

ABSTRACT
The DiGeorge/22q11-deletion syndrome (22q11DS), also known as velocardiofacial syndrome, is a congenital disease causing numerous structural and behavioral disorders, including cardiac outflow tract anomalies, craniofacial dysmorphogenesis, parathyroid and thymus hypoplasia, and mental disorders. It results from a unique chromosomal microdeletion on the 22q11.2 region in which the transcriptional activator TBX1 is decisive for the occurrence of the disease. During embryogenesis, Tbx1 is required for patterning of pharyngeal region giving rise to structures of the face, neck and chest. Genetic and developmental studies demonstrated that the severity and variability of the syndrome are determined by Tbx1 targets involved in pharyngeal neural crest cell migration and survival. Recently, we demonstrated that the chemokine Sdf1/Cxcl12 and its receptor Cxcr4 are genetically downstream of Tbx1 during pharyngeal development and that reduction of CXCR4 signaling results in defects which recapitulate the major morphological anomalies of 22q11DS, supporting the possibility of a pivotal role for the SDF1/CXCR4 axis in its etiology.
KEYWORDS: 22q11-deletion syndrome, CXCR4, Digeorge syndrome, neural crest, pharyngeal arch, SDF1, TBX1, velocardiofacial syndrome
DiGeorge/22q11-deletion syndrome: Clinical features and genetic basis
DiGeorge syndrome (DGS), also referred to as velocardiofacial syndrome, is a relatively-frequent congenital disease with an incidence estimated to 1:2000/1:7000. It represents a typical example of multiple-anomaly syndromes covering nearly every organ and system. 1 Of the diverse clinical features found in DGS, those that have attracted the greatest attention are the developmental and behavioral disorders. More than 70% of patients with DGS exhibit conotruncal heart anomalies, including defective outflow septation, outflow misalignment or mispatterning of the great arteries, as observed in tetralogy of Fallot, persistent truncus arteriosus, and double-outlet right ventricle. Craniofacial anomalies also concur to a significant proportion of DGS cases, with a characteristic facial appearance and palatal/velopharyngeal dysfunction with or without cleft. Numerous other structural anomalies have been described in DGS, in particular mispositioning of the vascular system in the pharynx, neck and chest, and hypoparathyroidism, responsible for severe hypocalcemia. Likewise, due to thymic hypoplasia, immune disorders, such as chronic respiratory infections, are also relatively common although severe immunodeficiencies are the exception. Dysphagia is often a serious complication of DGS, but the exact physiological causes for this anomaly are still debated. Finally, numerous psychiatric disorders, especially psychosis, schizophrenia, mental retardation and speech and language impairment, have been associated with DGS.
The wide phenotypic spectrum (more than 180 clinical features both physical and behavioral) exhibited by patients with DGS has long hampered the precise diagnosis of the pathology and the elucidation of its etiology. In addition, no single clinical feature occurs in 100% of cases and there are no reported cases of the syndrome that has all or even most of the clinical findings. 1 To date, the only reliable criterion to diagnose DGS cases is the hemizygous chromosomal microdeletion within the 22q11.2 region, and it is now widely accepted that this pathology is primarily defined on this genetic basis. 1,2 This is why hereafter, this syndrome will be referred to as 22q11-deletion syndrome (22q11DS). The 22q11.2 region harbors about 30 to 40 different genes, and genetic studies in mice carrying deletions in the region of synteny to the 22q11.2 region (chromosome 16 in the Df1/+ and LgDel/+ mouse strains) helped to identify Tbx1, a member of the T-box family of transcription factors, as one of the major genes responsible for the abnormalities observed. 3 Moreover, Tbx1−/− mice recapitulate many of the clinical features of patients with 22q11DS, including cleft palate, absent or reduced parathyroid and thymus as well as malformed outflow tract. 4 In human, TBX1 maps to the 22q11.2 region and its haploinsufficiency or sequence mutations have been found in patients with 22q11DS. 5 It should be emphasized though that many 22q11.2 deletion variants do not impact TBX1 and that TBX1 mutations in humans are primarily associated with cardiac manifestations, implying that TBX1 alone cannot account for the complexity of the 22q11DS spectrum.
Development of the pharyngeal region: A crosstalk between several embryonic tissues deregulated in 22q11DS
How can TBX1 mutation result in such a diversity of phenotypes and extreme variability in their occurrence? Part of the answer lies in the embryological origin of the tissues affected. Indeed, all the cardiac, facial, thymic and parathyroid defects concern a single broad region, termed pharyngeal arches, that is situated ventrally in the anterior part of the embryo. These transient structures are bilateral, segmented structures that develop during the initial steps of cephalic development to form, at the level of the hindbrain, 6 arches and pouches in a cranial-to-caudal order. Later during development, pharyngeal arches undergo extensive structural reorganizations and are at the origin of most tissues and structures of the face, neck, and chest, including the upper and lower jaws, the pharynx as well as components of the cardiac outflow tract and cephalic vasculature. Early during their development, Tbx1 is prominently expressed in pharyngeal arches and Tbx1−/− mice display severe pharyngeal defects. 4,6 Furthermore, Tbx1 is required at different times, in different tissues and at different levels to pattern all pharyngeal arch-derived structures. 7-11
Pharyngeal arches are of a mixed embryonic origin, with contributions of the 3 primary germ layers (ectoderm, endoderm, mesoderm) along with neural crest (NC) cells, thus raising the question of the identity of cells affected in DGS. While the pharyngeal endoderm provides precursors to craniofacial organs, the mesodermal contingent of the pharyngeal arches gives rise to facial and neck muscles as well as to most of the cardiac and vascular systems. Regarding NC cells, after migrating from the dorsal hindbrain toward the arches where they form the mesenchyme surrounding the arch arteries and the core mesoderm, they contribute to the skeletal structures of the face and, in the neck and chest, to the conotruncus, the parathyroid, part of the thymus and the peripheral nervous system. Quite remarkably, most tissues that are affected in the face and neck of patients with 22q11DS comprise at least partly NC-derived cells. Genetic studies in mouse provided evidence that defects in NC development can result in malformations reminiscent of 22q11DS. 12 However, it was the classical embryology techniques developed in avians that clearly demonstrated that 22q11DS results primarily from defective development of NC cells, making this pathology one of the most severe among the numerous neurocristopathies. Specific ablation of the NC population that invade the first 6 pharyngeal arches (hereafter called pharyngeal NC) was found to phenocopy many of the cardiocraniofacial anomalies found in patients with DGS. 13
Tbx1 orchestrates tissue interactions during pharyngeal development
Intriguingly, at least in chick and mouse, Tbx1 transcript are not found in NC cells themselves in pharyngeal arches. Rather, Tbx1 is expressed dynamically in the pharyngeal ectoderm and endoderm as well as in the mesoderm.8,14 In addition, loss of Tbx1 impairs NC cell migration and differentiation,15-17 indicating that it acts on NC development in a non-autonomous fashion. Since then, many studies focused on the signals elicited by Tbx1 that mediate interactions between pharyngeal NC cells and their surrounding tissues to uncover the molecular mechanisms underlying DGS.
The essential role of Tbx1 in pharynx morphogenesis relies on its ability to interact with crucial signaling pathways during development, such as the fibroblast growth factor (FGF), hedgehog and retinoic acid.2 Among them, FGF8 emerged as a key player for mediating Tbx1 effect on pharyngeal NC cells. Fgf8 is downregulated in the pharyngeal endoderm of Tbx1 mutants and mice heterozygous for both Fgf8 and Tbx1 have a greater penetrance of pharyngeal arch defects than Tbx1 heterozygotes.18 In addition, in Fgf8-hypomorphic mice, pharyngeal NC cells undergo massive cell death in the pharynx, suggesting that FGF8 plays a major role in maintaining NC cell survival during the late phases of arch colonization.19,20 Further experiments performed in chick demonstrated that FGF8 is chemotactic for NC cells migrating to the heart, 21 therefore raising the intriguing possibility that the pharyngeal NC defects observed in DGS may not be solely due to defective survival signals in the arches, but also to misregulation at earlier stages during their dorsoventral migration from the hindbrain to the arches. However, Fgf8 defects cannot by far account for the whole range of NC defects as it was shown that only cells that populate the heart (i.e. originating from the caudal hindbrain at the level of rhombomere 6-7 and invading archs 3-6) respond to FGF8 while those populating arch 2 from rhombomere 4 respond to VEGF. 21,22 If chemotaxis is truly involved in 22q11DS occurrence, then another factor attracting indistinctly all pharyngeal NC cells into arches 1-6 is to be identified. Beside FGF8 and VEGF, NC cells have been previously shown to respond to a variety of chemoattractants, including FGF2, semaphorins, and the Stromal-Derived Factor-1 (SDF1). 23
SDF1 chemokine signaling is required for pharyngeal NC development
SDF1 (also named CXCL12), a member of the chemokine family that is widely expressed during embryonic development, attracted our attention for several reasons. It is capable of driving oriented migration of various NC cell populations, either truncal or cranial, through binding to its CXCR4 receptor. 23,24 Loss of Sdf1 or Cxcr4 in mouse embryo causes ventricular septal defects similar to those observed in 22q11DS. 25,26 Interestingly, signaling by CXCR4 is deficient in brain cortical interneurons of the LgDel/+ and Df1/+ mouse strains, resulting in their abnormal migration, and this is believed to constitute the basis of the mental and behavioral disorders shown by patients with 22q11DS.27,28 Finally, in silico analysis revealed the presence of putative TBX1-binding sites in the Cxcr4 promoter. 29 We thus analyzed whether CXCR4 signaling could drive oriented migration of pharyngeal NC cells and investigated whether its misregulation could ultimately cause cardiocraniofacial defects associated with 22q11DS. 30,31
In a first study, we focused on NC cells that migrate from the posterior hindbrain to populate the heart and termed cardiac NC using the chick embryo as a model, taking advantage that in this species, the spatiotemporal course of NC cell migration has been fully documented and can be easily and specifically manipulated owing to numerous available molecular tools. 30 We found that cardiac NC cells express the CXCR4 receptor during their initial migration from the hindbrain to pharyngeal arches 3-6, and that Sdf1 exhibits a complementary pattern in the ectoderm along their migration route. We then performed functional experiments using electroporation in premigratory cardiac NC cells either of a miRNA, to knockdown Cxcr4 expression, or of a dominant-negative form of Cxcr4 in which the chemotactic, but not the survival, response to SDF1 was abolished. We also challenged cardiac NC cells with Sdf1 ectopically expressed in the neural tube. Cxcr4 loss-of function caused delayed migration and enhanced death (when miRNA to Cxcr4 was applied) of cardiac NC cells, whereas Sdf1 misexpression resulted in diversion of cardiac NC cells away from their normal migration pathway, confirming our hypothesis that SDF1 acts as a chemoattractant for cardiac NC. Finally, alterations of SDF1 signaling during migration led invariably at late stages of development to various cardiovascular defects commonly found in 22q11DS. These data therefore identify Sdf1 and its receptor Cxcr4 as candidate genes responsible for the cardiovascular congenital malformations of 22q11DS.
Using the same experimental approach, we then investigated whether this SDF1-mediated chemotactic mechanism applies to the other NC populations migrating more anteriorly through pharyngeal arches 1-2. 31 We found that, both in chick and mouse, Cxcr4 and Sdf1 exhibit the same dynamic spatiotemporal expression patterns as more caudally and that reduction of CXCR4 signaling causes early misrouting of pharyngeal NC cells and, later, dramatic morphological alterations in the mandibular skeleton, thymus and cranial sensory ganglia. Thus, the SDF1-CXCR4 axis fulfills almost all the requirements for being implicated in the morphological disorders observed in patients with 22q11DS. Moreover, we showed that, in the mouse embryo, Tbx1 is expressed in the same regions of the ectoderm and pharyngeal endoderm as Sdf1 during initial migration of NC cells under the ectoderm. In addition, we found a strong reduction of both Sdf1 and Cxcr4 expressions in the pharyngeal arches of Tbx1 mouse mutants compared to wild-type embryos. Our data therefore indicate that in mouse, Cxcr4 and Sdf1 are downstream of Tbx1 in the genetic cascade regulating pharyngeal arch development and suggest a model in which Tbx1 may regulate Sdf1 expression in the lateral ectoderm and pharyngeal endoderm and control the chemotactic guidance of pharyngeal NC cells toward the pharyngeal arches. This view is fully consistent with the recent findings showing functional defects of CXCR4 signaling in the brain of the LgDel/+ and Df/+1 mice. 27,28
How Tbx1 and SDF1 signaling interact during pharyngeal development?
A question remains as to how Tbx1 regulates Sdf1 and Cxcr4 expressions in cells. Although bioinformatic analyses in mouse identified putative binding sites for TBX1 in the Cxcr4 promoter, 29 their exclusive expression patterns in distinct compartments of the pharyngeal arches (the ectoderm and endoderm for Tbx1 and migrating NC cells for Cxcr4) clearly exclude a direct regulation. Conversely, the coincident expression of Sdf1 and Tbx1 in the lateral ectoderm and in the pharyngeal endoderm would rather be in favor of a possible direct regulation. However, if it exists, such a mechanism would be only transient and require additional co-factors as both genes exhibit poorly-overlaping expression patterns once NC cells colonize the arches. Moreover, previous studies failed to detect difference in the relative expression of Sdf1/Cxcl12 in Tbx1−/− versus Tbx1+/+ cells. 32 Therefore, more complex relationships are likely to occur between Tbx1, Sdf1 and its Cxcr4 receptor. A possible mechanism is that these genes are under the control of a common set of transcriptional regulators. For example, Tbx1 expression in the pharyngeal endoderm and mesoderm has been demonstrated to be regulated by the FOX transcription factors Foxa2, Foxc1 and Foxc2, 33 while genomic analyses revealed conserved binding sites for Foxa2 and Foxc2 in the proximal enhancer region of the Cxcr4 gene. 34 Alternatively, more indirect mechanisms may also operate. Thus, we found that the expression domain of Sdf1 in the ectoderm becomes gradually restricted with the lateral progression of the cardiac NC stream toward the pharyngeal arches, 30 suggesting that NC cells themselves provide the cues for repressing Sdf1 expression during migration. However, we also showed that if NC cells are prevented to migrate laterally along the ectoderm, Sdf1 becomes downregulated, indicating that its expression requires positive signals emanating from NC cells. Conversely, forced overexpression of Sdf1 is able to induce Cxcr4 expression. These observations suggest that Sdf1 and Cxcr4 cross-regulate each other in neighboring cells by a yet-undefined mechanism. This process may therefore explain why in Tbx1−/− embryos, expression of both Sdf1 and Cxcr4 would be down-regulated although they are not coexpressed in the same cell populations. Such a mechanism is believed to ensure a fast and robust regulation of chemotactic guidance of NC cells. Of interest, a similar loop of regulation has been proposed for FGF8 and its receptor during their migration toward the heart. 21
SDF1-CXCR4 signaling: A central role in 22q11DS defects?
In a pathology such as 22q11DS presenting a large spectrum of phenotypes with a variable incidence, it is of importance to determine whether candidate genes can explain the vast majority of the clinical features of the disease or only a few of them, thereby accounting for its diversity. While there is no doubt that Tbx1 is an essential gene in 22q11DS as its inactivation in mouse and haploinsufficiency in human cause a large array of velocardiofacial defects, the question is of a great relevance for its putative transcriptional downstream targets. As discussed above, Fgf8 has been found to drive chemotactic migration of NC cells to pharyngeal arches 3-6 but not of those invading arch 2. 21 In agreement with this observation, conditional deletion of Fgf8 in the arches results primarily in anomalies in the cardiac outflow tract, the parathyroid and the thymus, all derived from arches 3-6, 35 indicating that it is not involved in the occurrence of facial defects observed in 22q11DS. Moreover, targeted manipulation of Fgf8 expression in the Tbx1-positive cells in the mouse embryo was found to cause skeletal abnormalities and to alter patterning of the aortic arches but not the outflow tract, suggesting that Tbx1 and Fgf8 play independent roles in regulating outflow tract development. 36 Thus, Fgf8 may account mostly for the variable penetrance of the cardiovascular and glandular defects in 22q11DS and not for its overall phenotype. Likewise, the homeobox-containing transcription factor, Gbx2, another downstream effector of Tbx1, has been shown to be required in the pharyngeal ectoderm to drive NC cell migration into pharyngeal arch 3-6 via the Slit/Robo signaling pathway. 16 However, as for Fgf8, Gbx2 function is restricted to arch 3-6 and does not extend to more anterior regions, thereby accounting only partially for 22q11DS defects.
In striking contrast, our studies suggest that, unlike Fgf8 and Gbx2, the Sdf1-Cxcr4 signaling axis may play a broad role in governing migration and survival of most if not all NC cells invading pharyngeal arches 1-6. Accordingly, inhibition of CXCR4 activity or alterations in Sdf1 expression pattern both lead to a large spectrum of abnormalities in the cardiovascular, skeletal, neuronal and glandular derivatives of pharyngeal NC. 30 Moreover, we also observed considerable mispatterning of cranial sensory ganglia in embryos in which CXCR4 activity was abolished with variable phenotypes, such as missing, fused or misrouted cranial nerves, as also reported in the LgDel/+ mouse. 37 Importantly, these anomalies did not result from a direct effect on NC cells at the origin of the cranial sensory ganglia and nerves (these cells do not respond to SDF1 signals), but merely from the misrouting of pharyngeal NC cells (which do rely on SDF1 for oriented migration) to aberrant locations. Thus, the implication of the SDF1-CXCR4 signaling pathway in the occurrence of the diverse clinical features of 22q11DS may extend well beyond the sole NC derivatives in the pharyngeal arches and might concern tissues that do not rely directly on Tbx1 expression (Fig. 1). Given that CXCR4 signaling has also been shown to be defective in the brain of mouse models of DGS, 27,28 we propose that, although the 22q11.2 region deleted in patients harbors neither gene, the SDF1-CXCR4 signaling axis may play a pivotal role in 22q11DS etiology and account for its neural, behavioral, cardiovascular, immune, endocrine and facial aspects.
Figure 1.
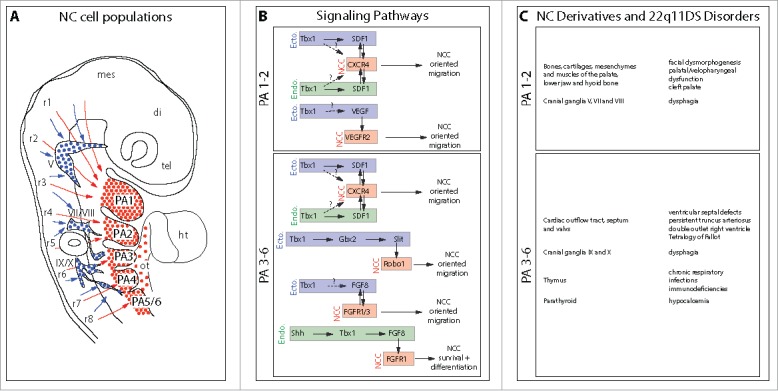
The SDF1/CXCR4 signaling pathway: A new player involved in DiGeorge/22q11-deletion syndrome malformations. A. Schematic representation of the different NC cell populations that colonize the pharyngeal arches, the cardiac outflow tract and the cranial ganglia. NC cells that express the CXCR4 receptor and respond to SDF1 signals allowing them to migrate along the superficial ectoderm and populate the arches and the cardiac outflow tract are indicated in red. Those which do not express CXCR4 and migrate ventrally to contribute to the cranial sensory ganglia are in blue. Arrows indicate the level of origin of NC cells along the rostro-caudal axis. tel, telencephalon; di, diencephalon; mes, mesencephalon; r1-r8, rhombomeres 1-8; V, trigeminal ganglion; VII/VIII, facial and vestibuloacoustic ganglia; IX/X, glossopharyngeal and vagal ganglia; PA1-6, pharyngeal arches 1-6; ht, heart; ot, outflow tract. B. Putative signaling pathways regulating migration, survival and differentiation of NC cells in PA1-2 and PA3-6, respectively. The tissular origin of the signal (enodermal or ectodermal) is indicated. Single arrows with plain line represent demonstrated (direct or indirect) regulations and double arrows indicate reciprocal regulations. Arrows with dashed line and associated with question marks represent hypothetical regulatory pathways. C. Lists of the main NC derivatives generated in PA1-2 and PA3-6, respectively, and of the corresponding disorders commonly observed in 22q11DS. Neural defects consecutive to CXCR4 signaling defects and causing mental retardation and other psychiatric disorders are not listed because, to our knowledge, they are not directly related with anomalies in pharyngeal NC cells.
In conclusion, our study illustrates the great utility and versatility of the avian system to investigate the function of candidate genes for complex human congenital diseases and to analyze in detail the developmental processes responsible for their occurrence. More specifically in the case of 22q11DS, the use of the chick embryo allowed establishment of the precise origin of the NC cell populations that populate the branchial arches and their contribution to the facial, pharyngeal and cardiac tissues, 13 to demonstrate the importance of guidance systems in the segregation of the different NC populations that arise at cranial levels and in their patterning into various derivatives, 22,30, 31 and to characterize the implication of several genes located in the 22q11.2 region, such as HIRA, Ufd1l, and DGRC6, in the conotruncal anomalies observed in 22q11DS. 38-40
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the CNRS, the Université Pierre et Marie Curie and the Association Française contre les myopathies (grant No. 11405). Sophie Escot was a recipient of a doctoral fellowship from the Ministère de l'Enseignement Supérieur et de la Recherche.
References
- [1].Shprintzen RJ, Higgins AM, Antshel K, Fremont W, Roizen N, Kates W. Velo-cardio-facial syndrome. Curr Opin Pediatr 2005; 17:725-30; PMID:16282778; http://dx.doi.org/ 10.1097/01.mop.0000184465.73833.0b [DOI] [PubMed] [Google Scholar]
- [2].Scambler PJ. 22q11 deletion syndrome: a role for TBX1 in pharyngeal and cardiovascular development. Pediatr Cardiol 2010; 31:378-90; PMID:20054531; http://dx.doi.org/ 10.1007/s00246-009-9613-0 [DOI] [PubMed] [Google Scholar]
- [3].Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah YC, Rosenblatt HM, Bradley A, Baldini A. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 1999; 401:379-83; PMID:10517636 [DOI] [PubMed] [Google Scholar]
- [4].Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet 2001; 27:286-91; PMID:11242110; http://dx.doi.org/ 10.1038/85845 [DOI] [PubMed] [Google Scholar]
- [5].Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al.. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001; 104:619-29; PMID:11239417; http://dx.doi.org/ 10.1016/S0092-8674(01)00247-1 [DOI] [PubMed] [Google Scholar]
- [6].Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, et al.. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001; 410:97-101; PMID:11242049; http://dx.doi.org/ 10.1038/35065105 [DOI] [PubMed] [Google Scholar]
- [7].Zhang Z, Baldini A. In vivo response to high-resolution variation of Tbx1 mRNA dosage. Hum Mol Genet 2008; 17:150-7; PMID:17916582; http://dx.doi.org/ 10.1093/hmg/ddm291 [DOI] [PubMed] [Google Scholar]
- [8].Zhang Z, Cerrato F, Xu H, Vitelli F, Morishima M, Vincentz J, Furuta Y, Ma L, Martin JF, Baldini A, et al.. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development 2005; 132:5307-15; PMID:16284121; http://dx.doi.org/ 10.1242/dev.02086 [DOI] [PubMed] [Google Scholar]
- [9].Zhang Z, Huynh T, Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development 2006; 133:3587-95; PMID:16914493; http://dx.doi.org/ 10.1242/dev.02539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Arnold JS, Werling U, Braunstein EM, Liao J, Nowotschin S, Edelmann W, Hebert JM, Morrow BE. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development 2006; 133:977-87; PMID:16452092; http://dx.doi.org/ 10.1242/dev.02264 [DOI] [PubMed] [Google Scholar]
- [11].Yamagishi H, Srivastava D. Unraveling the genetic and developmental mysteries of 22q11 deletion syndrome. Trends Mol Med 2003; 9:383-9; PMID:13129704; http://dx.doi.org/ 10.1016/S1471-4914(03)00141-2 [DOI] [PubMed] [Google Scholar]
- [12].Epstein JA. Developing models of Digeorge syndrome. Trends Genet 2001; 17:S13-S7; PMID:11585671; http://dx.doi.org/ 10.1016/S0168-9525(01)02450-7 [DOI] [PubMed] [Google Scholar]
- [13].Hutson MR, Kirby ML. Model systems for the study of heart development and disease: cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol 2007; 18:101-10; PMID:17224285; http://dx.doi.org/ 10.1016/j.semcdb.2006.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Garg V, Yamagishi C, Hu T, Kathiriya IS, Yamagishi H, Srivastava D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev Biol 2001; 235:62-73; PMID:11412027; http://dx.doi.org/ 10.1006/dbio.2001.0283 [DOI] [PubMed] [Google Scholar]
- [15].Kochilas L, Merscher-Gomez S, Lu MM, Potluri V, Liao J, Kucherlapati R, Morrow B, Epstein JA. The role of neural crest during cardiac development in a mouse model of DiGeorge syndrome. Develop Biol 2002; 251:157-66; PMID:12413905; http://dx.doi.org/ 10.1006/dbio.2002.0819 [DOI] [PubMed] [Google Scholar]
- [16].Calmont A, Ivins S, Van Bueren KL, Papangeli I, Kyriakopoulou V, Andrews WD, Martin JF, Moon AM, Illingworth EA, Basson MA, et al.. Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development 2009; 136:3173-83; PMID:19700621; http://dx.doi.org/ 10.1242/dev.028902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet 2002; 11:915-22; PMID:11971873; http://dx.doi.org/ 10.1093/hmg/11.8.915 [DOI] [PubMed] [Google Scholar]
- [18].Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development 2002; 129:4605-11; PMID:12223416 [DOI] [PubMed] [Google Scholar]
- [19].Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development 2002; 129:4613-25; PMID:12223417 [DOI] [PubMed] [Google Scholar]
- [20].Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development 2002; 129:4591-603; PMID:12223415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sato A, Scholl AM, Kuhn EN, Stadt HA, Decker JR, Pegram K, Hutson MR, Kirby ML. FGF8 signaling is chemotactic for cardiac neural crest cells. Dev Biol 2011; 354:18-30; PMID:21419761; http://dx.doi.org/ 10.1016/j.ydbio.2011.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McLennan R, Teddy JM, Kasemeier-Kulesa JC, Romine MH, Kulesa PM. Vascular endothelial growth factor (VEGF) regulates cranial neural crest migration in vivo. Dev Biol 2010; 339:114-25; PMID:20036652; http://dx.doi.org/ 10.1016/j.ydbio.2009.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kulesa PM, Bailey CM, Kasemeier-Kulesa JC, McLennan R. Cranial neural crest migration: New rules for an old road. Dev Biol 2010:534-554; PMID:21146519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kasemeier-Kulesa JC, McLennan R, Romine MH, Kulesa PM, Lefcort F. CXCR4 controls ventral migration of sympathetic precursor cells. J Neurosci 2010; 30:13078-88; PMID:20881125; http://dx.doi.org/ 10.1523/JNEUROSCI.0892-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, Bronson RT, Springer TA. Impaired B-lymphopoieisis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF1-deficient mice. Proc Natl Acad Sci USA 1998; 95:9448-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998; 393:595-9; PMID:9634238; http://dx.doi.org/ 10.1038/31269 [DOI] [PubMed] [Google Scholar]
- [27].Meechan DW, Tucker ES, Maynard TM, LaMantia AS. Cxcr4 regulation of interneuron migration is disrupted in 22q11.2 deletion syndrome. Proc Natl Acad Sci U S A 2012; 109:18601-6; PMID:23091025; http://dx.doi.org/ 10.1073/pnas.1211507109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Toritsuka M, Kimoto S, Muraki K, Landek-Salgado MA, Yoshida A, Yamamoto N, Horiuchi Y, Hiyama H, Tajinda K, Keni N, et al.. Deficits in microRNA-mediated Cxcr4/Cxcl12 signaling in neurodevelopmental deficits in a 22q11 deletion syndrome mouse model. Proc Natl Acad Sci U S A 2013; 110:17552-7; PMID:24101523; http://dx.doi.org/ 10.1073/pnas.1312661110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Castellanos R, Xie Q, Zheng D, Cvekl A, Morrow BE. Mammalian TBX1 preferentially binds and regulates downstream targets via a tandem T-site repeat. PLoS One 2014; 9:e95151; PMID:24797903; http://dx.doi.org/ 10.1371/journal.pone.0095151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Escot S, Blavet C, Hartle S, Duband JL, Fournier-Thibault C. Misregulation of SDF1-CXCR4 signaling impairs early cardiac neural crest cell migration leading to conotruncal defects. Circ Res 2013; 113:505-16; PMID:23838132; http://dx.doi.org/ 10.1161/CIRCRESAHA.113.301333 [DOI] [PubMed] [Google Scholar]
- [31].Escot S, Blavet C, Faure E, Zaffran S, Duband JL, Fournier-Thibault C. Disruption of CXCR4 signaling in pharyngeal neural crest cells causes DiGeorge syndrome-like malformations. Development 2016; 143:582-8; PMID:26755698; http://dx.doi.org/ 10.1242/dev.126573 [DOI] [PubMed] [Google Scholar]
- [32].van Bueren KL, Papangeli I, Rochais F, Pearce K, Roberts C, Calmont A, Szumska D, Kelly RG, Bhattacharya S, Scambler PJ. Hes1 expression is reduced in Tbx1 null cells and is required for the development of structures affected in 22q11 deletion syndrome. Dev Biol 2010; 340:369-80; PMID:20122914; http://dx.doi.org/ 10.1016/j.ydbio.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yamagishi H, Maeda J, Hu T, McAnally J, Conway SJ, Kume T, Meyers EN, Yamagishi C, Srivastava D. Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev 2003; 17:269-81; PMID:12533514; http://dx.doi.org/ 10.1101/gad.1048903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Katoh M, Katoh M. Integrative genomic analyses of CXCR4: transcriptional regulation of CXCR4 based on TGFbeta, Nodal, Activin signaling and POU5F1, FOXA2, FOXC2, FOXH1, SOX17, and GFI1 transcription factors. Int J Oncol 2010; 36:415-20; PMID:20043076 [DOI] [PubMed] [Google Scholar]
- [35].Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development 2006; 133:2419-33; PMID:16720879; http://dx.doi.org/ 10.1242/dev.02367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Vitelli F, Zhang Z, Huynh T, Sobotka A, Mupo A, Baldini A. Fgf8 expression in the Tbx1 domain causes skeletal abnormalities and modifies the aortic arch but not the outflow tract phenotype of Tbx1 mutants. Dev Biol 2006; 295:559-70; PMID:16696966; http://dx.doi.org/ 10.1016/j.ydbio.2006.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Karpinski BA, Maynard TM, Fralish MS, Nuwayhid S, Zohn IE, Moody SA, LaMantia AS. Dysphagia and disrupted cranial nerve development in a mouse model of DiGeorge (22q11) deletion syndrome. Dis Model Mech 2014; 7:245-57; PMID:24357327; http://dx.doi.org/ 10.1242/dmm.012484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Farrell MJ, Stadt H, Wallis KT, Scambler P, Hixon RL, Wolfe R, Leatherbury L, Kirby ML. HIRA, a DiGeorge syndrome candidate gene, is required for cardiac outflow tract septation. Circ Res 1999; 84:127-35; PMID:9933243; http://dx.doi.org/ 10.1161/01.RES.84.2.127 [DOI] [PubMed] [Google Scholar]
- [39].Yamagishi C, Hierck BP, Gittenberger-De Groot AC, Yamagishi H, Srivastava D. Functional attenuation of UFD1l, a 22q11.2 deletion syndrome candidate gene, leads to cardiac outflow septation defects in chicken embryos. Pediatr Res 2003; 53:546-53; PMID:12612215; http://dx.doi.org/ 10.1203/01.PDR.0000055765.11310.E3 [DOI] [PubMed] [Google Scholar]
- [40].Hierck BP, Molin DG, Boot MJ, Poelmann RE, Gittenberger-de Groot AC. A chicken model for DGCR6 as a modifier gene in the DiGeorge critical region. Pediatr Res 2004; 56:440-8; PMID:15333760; http://dx.doi.org/ 10.1203/01.PDR.0000136151.50127.1C [DOI] [PubMed] [Google Scholar]