address the challenge of designing variants with a large net stabilizing effect, yet without modifying their function, our computational process began by scanning the natural sequence diversity. For every target wild-type sequence, we generated a sequence alignment, from which we computed a position-specific substitution matrix (PSSM) (Altschul et al., 2009); the PSSM represents the log-likelihood of observing any of the 20 amino acids at each position. At every amino acid position, “allowed” mutations were defined as those with a favorable PSSM score (≥0). The rationale for restricting the allowed sequence space through the alignment scan is that, in general, deleterious mutations are purged by natural selection; purging is not absolute, however, and less favorable amino acids may occur at certain positions. Indeed, mutation to the most frequently observed amino acid often increases stability (the consensus effect) (Lehmann et al., 2000, Magliery, 2015, Steipe et al., 1994). However, our approach does not implement the consensus per se. Foremost, the alignment scan eliminates mutations that are rare or never seen in the natural diversity, rather than strictly selecting the most frequent amino acid. Next, we applied Rosetta computational mutation scanning (Whitehead et al., 2012), wherein each “allowed” mutation from the previous step was singly modeled against the background of the wild-type structure, and the energy difference between the wild-type and the single-point mutant was calculated (ΔΔGcalc). By computing the effects of each mutation singly, rather than in combination with others, we restricted design choices to mutations that were likely to make additive contributions to stability, rather than nonadditive and context-dependent contributions, thereby minimizing the risk of false positives. We thereby defined the space of “potentially stabilizing” mutations as mutations with ΔΔGcalc ≤−0.45 Rosetta energy units (R.e.u.); we chose this cutoff rather than 0 R.e.u. to further lower the risk of false positives, as indicated by the systematic evaluation described below. As a final step, we used Rosetta combinatorial sequence design to find an optimal combination of mutations within the space of potentially stabilizing mutations. Additionally, we used lower ΔΔGcalc cutoffs to select potentially stabilizing mutations prior to combinatorial sequence design, thereby generating several designs for experimental testing (see Data S1 available online).
The choice of mutations at Gly416 in human acetylcholinesterase (hAChE) illustrates the role of these two filters (alignment scan and computational mutation scanning) in pruning false positives (Figure 1A). Position 416 is located on a partially exposed helical surface, where the small and flexible amino acid Gly is likely to destabilize hAChE. Indeed, in the alignment of AChE homologs, Gly is infrequent, and His is the most prevalent amino acid. Modeling shows, however, that in the specific context of hAChE, His adopts a strained side-chain conformation; in contrast, Gln, the third most prevalent amino acid, is predicted to be most stabilizing owing to its high helical propensity and favorable hydrogen bonding with Tyr503. The combined filter therefore favors Gln over His for downstream design calculations.
Figure 1.

Eliminating Potentially Destabilizing Mutations through Homologous-Sequence Analysis and Computational Mutation Scanning
(A) (Left) Sequence logo for hAChE position Gly416. Letter height represents the respective amino acid’s frequency in an alignment of homologous AChE sequences. The evolutionarily “allowed” sequence space (PSSM scores ≥0) at position 416 includes the nine amino acids shown. (Right) Structural models of mutations to the evolutionarily favored amino acid His, and to Gln, which is favored by Rosetta energy calculations. The His side chain is strained due to its proximity to the bulky Tyr503 aromatic ring, whereas the Gln side chain is relaxed and forms a favorable hydrogen bond with Tyr503 (dashed line).
(B) Computational mutation scanning correctly identifies all of the destabilizing mutations and ⅔ of the stabilizing mutations in yeast triosephosphate isomerase (TIM). Sullivan et al. (2012) used consensus design to predict 23 mutations that would stabilize yeast TIM and measured each mutant’s Tm difference relative to wild-type (ΔTm). We used computational mutation scanning to predict the effects of each mutation on stability (ΔΔGcalc). Four mutations were experimentally found to be highly deleterious, resulting in no functional expression, and therefore the Tm for these could not be measured (open circles); these four mutations have highly unfavorable ΔΔGcalc values (>4.6 Rosetta energy units; R.e.u.).
See also Table S1.
To systematically evaluate the ability of our filtering method to identify stabilizing mutations, we compared its output to published experimental data on the stability effects of single-point mutations in the enzymes fungal endoglucanase Cel5A and yeast triosephosphate isomerase (TIM) (Figure 1B; Table 1) (Sullivan et al., 2012, Trudeau et al., 2014). The two filters (PSSM and Rosetta mutational scanning using ΔΔGcalc ≤−0.45 R.e.u. as cutoff) eliminated all the severely destabilizing mutations (and 99.6% of all destabilizing mutations) and retained one-third and two-thirds of the experimentally verified stabilizing mutations in Cel5A and TIM, respectively. Thus, for both enzymes, our method identified a large fraction of the known stabilizing mutations, while excluding false positives almost entirely. The energy cutoff emphasizes the importance of minimizing false-positive predictions, that is, mutations that are experimentally destabilizing but are computationally assessed as favorable; for instance, in the case of Cel5A, using ΔΔGcalc ≤0 R.e.u. as cutoff would correctly predict four additional stabilizing mutations but would introduce eight more destabilizing mutations. False-negative predictions—that is, mutations that are stabilizing in experiment but are considered destabilizing by modeling—could not be attributed to a single modeling error (Table S1). Specifically, out of 22 false negatives in Cel5A, eight were filtered because they are unlikely according to the sequence alignment (PSSM score <0), and four showed ΔΔGcalc in the range −0.45–0 R.e.u., thereby missing the energy threshold by a small margin. Of the ten remaining false negatives, three occurred at core positions, where energy calculations typically penalize mutations, and two were mutations to proline. Improvements in the energy function and the conformation-relaxation procedure may in the future increase accuracy, and the analysis reported here provides a benchmark for such improvements.
Table 1.
Computational Mutation Scanning versus Experimental Point Mutations in Fungal Endoglucanase 5A
| Mutations | Stabilizinga | Destabilizinga | Total |
|---|---|---|---|
| True predictionsb | 12 (35%) | 230 (99.6%) | 242 |
| False predictionsb | 22 (65%) | 1 (0.4%) | 23 |
| Total | 34 | 231 | 265 |
Mutations were experimentally assayed by heat inactivation (Trudeau et al., 2014; raw data kindly provided by D. Trudeau).
Mutations were predicted as stabilizing if they exhibited ΔΔGcalc<−0.45 R.e.u. and destabilizing otherwise.
Results
Designed hAChE with Nearly 2,000-Fold Higher Bacterial Expression and Intact Activity
We applied our design strategy to human acetylcholinesterase (hAChE), a 60 kDa enzyme responsible for terminating synaptic transmission at cholinergic synapses by rapidly hydrolyzing the neurotransmitter acetylcholine (ACh) (Sussman et al., 1991). Although AChE is potentially useful for detection and detoxification of organophosphate nerve agents and pesticides, hAChE is currently produced by costly procedures involving purification from blood erythrocyte membranes, or using plant or mammalian expression systems. Previous attempts to express hAChE in bacteria resulted in extremely low levels of soluble and active protein (Fischer et al., 1993). AChE’s active site is located at the bottom of a deep gorge that penetrates half way (20 Å) into the enzyme, and mutations along the gorge can reduce ACh hydrolysis rates by up to 1,000-fold (Ordentlich et al., 1995). To increase the stability and expression levels of hAChE without altering its activity, we imposed a further restriction on the allowed sequence space of the newly designed hAChE: in all Rosetta modeling simulations, the side-chain conformations of amino acids within 8 Å of the reversible inhibitor E2020, which spans the full length of the active-site gorge (Cheung et al., 2012), had to remain as in the native hAChE structure (Table S2). The latter restriction, in combination with the two above-described filters, dramatically reduced the sequence space available for design. The theoretical sequence space for hAChE, a 550-residue enzyme, is 10750, a formidable number inaccessible even for advanced modeling algorithms. The size of the reduced sequence space, by contrast, was 1031 sequences, equivalent to complete computational design of a 24-amino acid peptide, a challenge solved already in the 1990s (Dahiyat and Mayo, 1997). We noted that the reduced sequence space led to convergence of combinatorial sequence optimization to identical, or nearly identical, sequences for any given ΔΔGcalc cutoff; this convergence, which is not usual in computational design (Fleishman et al., 2011), is a prerequisite for reproducibility and usage by nonexperts.
Given AChE’s large size, we designed five alternatives using different ΔΔGcalc thresholds, with 17–67 mutations relative to hAChE (Table S3; Data S2), and subjected them to experimental testing. Synthetic genes encoding wild-type hAChE and the five designs were optimized for bacterial translation efficiency, fused to the C terminus of thioredoxin, and expressed in E. coli ShuffleT7express cells to facilitate disulfide-bond formation. In SDS-PAGE gels of supernatant fractions from bacterial lysates, the AChE band overlapped with other bands, precluding visual quantification. Nevertheless, because the designed variants’ specific activity was found to be nearly identical to the wild-type’s (Table 2), we could quantify the relative levels of soluble and active enzyme by measuring AChE activity in crude lysates. The five designs showed ≥100-fold higher lysate ACh-hydrolysis rates compared to hAChE, with the best design, dAChE4 (Figure 2A; 51 mutations), exhibiting an almost 2,000-fold higher rate (Figure 2B). Furthermore, due to its extremely low soluble expression, hAChE could not be purified from crude cell lysates, whereas the designed variants were readily purified to homogeneity, with dAChE4 yields in standard shaker flasks approaching 2 mg protein per liter of bacterial culture (Table S4).
Table 2.
Stability and Kinetic Parameters of Human and Designed AChE Variants
| AChE Variant | Muta | Normalized Activityb | Inactivation Temperature (°C)c |
ki (SP-VX) × 107 (M−1min−1) | ACh Hydrolysis |
|||
|---|---|---|---|---|---|---|---|---|
| Lysate | Purified | KM (mM) | kcat × 105 (min−1) | kcat/KM × 109 (M−1min−1) | ||||
| hAChE (HEK293) | – | – | – | 50.6 ± 0.3 | 7.92 ± 0.15 | 0.09 ± 0.01 | 3.8 ± 0.2 | 4.4 ± 0.6 |
| hAChE (bacterial) | – | 1 | 44 ± 0.4 | NDd | NDd | NDd | NDd | NDd |
| dAChE1 | 17 | 119 ± 20 | 60.5 ± 0.4 | NDd | 6.48 ± 0.71 | 0.050 ± 0.006 | 4.4 ± 0.1 | 8.7 ± 1.1 |
| dAChE2 | 30 | 280 ± 40 | 61.5 ± 0.5 | 67.1 ± 0.3 | NDd | NDd | NDd | NDd |
| dAChE3 | 42 | 308 ± 44 | 62.3 ± 0.2 | 69.4 ± 0.3 | 2.65 ± 0.52 | 0.177 ± 0.010 | 4.4 ± 0.1 | 2.5 ± 0.2 |
| dAChE4 | 51 | 1770 ± 258 | 62.3 ± 0.3 | 66.2 ± 1.2 | 7.60 ± 0.34 | 0.071 ± 0.007 | 2.73 ± 0.07 | 3.9 ± 0.4 |
| dAChE5 | 67 | 637 ± 134 | 61.1 ± 0.2 | 68.5 ± 0.6 | 6.47 ± 0.82 | 0.104 ± 0.010 | 3.16 ± 0.01 | 3.0 ± 0.3 |
Number of amino acid mutations relative to wild-type hAChE.
Activity in crude lysates of cells expressing the AChE variants from 250 ml E. coli cultures.
The temperature at which 50% of activity was retained. Enzyme samples were incubated for 30 min at varying temperatures and tested for AChE activity after cooling.
Not determined.
Figure 2.

Design of a Stable hAChE variant and Its Functional Expression in Bacteria
(A) The structural underpinnings of stabilization in the designed variant dAChE4. Wild-type hAChE is shown in blue and 51 mutated positions, which are distributed throughout dAChE4, are indicated by orange spheres. Thumbnails highlight stabilizing effects of selected mutations.
(B) Bacterial lysate activity levels of designed AChEs normalized to hAChE activity. Crude lysates were derived from 250 ml flasks (medium scale) or 0.5 ml E. coli cultures grown in a 96-well plate (small scale). The higher activity levels in the designed variants reflect higher levels of soluble, functional enzyme.
(C) Designed AChE variants (colored lines) show higher resistance to heat inactivation compared to hAChE (black). Residual activities following incubation at different temperatures were measured in bacterial lysates and normalized to the activity in nontreated lysates.
(D) Sub-Ångstrom accuracy in alignment of key residues in the vicinity of the catalytic triad in the crystallographic structure of dAChE4 (PDB: 5HQ3, yellow) compared to hAChE (PDB: 4EY4, green).
See also Figure S1 and Tables S2–S5.
The designed AChE mutations are scattered throughout the enzyme and show typical characteristics of stabilizing mutations, including improved core packing, higher backbone rigidity, and increased surface polarity (Figure 2A). In agreement with the design strategy and the higher levels of soluble and functional enzyme, we observed increased resistance to heat inactivation of up to 20°C relative to the E. coli-expressed hAChE (Figure 2C) and to hAChE expressed in mammalian cells (Table 2). The designs hydrolyzed ACh at rates that are within a 2-fold margin of hAChE rates, and displayed inactivation-rate constants by the nerve agent VX that are nearly identical to hAChE (the largest deviation was observed for dAChE3, which exhibited a 2.5-fold lower inactivation rate; Table 2).
The above observations of nearly identical activity profiles of the designed and wild-type AChEs suggested that the designed enzymes’ active sites are essentially identical to that of hAChE. To verify this, we conducted crystallization trials on dAChE4, the design exhibiting the highest bacterial-expression yields. We noted that, in contrast to various natural AChEs studied by us, large crystals formed already within a few days and more reproducibly. dAChE4’s structure was solved at 2.6Å resolution, thus yielding, to the best of our knowledge, the first structure of an AChE expressed in a prokaryote (Table S5). dAChE4’s structure is very similar to that of wild-type hAChE, with a 0.7 Å root-mean-square deviation (rmsd) over Cα atoms. Residues at the catalytic gorge aligned particularly well, with an all-atom rmsd of only 0.125Å (Figure 2D). Thus, despite 51 mutations relative to wild-type, ∼2,000-fold gain in bacterial expression levels, and 20°C higher heat tolerance, dAChE4 is virtually indistinguishable in its active site from hAChE (differences in other parts of the enzyme are shown in Figure S1 and online at http://proteopedia.org/w/Journal:Molecular_Cell:1). dAChE4 can therefore serve in future structural studies of inhibitors that target the AChE active site.
Other Designed Enzymes Show High Stability and/or Soluble Expression
We applied our algorithm to two other human enzymes—the histone deacylase SIRT6, the human DNA methyltransferase Dnmt3a, and a bacterial phosphotriesterase dubbed PTE. Being the only enzyme known to have actually evolved to degrade organophosphates (OPs), PTE is of considerable biotechnological potential for decontamination and detoxification of OPs, including nerve agents. However, PTE detoxifies most nerve agents at rates too slow for practical applications. Wild-type PTE was previously engineered for higher expression (Roodveldt and Tawfik, 2005) and hydrolysis rates toward various OPs (Bigley et al., 2015, Cherny et al., 2013). Introduction of function-altering mutations, however, destabilized the enzyme, as is often the case for laboratory-evolved enzymes (Tokuriki et al., 2008); thus, stabilization is a prerequisite for further engineering (Bloom et al., 2006). We designed three alternatives using different ΔΔGcalc thresholds, encoding 9–28 mutations relative to wild-type PTE (Table S3), and subjected them to experimental testing. Synthetic genes encoding the three designs and PTE-S5, a published variant encoding three mutations, which displays ∼20-fold higher expression levels compared to wild-type (Roodveldt and Tawfik, 2005), were optimized for bacterial translation efficiency, fused to maltose-binding protein, and expressed in E. coli GG48 cells. All three designs displayed increased levels of soluble, functional enzyme compared to PTE-S5, which already displays higher expression levels than wild-type (Table 3). Two of the three designs showed ∼10°C higher tolerance to heat inactivation relative to PTE-S5, with no significant change in activity toward the OP substrate paraoxon (Table 3; Figure S2A). Another noteworthy outcome of stabilization design was increased metal affinity. PTE is a metalloenzyme bearing two active-site metals, typically Zn+2 (Benning et al., 2001). Directed evolution of wild-type PTE for higher expression (PTE-S5) led to a significant decrease in metal affinity—a major practical drawback for applications in conditions in which Zn+2 cannot be supplemented. The designed variant dPTE2, which contained 19 mutations and exhibited the highest tolerance to heat inactivation, also exhibited a marked increase in metal affinity compared to PTE-S5, approaching the affinity of wild-type PTE (Figures 3A and S2B; Table 3).
Table 3.
Stability and Kinetic Parameters of PTE Variants
| Variant | Muta | Normalized Activityb | Inactivation Temperature (°C) |
T1/2 Chelatorc (min) | KM (mM)d | kcat × 105 (min-1) d | kcat/KM × 109 (min-1M-1) d | |
|---|---|---|---|---|---|---|---|---|
| Lysate | Purified | |||||||
| PTE-S5 | 3 | 1 | 50.9 ± 0.7 | 52.4 ± 0.2 | 7.5 ± 0.3 | 0.101 ± 0.023 | 0.970 ± 0.076 | 0.96 ± 0.33 |
| dPTE1 | 9 | 2.0 | 54.7 ± 3.2 | NDe | NDe | NDe | NDe | NDe |
| dPTE2 | 19 | 6.1 | 59.2 ± 0.7 | 62.0 ± 0.2 | 51.2 ± 5.1 | 0.060 ± 0.014 | 0.70 ± 0.05 | 1.17 ± 0.35 |
| dPTE3 | 28 | 2.3 | 47.0 ± 1.3 | NDe | NDe | NDe | NDe | NDe |
Number of mutations relative to wild-type PTE.
Fold increase in activity in crude E. coli lysates relative to PTE-S5.
Chelator, 50 μM 1,10-phenanthroline.
Kinetic parameters for paraoxon.
ND, not determined.
Figure 3.

Higher Expression, Stability, and Activity of Designed Variants of PTE, SIRT6, Dnmt3a, and Myoc-OLF
(A) Compared to the previously engineered variant PTE-S5 (Roodveldt and Tawfik, 2005), the design dPTE2 shows higher resistance to inactivation by the metal chelator 1,10-phenanthroline (50 μM), indicating higher metal affinity and stability.
(B) (Upper panel) The previously engineered E1 variant of hSIRT6 shows a 3-fold decline in in vivo expression levels, whereas following computational design, dSIRT6-E1 (denoted as dE1) recapitulates hSIRT6’s expression levels (western blot quantified using ImageJ). Actin expression levels are provided as control. (Lower panel) dE1 exhibits higher in vivo histone H3 Lys56-deacylation activity compared to hSIRT6. HY denotes a loss-of-function mutant of hSIRT6; H3 expression levels are provided as control.
(C) The designed variant dDnmt3A shows 10-fold higher DNA-methylation compared to hDnmt3a as determined by the levels of incorporation of H3-methyl groups in the presence of equal enzyme concentrations.
(D) dMyoc-OLF is more thermostable than hMyoc-OLF, and addition of Ca+2 further stabilizes it.
(E) Size-exclusion chromatography of MBP-fused hMyoc-OLF and dMyoc-OLF indicated a significant aggregated fraction in hMyoc-OLF, as previously described (Burns et al., 2010), and a minor aggregated fraction in dMyoc-OLF. (Inset) SDS-PAGE analysis of dMyoc-OLF. Lane 1, molecular weight standards (kDa); lanes 2–6, size exclusion fractions as labeled in chromatogram.
See also Figures S2 and S3 and Tables S2, S3, and S6.
SIRT6 is an ADP-ribosylase and NAD+-dependent deacylase that removes acyl groups from acylated lysines, thereby regulating several essential cellular processes (Kugel and Mostoslavsky, 2014). The low bacterial-expression levels of human SIRT6 (hSIRT6), and its weak deacylation activity relative to SIRT1, limit its study. The designed variant (dSIRT6) contained 11 mutations relative to hSIRT6 (Tables S2 and S3). It was expressed in E. coli and purified to homogeneity with yields of 20 mg per liter culture, an ∼5-fold increase relative to hSIRT6, and its deacylase activity was 60% higher than hSIRT6’s (Figures S3A and S3B). We subsequently incorporated the design mutations on the background of an engineered hSIRT6 variant dubbed E1, which contains three mutations relative to hSIRT6 (our unpublished data). E1 exhibits increased deacylation activity compared to hSIRT6 but 3-fold lower expression levels in human cell lines. In contrast, the designed variant dE1 recapitulates hSIRT6’s expression levels in human cell lines while maintaining high deacylation activity (Figure 3B). Given the beneficial effects of SIRT6 overexpression on longevity in mice (Kanfi et al., 2012), our designed mutant can be used to establish a correlation between SIRT6’s deacylation rates and its physiological function.
In the case of human Dnmt3a (hDnmt3a), soluble and active fractions of the human enzyme can be obtained by E. coli expression, but enzyme activity is very low. We tested one design dDnmt3a containing 14 mutations relative to hDnmt3a (Tables S2 and S3). In contrast to all other designs, the designed dDnmt3a showed significantly lower expression levels than hDnmt3a in E. coli (Figures S3C and S3D). Nonetheless, it exhibited nearly 10-fold higher specific activity (Figure 3C), suggesting that hDnmt3a preparations contain a significant fraction of soluble but inactive or poorly active enzyme. Therefore, although Dnmt3a failed to meet our design method’s success criteria (higher stability and soluble expression), it significantly increased the protein’s functional yield.
A Webserver for Protein Stabilization
Encouraged by the consistently positive results presented above, we implemented the algorithm as a webserver, called the Protein Repair One Stop Shop (PROSS, http://pross.weizmann.ac.il). Following their request for assistance in solving a critical expression and stability question regarding the human myocilin OLF domain (hMyoc-OLF), two of the authors (S.E.H. and R.L.L.) were granted unsupervised access to the webserver. OLF domains are found in extracellular proteins of multicellular organisms. A number of OLF domains have been implicated in human disease, but the function(s) of most OLF domains remains elusive. The best-studied OLF domain is that of hMyoc-OLF, in which more than 100 nonsynonymous mutations are implicated in inherited forms of open-angle glaucoma. Mutations documented in patients lead to destabilized myocilin protein that forms cytotoxic aggregates (Burns et al., 2010, Yam et al., 2007). S.E.H. and R.L.L. therefore posited that mutations that confer enhanced stability might reduce hMyoc-OLF’s propensity to misfold at physiological temperatures.
Three hMyoc-OLF structures (PDB: 4WXQ, 4WXS, and 4WXU) were submitted to PROSS, producing seven designs for each structure with 5–25 mutations each. From all suggested point mutations, S.E.H. and R.L.L. manually derived one variant (dMyoc-OLF) comprising 21 mutations. Although hMyoc-OLF binds Ca+2 (Donegan et al., 2012), the physiological role of Ca+2 binding is still unknown; nevertheless, the Ca+2-binding positions remained unchanged, reflecting the high sequence and structural constraints acting on them. Furthermore, none of the design mutations have been implicated in human disease, supporting the notion that the disease-associated mutations are destabilizing.
dMyoc-OLF and hMyoc-OLF were expressed in E. coli fused to maltose-binding protein (MBP-OLF). The designed variant gave an order of magnitude higher yield than hMyoc-OLF (Table S6). In addition, whereas the purified hMyoc-OLF comprises a mixture of a soluble MBP-OLF-aggregate and the properly folded monomer (Burns et al., 2010), the designed variant was almost exclusively monomeric, and also exhibited a 16.8°C higher melting temperature (Tm) than hMyoc-OLF (Figures 3D and 3E). This is the highest Tm measured so far for Myoc-OLF, nearly the same as that of the gliomedin OLF domain that lacks a Ca+2-binding site (Hill et al., 2015). Nonetheless, as indicated by the Tm increase in the presence of Ca+2 (Figure 3D; Table S6), Ca+2 binding was retained in dMyoc-OLF. The designed variant dMyoc-OLF is therefore a starting point for detailed OLF structure-function analysis.
Discussion
Most existing stability-design methods focus on one element or another of protein stability. For instance, some methods introduce disulfides or prolines, while others optimize core packing (Borgo and Havranek, 2012, Korkegian et al., 2005) and increase surface polarity (Magliery, 2015) and charge (Lawrence et al., 2007). Our design algorithm, by contrast, selects for all amino acid mutations that optimize the protein’s computed energy, subject to constraints inferred from homologous sequences. It thereby generates designs that improve a range of molecular parameters that are associated with stability (Table 4) while preserving function. We find, furthermore, that in the various proteins studied here, the algorithm implemented solutions that appear to selectively tackle specific defects of each protein. For instance, 17 of the 51 mutations in design dAChE4 impact core positions, a very large number compared to previous design studies that targeted protein cores (Borgo and Havranek, 2012, Korkegian et al., 2005). Indeed, natural AChE structures contain unusually large core cavities (Koellner et al., 2000). In design dPTE2, by contrast, only two core mutations were implemented, and the net negative charge increased by 13 units, shifting the isoelectic point (pI) from a near-neutral value of 6.7 for the wild-type to 5.0; indeed, near-neutral pI is often associated with poor solubility, and surface “supercharging” has been used by others to improve thermal stability (Lawrence et al., 2007). The stability considerations, however, do not come at the expense of functional constraints. In Dnmt3a, for example, the positive net charge and basic pI are fully conserved as expected for a DNA-binding protein. We conclude that the combination of evolutionary constraints, selection of mutations that individually contribute to computed stability, and combinatorial sequence optimization within the space of these mutations can address a broad range of stability defects in various proteins without affecting their original molecular functions.
Table 4.
Structure and Sequence Features of Designed Variants Compared to Their Wild-Type Counterparts
| Design Variant | aaa | Mutb | Corec | Salt Bridgesd | H Bondsd | X → Proe | Charge N/Pd,f | pIg |
|
|---|---|---|---|---|---|---|---|---|---|
| des | WT | ||||||||
| dAChE1 | 530 | 17 (3%) | 9 (53%) | 3 | 14 | 1 | 1/2 | 5.5 | 5.5 |
| dAChE2 | 30 (6%) | 15 (50%) | 4 | 11 | 3 | 4/4 | 5.5 | ||
| dAChE3 | 42 (8%) | 16 (38%) | 5 | 18 | 6/6 | 5.5 | |||
| dAChE4 | 51 (10%) | 17 (33%) | 3 | 10 | 7/8 | 5.5 | |||
| dAChE5 | 67 (13%) | 19 (28%) | 10 | 15 | 5 | 7/9 | 5.6 | ||
| dPTE1 | 331 | 9 (3%) | 0 (0%) | 3 | 5 | 0 | 6/−2 | 5.3 | 6.7 |
| dPTE2 | 19 (6%) | 2 (11%) | 8 | 9 | 11/−2 | 5.0 | |||
| dPTE3 | 28 (9%) | 5 (18%) | 12 | 7 | 16/−3 | 4.8 | |||
| dSIRT6 | 280 | 11 (4%) | 1 (9%) | 0 | 4 | 1 | 0/0 | 8.3 | 8.3 |
| dDnmt3 | 272 | 14 (5%) | 0 (0%) | 4 | 7 | 1 | 1/1 | 8.4 | 8.6 |
| dMyoc-OLF | 259 | 21 (8%) | 2 (10%) | 2 | 4 | 0 | −2/1 | 5.4 | 5.0 |
Number of amino acids in the PDB structure (per monomer); the design algorithm ignores all protein segments that are absent from the structure.
Number of amino acid mutations in the design compared to wild-type; in brackets, as percentage of total number of amino acids.
Number of mutations in protein core. In brackets, as percentage of total number of mutations.
Difference compared to wild-type structure. Positive values indicate higher number in design than wild-type.
Number of mutations to proline.
Mutations to negative (D/E) and positive (K/R) amino acids are indicated to the left and right of the slash, respectively.
Isoelectric point: des refers to designed variant, and WT refers to wild-type.
The case studies discussed here represent a diverse group of proteins belonging to different fold families (TIM barrel, β-propeller, and α/β hydrolase fold) and displaying different activities. In each case, the wild-type protein and variants previously engineered using traditional methods suffer from low stability, as manifested by intolerance to high temperature and function-modifying mutations or by low solubility, heterologous expression, cofactor affinity, or specific activity. That a single fully automated method that only considers phylogenetic constraints and optimizes the native state’s energy is able to address these diverse challenges without sacrificing function demonstrates that the underlying source of these problems is the native state’s marginal stability. Furthermore, the stability-design method has the potential to optimize proteins used as scaffolds for de novo binder or enzyme design, where low stability and expression levels have been limiting (Fleishman et al., 2011, Khersonsky et al., 2011).
Limitations
The stability design algorithm requires a few dozen unique sequence homologs and an atomic structure of the target protein. With the burgeoning of sequence databases, the first requirement is likely to be met with few exceptions; the second, however, may be an impediment, particularly since low protein yields and stability negatively impact structure determination. To expand beyond experimentally determined structures, homology models may be used, and it remains to be seen whether they provide sufficient accuracy for atomistic stability-design calculations.
Experimental Procedures
Computational Procedures
Phylogenetic Sequence Constraints
For every query sequence, homologous sequences were collected using the BLASTP algorithm (Altschul et al., 1990) on the nonredundant (nr) database, with the maximum number of hits set to 500 and the e value to 10−4. All other parameters were set to default values. Hits were excluded if they covered less than 60% of the query sequence or if their sequence identity to the query sequence was lower than 30% (28% for PTE due to the low diversity of homologs at >30% sequence identity). The remaining sequences were clustered using cd-hit (Li and Godzik, 2006), with a clustering threshold of 90% and default parameters.
MUSCLE (Edgar, 2004) was used with default parameters to derive a multiple sequence alignment (MSA) from the clustered sequences. Gaps in the alignment between the query and any homolog often occur within loop regions and may reflect differences in the local backbone conformation. To reduce alignment uncertainty, we detected secondary-structure elements in the query protein structure (using DSSP [Kabsch and Sander, 1983]) and eliminated subsequences in homologs that contained gaps in loop segments that intervene between any two secondary-structure elements relative to the query. In effect, for every loop region this procedure generated a specific alignment that only comprised homologous sequences with no insertions or deletions relative to the query. Since this feature of the stability-design algorithm had not yet been implemented when we designed AChE and Dnmt3a, these two proteins were designed with default MUSCLE alignments, containing information from all homologs in loop regions.
Given the alignments of each query sequence, we computed a position-specific scoring matrix (PSSM) using the PSI-BLAST algorithm (Altschul et al., 2009). The PSSM represents the log probability of observing each of the 20 amino acids at each position in the query. Non-negative PSSM scores are considered likely to occur in evolution and define the space of allowed mutations.
Structure-Based Constraints
To prevent activity loss, residues within 8 Å of small-molecule ligands observed in the active site (hAChE and Dnmt3a), or within 5 Å of metal cofactors (PTE), DNA chains (Dnmt3a) and protein ligands (SIRT6) were held fixed throughout all Rosetta simulations. In homo-oligomer structures (PTE and AChE), residues within 5Å of the oligomer interface were held fixed. For PTE and SIRT6, some of the active-site constraints were defined using an alignment to another, ligand-bound structure, namely residues within 8Å of DPJ in 4NP7 for PTE, and residues within 5Å of chain F in 3ZG6 for SIRT6 (Table S2). Two positions on either side of missing density and residues at the termini (if the termini made no interactions with other parts of the protein) were not allowed to mutate. For hSIRT6, four residues that were found to increase activity in previous laboratory-evolution experiments were modeled prior to simulation, and not allowed to mutate (see Table S2). In the PROSS webserver, active-site constraints follow the same thresholds defined above, and the user may specify any other residue to be held fixed during the simulations through an interactive HTML form.
Rosetta Structure Modeling and Design
See Data S1 online for RosettaScripts, flags, and command lines.
Two energy functions were alternated throughout the simulations: the all-atom “hard-repulsive” Rosetta energy function (talaris2014) (O’Meara et al., 2015), which is dominated by van der Waals, hydrogen bonding, implicit solvation, and Coulomb electrostatics with a distance-dependent dielectric coefficient, and a “soft-repulsive” version of this energy function (soft-rep), where van der Waals overlaps and residue conformational strain are downweighted. The two energy functions were augmented with harmonic backbone coordinate restraints set to the coordinates observed in the PDB structure, and with a term that favors mutations with a higher PSSM score.
We refined each PDB structure by four iterations of side-chain packing (except in active-site residues) and side-chain and backbone minimization, saving the minimum-energy structure. For hAChE, the largest protein tested here, refinement calculations took, on average, 2.5 hr per trajectory (Data S1).
Computational mutation scanning was applied to the refined structure using the FilterScan mover in Rosetta (Whitehead et al., 2012). At every position, each allowed mutation (that is, every amino acid identity with PSSM score ≥0) was modeled singly against the background of the refined structure. Protein sidechains within 8 Å of the modeled mutation were repacked, and side-chain and constrained backbone minimization were used to accommodate the mutation. The energy difference between the refined structure and the optimized configuration of the single-point mutant was calculated using talaris2014 (O’Meara et al., 2015). Seven energy thresholds were used to define different mutation spaces (−0.45, −0.75, −1.0, −1.25, −1.5, −1.8, and −2.0 R.e.u.). Mutation scanning is the most time-consuming step in the calculation, taking between 30 s and 6 min per position (1 min per position on average for hAChE) using a standard CPU (Data S1). The procedure can be parallelized using a computer cluster, as implemented in the PROSS webserver.
For Dnmt3a, nine mutations were eliminated from the allowed sequence space since they showed exceptionally high and unexpected changes in their Rosetta backbone-omega energy, probably due to the poor crystallographic resolution of the input PDB structure (Table S2).
For each of the sequence spaces of potentially stabilizing mutations, combinatorial sequence optimization was implemented. Starting from the refined structure, we imposed coordinate restraints relative to the coordinates in the PDB file, and implemented four iterations of sequence design, side-chain, and backbone minimization, while alternating soft and hard repulsive potentials. For AChE, each design trajectory took ∼2.5 hr on average (Data S1).
Design Model Analysis
Sequence and structural features of designed variants were compared to the sequence and structure of the wild-type protein. The differences in the number of salt bridges and hydrogen bonds were evaluated using the Rosetta FeaturesReporter suite (Leaver-Fay et al., 2013, O’Meara et al., 2015), with a salt-bridge interaction defined as two counter charges within less than 5.5Å. Buried residues were defined as residues with >22 and >75 neighboring nonhydrogen atoms within 10 Å and 12 Å, respectively. Protein isoelectric points (pI) were calculated using the ExPASy webserver (http://web.expasy.org/compute_pi).
For experimental procedures, see Supplemental Experimental Procedures online.
For DNA and protein sequences, see Data S2.
Author Contributions
A.G., D.S.T., and S.J.F. conceived the idea. A.G. and S.J.F. developed the algorithm. A.G. and J.P. developed the automated PROSS webserver. A.G. designed variants of AChE, PTE, Dnmt3a, and SIRT6. S.E.H. and R.L.L designed dMyoc-OLF. Experimental characterization was by M.G. and Y.A. (AChE), M.G. (PTE), S.E.H. (Myoc-OLF), O.G. (SIRT6), and P.L. (Dnmt3a). T.U. purified dAChE4 for crystallization, and O.D. solved its structure, with input from J.A.S., I.S., and S.J.F. The results were discussed and the manuscript written by A.G., D.S.T., and S.J.F. with input from all authors.
Acknowledgments
We thank David Schreiber for analyzing structural features of design models; Ravit Netzer, Gideon Lapidoth, and Christoffer Norn for suggestions; and our collaborators for testing the PROSS webserver. Research in the Fleishman laboratory is supported by the Israel Science Foundation (ISF) through an individual grant, the Center for Research Excellence in Structural Cell Biology, and the joint ISF-UGC program, a Starter’s Grant from the European Research Council, a Career Development Award from the Human Frontier Science Program and a Marie Curie Reintegration Grant, the Minerva Foundation, an Alon Fellowship, and a charitable donation from Sam Switzer and Family. Funding by a DTRA project grant (HDTRA1-11-C-0026) to D.S.T. and NIH (R01EY021205) to RLL are gratefully acknowledged. S.J.F. is a Martha S. Sagon Career Development Chair. D.S.T. is the Nella and Leon Benoziyo Professor of Biochemistry. The collaboration between the Fleishman and Tawfik laboratories is also supported by the Rothschild-Caesaria Foundation.
Published: July 14, 2016
Footnotes
Supplemental Information includes three figures, six tables, two data sets, and Supplemental Experimental Procedures and can be found with this article at http://dx.doi.org/10.1016/j.molcel.2016.06.012.
Contributor Information
Dan S. Tawfik, Email: dan.tawfik@weizmann.ac.il.
Sarel J. Fleishman, Email: sarel@weizmann.ac.il.
Supplemental Information
References
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Gertz E.M., Agarwala R., Schäffer A.A., Yu Y.K. PSI-BLAST pseudocounts and the minimum description length principle. Nucleic Acids Res. 2009;37:815–824. doi: 10.1093/nar/gkn981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benning M.M., Shim H., Raushel F.M., Holden H.M. High resolution X-ray structures of different metal-substituted forms of phosphotriesterase from Pseudomonas diminuta. Biochemistry. 2001;40:2712–2722. doi: 10.1021/bi002661e. [DOI] [PubMed] [Google Scholar]
- Bigley A.N., Mabanglo M.F., Harvey S.P., Raushel F.M. Variants of phosphotriesterase for the enhanced detoxification of the chemical warfare agent VR. Biochemistry. 2015;54:5502–5512. doi: 10.1021/acs.biochem.5b00629. [DOI] [PubMed] [Google Scholar]
- Bloom J.D., Labthavikul S.T., Otey C.R., Arnold F.H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. USA. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgo B., Havranek J.J. Automated selection of stabilizing mutations in designed and natural proteins. Proc. Natl. Acad. Sci. USA. 2012;109:1494–1499. doi: 10.1073/pnas.1115172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns J.N., Orwig S.D., Harris J.L., Watkins J.D., Vollrath D., Lieberman R.L. Rescue of glaucoma-causing mutant myocilin thermal stability by chemical chaperones. ACS Chem. Biol. 2010;5:477–487. doi: 10.1021/cb900282e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny I., Greisen P., Jr., Ashani Y., Khare S.D., Oberdorfer G., Leader H., Baker D., Tawfik D.S. Engineering V-type nerve agents detoxifying enzymes using computationally focused libraries. ACS Chem. Biol. 2013;8:2394–2403. doi: 10.1021/cb4004892. [DOI] [PubMed] [Google Scholar]
- Cheung J., Rudolph M.J., Burshteyn F., Cassidy M.S., Gary E.N., Love J., Franklin M.C., Height J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012;55:10282–10286. doi: 10.1021/jm300871x. [DOI] [PubMed] [Google Scholar]
- Dahiyat B.I., Mayo S.L. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- Donegan R.K., Hill S.E., Turnage K.C., Orwig S.D., Lieberman R.L. The glaucoma-associated olfactomedin domain of myocilin is a novel calcium binding protein. J. Biol. Chem. 2012;287:43370–43377. doi: 10.1074/jbc.M112.408906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M., Ittah A., Liefer I., Gorecki M. Expression and reconstitution of biologically active human acetylcholinesterase from Escherichia coli. Cell. Mol. Neurobiol. 1993;13:25–38. doi: 10.1007/BF00712987. [DOI] [PubMed] [Google Scholar]
- Fleishman S.J., Whitehead T.A., Ekiert D.C., Dreyfus C., Corn J.E., Strauch E.M., Wilson I.A., Baker D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S.E., Donegan R.K., Nguyen E., Desai T.M., Lieberman R.L. Molecular details of olfactomedin domains provide pathway to structure-function studies. PLoS ONE. 2015;10:e0130888. doi: 10.1371/journal.pone.0130888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacak R., Leaver-Fay A., Kuhlman B. Computational protein design with explicit consideration of surface hydrophobic patches. Proteins. 2012;80:825–838. doi: 10.1002/prot.23241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kanfi Y., Naiman S., Amir G., Peshti V., Zinman G., Nahum L., Bar-Joseph Z., Cohen H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- Khersonsky O., Röthlisberger D., Wollacott A.M., Murphy P., Dym O., Albeck S., Kiss G., Houk K.N., Baker D., Tawfik D.S. Optimization of the in-silico-designed kemp eliminase KE70 by computational design and directed evolution. J. Mol. Biol. 2011;407:391–412. doi: 10.1016/j.jmb.2011.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koellner G., Kryger G., Millard C.B., Silman I., Sussman J.L., Steiner T. Active-site gorge and buried water molecules in crystal structures of acetylcholinesterase from Torpedo californica. J. Mol. Biol. 2000;296:713–735. doi: 10.1006/jmbi.1999.3468. [DOI] [PubMed] [Google Scholar]
- Korkegian A., Black M.E., Baker D., Stoddard B.L. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugel S., Mostoslavsky R. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem. Sci. 2014;39:72–81. doi: 10.1016/j.tibs.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S., Phillips K.J., Liu D.R. Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A., O’Meara M.J., Tyka M., Jacak R., Song Y., Kellogg E.H., Thompson J., Davis I.W., Pache R.A., Lyskov S. Scientific benchmarks for guiding macromolecular energy function improvement. Methods Enzymol. 2013;523:109–143. doi: 10.1016/B978-0-12-394292-0.00006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M., Pasamontes L., Lassen S.F., Wyss M. The consensus concept for thermostability engineering of proteins. Biochim. Biophys. Acta. 2000;1543:408–415. doi: 10.1016/s0167-4838(00)00238-7. [DOI] [PubMed] [Google Scholar]
- Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Magliery T.J. Protein stability: computation, sequence statistics, and new experimental methods. Curr. Opin. Struct. Biol. 2015;33:161–168. doi: 10.1016/j.sbi.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara M.J., Leaver-Fay A., Tyka M.D., Stein A., Houlihan K., DiMaio F., Bradley P., Kortemme T., Baker D., Snoeyink J., Kuhlman B. Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J. Chem. Theory Comput. 2015;11:609–622. doi: 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordentlich A., Barak D., Kronman C., Ariel N., Segall Y., Velan B., Shafferman A. Contribution of aromatic moieties of tyrosine 133 and of the anionic subsite tryptophan 86 to catalytic efficiency and allosteric modulation of acetylcholinesterase. J. Biol. Chem. 1995;270:2082–2091. doi: 10.1074/jbc.270.5.2082. [DOI] [PubMed] [Google Scholar]
- Potapov V., Cohen M., Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng. Des. Sel. 2009;22:553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- Roodveldt C., Tawfik D.S. Directed evolution of phosphotriesterase from Pseudomonas diminuta for heterologous expression in Escherichia coli results in stabilization of the metal-free state. Protein Eng. Des. Sel. 2005;18:51–58. doi: 10.1093/protein/gzi005. [DOI] [PubMed] [Google Scholar]
- Steipe B., Schiller B., Pluckthun A., Steinbacher S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 1994;15:188–192. doi: 10.1006/jmbi.1994.1434. [DOI] [PubMed] [Google Scholar]
- Sullivan B.J., Nguyen T., Durani V., Mathur D., Rojas S., Thomas M., Syu T., Magliery T.J. Stabilizing proteins from sequence statistics: the interplay of conservation and correlation in triosephosphate isomerase stability. J. Mol. Biol. 2012;420:384–399. doi: 10.1016/j.jmb.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman J., Harel M., Frolow F., Oefner C., Goldman A., Toker L., Silman I. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science. 1991;253:872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- Tokuriki N., Stricher F., Serrano L., Tawfik D.S. How protein stability and new functions trade off. PLoS Comput. Biol. 2008;4:e1000002. doi: 10.1371/journal.pcbi.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau D.L., Lee T.M., Arnold F.H. Engineered thermostable fungal cellulases exhibit efficient synergistic cellulose hydrolysis at elevated temperatures. Biotechnol. Bioeng. 2014;111:2390–2397. doi: 10.1002/bit.25308. [DOI] [PubMed] [Google Scholar]
- Whitehead T.A., Chevalier A., Song Y., Dreyfus C., Fleishman S.J., De Mattos C., Myers C.A., Kamisetty H., Blair P., Wilson I.A., Baker D. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 2012;30:543–548. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijma H.J., Floor R.J., Jekel P.A., Baker D., Marrink S.J., Janssen D.B. Computationally designed libraries for rapid enzyme stabilization. Protein Eng. Des. Sel. 2014;27:49–58. doi: 10.1093/protein/gzt061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam G.H.-F., Gaplovska-Kysela K., Zuber C., Roth J. Aggregated myocilin induces russell bodies and causes apoptosis: implications for the pathogenesis of myocilin-caused primary open-angle glaucoma. Am. J. Pathol. 2007;170:100–109. doi: 10.2353/ajpath.2007.060806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Arnold F.H. Directed evolution converts subtilisin E into a functional equivalent of thermitase. Protein Eng. 1999;12:47–53. doi: 10.1093/protein/12.1.47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.