

Supplemental Digital Content is available in the text.
Keywords: cathepsin S, HDAC6, neointimal formation, vascular remodeling
Abstract
Objective—
Cathepsin S (CatS) participates in atherogenesis through several putative mechanisms. The ability of cathepsins to modify histone tail is likely to contribute to stem cell development. Histone deacetylase 6 (HDAC6) is required in modulating the proliferation and migration of various types of cancer cells. Here, we investigated the cross talk between CatS and HADC6 in injury-related vascular repair in mice.
Approach and Results—
Ligation injury to the carotid artery in mice increased the CatS expression, and CatS-deficient mice showed reduced neointimal formation in injured arteries. CatS deficiency decreased the phosphorylation levels of p38 mitogen-activated protein kinase, Akt, and HDAC6 and toll-like receptor 2 expression in ligated arteries. The genetic or pharmacological inhibition of CatS also alleviated the increased phosphorylation of p38 mitogen-activated protein kinase, Akt, and HDAC6 induced by platelet-derived growth factor BB in cultured vascular smooth muscle cells (VSMCs), and p38 mitogen-activated protein kinase inhibition and Akt inhibition decreased the phospho-HDAC6 levels. Moreover, CatS inhibition caused decrease in the levels of the HDAC6 activity in VSMCs in response to platelet-derived growth factor BB. The HDAC6 inhibitor tubastatin A downregulated platelet-derived growth factor–induced VSMC proliferation and migration, whereas HDAC6 overexpression exerted the opposite effect. Tubastatin A also decreased the intimal VSMC proliferation and neointimal hyperplasia in response to injury. Toll-like receptor 2 silencing decreased the phosphorylation levels of p38 mitogen-activated protein kinase, Akt, and HDAC6 and VSMC migration and proliferation.
Conclusions—
This is the first report detailing cross-interaction between toll-like receptor 2–mediated CatS and HDAC6 during injury-related vascular repair. These data suggest that CatS/HDAC6 could be a potential therapeutic target for the control of vascular diseases that are involved in neointimal lesion formation.
The process of neointima formation is common to various forms of vascular diseases, such as atherosclerosis, restenosis, and transplant vasculopathy.1,2 In response to vascular injury, the migration of vascular smooth muscle cells (VSMCs) from the media to the intima and the proliferation of intimal VSMCs are key events in restenotic lesion development.3 One essential factor required for VSMC migration is degradation of the basement membrane and surrounding extracellular matrix.4 Cathepsins are a family of cysteine proteases localized in lysosomes.5–7 Over the past decade, emerging data revealed unexpected roles of cathepsins in pathological conditions, such as metabolic disorder and atherosclerosis-based cardiovascular disease.8–10 Among cathepsin family, cathepsin S (CatS) was the first cathepsins found to be expressed in human atherosclerotic lesions.11 Previous studies showed that vascular atherosclerotic lesions associated with the injury overexpress the elastolytic and collagenolytic CatS but show no change in cystatin C expression, their endogenous inhibitor,4,12 suggesting a shift in the balance between cathepsins and their inhibitor that favors the remodeling of cardiovascular wall.
Histone deacetylases (HDACs) are a family of enzymes that remove acetyl groups from lysine residues of histone proteins and nonhistone proteins, a modification that results in epigenetic modulation of gene expression.13,14 Previous study reported that HDACs play a crucial role in the development of proliferative vascular diseases, including atherosclerosis and restenosis.13 Histone acetyltransferases and HDACs have been shown to regulate the expression of inflammatory and other genes involved in VSMC functions.15 Findeisen et al reported that short interfering RNA–mediated knockdown of HDAC 1, 2, or 3 prevented mitogen-induced SMC proliferation and that the pharmacological inhibition of HDAC decreased neointima formation.13 Yan et al found that HDACs modulate VSMC migration induced by cyclic mechanical strain.16 HDAC4 was recently reported to control neointimal hyperplasia via the stimulation of the proliferation and migration of VSMCs.17 HDAC6 has been found to play a pivotal role in modulating the migration and proliferation of cancer cells,18 and normal cells, including fibroblasts and epithelial cells.19 On the other hand, over the last couple of years’ researches highlighted that the cathepsin family is closely related to many critical signaling pathways, such as the Notch signaling pathway (cathepsin K),9 the peroxisome proliferator–activated receptor γ pathway (CatS),20 and histone H3 (cathepsin L).21 However, the relationship between CatS activity and the HDAC6 activation in the cellular events and injury-induced vascular repair is unclear.
In this study, we examined the hypothesis that CatS activity could modulate HDAC6 activation to stimulate VSMC migration and proliferation in vascular remodeling and neointimal hyperplasia in response to injury. We believed that this newly discovered CatS–HDAC6 interaction-mediated cellular mechanism has key roles in the hyperproliferative restenosis associated with endovascular treatments.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Reduced Neointimal Formation in the CatS−/− Mice
The ligation injury induced CatS expression in carotid arteries. The relative mRNA levels of CatS on day 1, 2, 4, 14, and 28 after ligation were increased by 25-fold over those of the uninjured control vessels of CatS+/+ mice (Figure IA in the online-only Data Supplement). CatS protein expression was also increased in the injured arteries by 4-fold (Figure IB in the online-only Data Supplement). As shown in Figure 1A and 1D, marked intimal lesion formation and long lesion lengths were observed in the samples from CatS+/+ mice at day 28 after the ligation injury. Much less intimal lesion formation was observed in the samples from the CatS−/− mice. The quantitative measurements revealed significantly lower levels of intimal areas from CatS−/− mice compared with those from the CatS+/+ mice (Figure 1B and 1E), but no significant difference in media thickness was observed between the CatS+/+ and CatS−/− mice (32.3±1.7 versus 29.1±1.4×103 /μm2; P=0.182). Therefore, the ratio of intimal to medial area was higher in the CatS+/+ mice (Figure1C). The proliferating cell nuclear antigen staining showed a low level of proliferative activity in both the intima and media of injured carotid arteries from CatS−/− mice compared with those of CatS+/+ mice (Figure 1F and 1G). As shown in Figure IC and ID in the online-only Data Supplement, we could not detect the expression of CatS gene and protein in the tail tissues and in the cultured VSMCs of CatS−/− mice.
Figure 1.
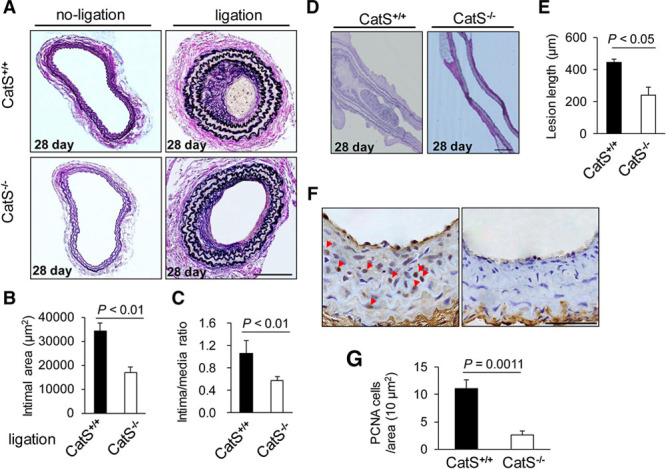
CatS−/− reduces neointimal formation in ligation-injured carotid arteries of mice. A, Representative images (van Geison staining) and quantitative data of hematoxylin and eosin (H&E) staining on cross-sections from injured carotid arteries of CatS+/+ and CatS−/− mice at day 28. Scale bar: 100 μm. B and C, The intimal area and intima/media ratio of injured arteries on day 28 were lower in CatS−/− mice. Data are mean±SEM (n=8), Student unpaired t test. D and E, Representative images (van Geison staining) and quantitative data of H&E on longitudinal sections from injured carotid arteries of CatS+/+ and CatS−/− mice. Scale bar: 200 μm. Data are mean±SEM (n=12 and 10, respectively), Student unpaired t test. F and G, Representative images and quantitative data of proliferating cell nuclear antigen (PCNA) staining on cross-sections from injured carotid arteries of CatS+/+ and CatS−/− mice. Scale bar: 50 μm. Data are mean±SEM (n=6), Student unpaired t test. Triangles indicate PCNA-stained cells. CatS indicates cathepsin S.
The macrophage activation–related release of inflammatory chemokines is an important hallmark of human and animal vascular repair and is mediated by a toll-like receptor (TLR) signaling pathway in cardiovascular disease.10,22 Here, we evaluated TLRs and inflammatory chemokine expressions. The quantitative polymerase chain reaction revealed that compared with the CatS+/+ mice, the lesions in CatS−/− mice that received a ligation injury had lower mRNA levels of TLR2, as well as monocyte chemoattractant protein-1, whereas TLR4 exhibited no significant difference (Figure IIA–IIC in the online-only Data Supplement). As shown in Figure IIF in the online-only Data Supplement, the TLR2-positive cells were higher in the neointima of the injured vessels from CatS+/+ mice on days 4 than in that of CatS−/− mice. There was also no significant difference in the cathepsin K or cystatin C mRNA expressions between the CatS+/+ and CatS−/− mice (Figure IID and IIE in the online-only Data Supplement).
Reduced Levels of Phospho-HDAC6 in CatS−/− Mice
Representative immunoblots showed that the level of phospho-HDAC6 (p-HDAC6) was increased in the injured arteries of CatS+/+ mice, and this increased expression was ablated in the CatS−/− mice on day 1 after ligation injury (Figure 2A and 2B). However, there are no significant differences in the total HDAC6 protein or HDAC6 mRNA levels between the injured and uninjured arteries of CatS+/+ mice (Figure 2A; Figure IIIA and IIIB in the online-only Data Supplement). With the exception of HDAC4, HDAC5, HDAC8, and HDAC9, we also observed that there were no between-group differences in other HDAC family members (including HDAC1, HDAC2, HDAC3, HDAC6, and HDAC7). As shown in Figure 2A, 2C, and 2D, we observed lower levels of phospho-p38 mitogen-activated protein kinase (p-p38MAPK) and p-Akt proteins in the injured arteries of CatS−/− mice. On operative day 1, compared with uninjured arteries, we observed an increase in the levels of hypoxia-inducible factor-1α (HIF-1α) gene in the injured arteries of CatS+/+ mice, indicating that ligation injury contributes to vascular local hypoxia (Figure IIG in the online-only Data Supplement). CatS deficiency caused decrease in HIF-1α gene change (Figure IIH in the online-only Data Supplement). The levels of plasma platelet-derived growth factor-BB (PDGF-BB) were increased on day 4 after ligation in the CatS+/+ mice and regressed on day 28 (Table I in the online-only Data Supplement). Interestingly, we observed that this increased expression of plasma PDGF was blunted in the CatS−/− mice on day 4, but there was no difference on day 28 between the 2 genotype groups.
Figure 2.
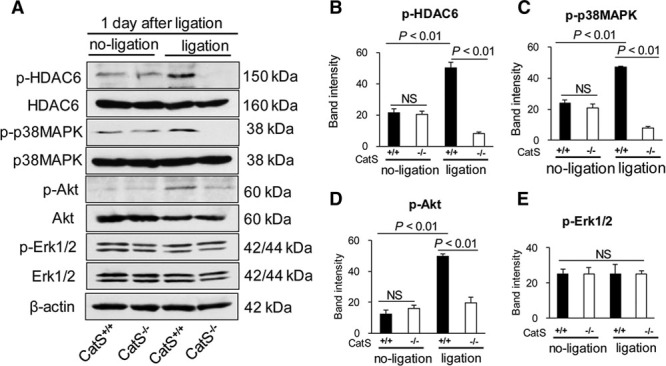
The levels of targeted protein in the ligation-injured arteries of the 2 experimental groups. On day 1 after injury, equal amounts of total protein extraction were immunoblotted using p-HDAC6, t-HDAC6, p-p38MAPK, t-p38MAPK, p-Akt, t-Akt, p-Erk1/2, t-Erk1/2, and β-actin antibodies. Representative immunoblots (A) and quantitative data (B–D) show reduced levels of p-HDAC6, p-p38MAPK, and p-Akt proteins in the injured arteries of CatS−/− mice, but show no changes in the levels of p-Erk1/2 between the CatS+/+ mice and CatS−/− mice (A and E). Data are mean±SEM of 4 independent experiments, Student unpaired t test or ANOVA and Bonferroni post hoc tests. ANOVA indicates analysis of variance; CatS, cathepsin S; ERK1/2, extracellular signal-regulated protein kinases 1/2; HDAC6, histone deacetylase 6; p-HDAC6, phospho-histone deacetylase 6; and p-p38MAPK, phospho-p38 mitogen-activated protein kinase.
HDAC6 Inhibition With Tubastatin A Decreased the Mitogen-Induced VSMC Proliferation and Migration and Induced Cell-Cycle Arrest
As shown in Figure 3A, the HDAC6-specific inhibitor tubastatin A reduced the PDGF-BB–induced proliferation of VSMCs at 48 hours after incubation. We also observed that tubastatin A reduced the PDGF-BB–induced migration of VSMCs at 36 hours after incubation (Figure 3B and 3C). To further explore the mechanisms by which HDAC inhibition prevents VSMC proliferation, we determined the cell-cycle distribution using flow cytometry. As shown in Figure 3D–3G, tubastatin A reduced the proportion of cells at S and G2/M stage. As anticipated, tubastatin A mitigated PDGF-induced HDAC6 phosphorylation in cultured VSMCs (Figure IV in the online-only Data Supplement). Moreover, HDAC6 silencing mitigated VSMC proliferation and migration in response to PDGF-BB (Figure V in the online-only Data Supplement). Thus, these findings indicate that HDAC6 activity may play a pivotal role in VSMC proliferation and migration, which are main processes of vascular remodeling. However, tubastatin A exhibited no effect on proliferation (0.11±0.02 versus 0.13±0.02; P=0.46) and migration (7.3±0.9 versus 9.1±1.0; P=0.81; n= 7 for each) in cultured VSMCs without PDGF-BB stimulation.
Figure 3.
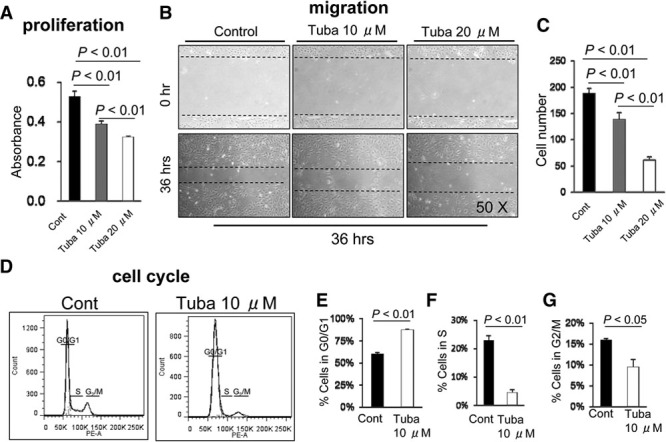
Histone deacetylase 6 (HDAC6) inhibition suppressed the growth factor–stimulated vascular smooth muscle cell (VSMC) proliferation and migration and blocked cell-cycle progression in vitro. A, VSMCs were grown in a 96-well plate in the absence or presence of tubastatin A (Tuba, 10 μM or 20 μM) with platelet-derived growth factor BB (PDGF-BB; 20 ng/mL) for 48 h. Data are mean±SEM (n=7). Representative images (B) and quantitative data (C) of effect of tubastatin A on the PDGF-BB–induced migration of VSMCs. Mouse VSMCs were scratched with a 1-mL pipet tip and cultured for 36 h in DMEM containing PDGF-BB (20 ng/mL) with or without tubastatin A (Tuba, 10 μM and 20 μM; n=7). The dotted lines define the areas lacking cells; ANOVA and Bonferroni post hoc tests. D-G, Representative images (D) and the quantitative data (E,F, and G) show the distribution of cells in G1 (E), S (F), and M (G) stage expressed as percentage of total cells. Data are mean±SEM (n=4), Student unpaired t test.
We also observed that pharmacological intervention targeted toward CatS mitigated VSMC proliferation and migration in response to PDGF-BB or 2% FBS (Figure VIA, VIC, and VID in the online-only Data Supplement). Likewise, CatS deficiency impaired VSMC proliferation in response to PDGF-BB (Figure VIB in the online-only Data Supplement). Compared with the CatS+/+ explants, the sprouting VSMC numbers and areas were markedly decreased in the aorta explants of the CatS−/− mice during the follow-up periods (Figure VII in the online-only Data Supplement). These data showed that both CatS and HDAC6 play an import role in VSMC proliferation and migration. CatS inhibitor had no effect on migration (4.0±1.2 versus 5.7±0.9; P=0.38) and proliferation (0.09±0.01 versus 0.10±0.00; P=0.91; n=5 for each) in cultured VSMCs without PDGF-BB stimulation. In vivo, CatS inhibitor also had no effects on the neointima areas (14 971±2371 versus 15 747±2101 μm2; P=0.23) and the ratio of intima to media (0.48±0.07 versus 0.51±0.08; P=0.66; n= 6 for each) in CatS deficiency mice.
HDAC6 Inhibition Reduced the Neointima Formation After Vascular Injury
Compared with the mice treated with vehicle, HDAC6 inhibition with tubastatin A reduced the neointimal formation in the ligation-injured arteries of CatS+/+ mice as determined by hematoxylin and eosin staining (Figure 4A and 4C). On day 14 after the surgery, we detected double-positive staining of bromodeoxyuridine and hematoxylin for proliferative VSMCs in the neointima of injured arteries from the CatS+/+ mice. The immunostaining of these cells was greatly decreased in the tubastatin A–treated mice (Figure 4B and 4D), indicating that HDAC6 activity plays a role in the process of proliferation of VSMCs. We also observed a decreased level of plasma HDAC6 activity in tubastatin A–treated group (Figure 4E). However, tubastatin exhibited no beneficial effects on neointima areas (15 392±1736 versus 16 079±2383 μm2; P=0.87) and the ratio of intima to media (0.52±0.10 versus 0.49±0.11; P=0.71; n= 6 for each) in CatS deficiency mice.
Figure 4.
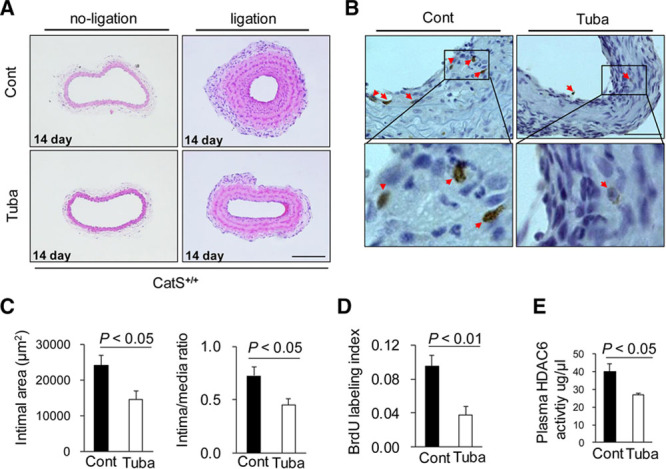
Histone deacetylase 6 (HDAC6) inhibition reduced the neointima formation after vascular injury. After tubastatin A (Tuba) was intraperitoneally administered to 10-week-old mice at a dose of 10 mg/kg every day for 2 weeks (n=6), the carotid artery was harvested and set in 5-μm paraffin sections. Representative images (A) and quantitative data (C) of hematoxylin and eosin (H&E) staining on cross-sections from injured carotid arteries of CatS+/+ mice treated with or without tubastatin A (Tuba). Scale bar: 100 μm. The intimal area and intima/media ratio of injured arteries were much lower in the tubastatin A–treated mice. Data are mean±SEM, Student unpaired t test. Representative images (B) of the bromodeoxyuridine (BrdU) staining on cross-sections from injured carotid arteries and quantitative data (D) of the BrdU labeling index. The BrdU labeling index was calculated by dividing the number of BrdU-labeled cells by the number of total cells in the neointima (n=5, each group). Triangles indicate BrdU-labeled cells. Tubastatin A reduced the plasma HDAC6 activity (E) as compared with control group (n=5, each group). Data are mean±SEM, Student unpaired t test. CatS indicates cathepsin S.
CatS Regulates HDAC6 Phosphorylation via the p38MAPK/Akt Signaling Pathway in Cultured VSMCs
We have observed a reduced expression of p-HDAC6 in the injured vessels of CatS−/− mice compared with those of CatS+/+ mice (Figure 2A and 2B). Here, we investigated how CatS regulates p-HDAC6 in cultured VSMCs. First, we observed that PDGF-BB increased the phosphorylation levels of HDAC6, p38MAPK, Akt, and extracellular signal-regulated protein kinases 1/2 (Erk1/2), with the peak at 10 to 30 minutes (Figure VIIIA and VIIIB in the online-only Data Supplement) in CatS+/+ VSMCs. Interestingly, the increased levels of p-HDAC6, p-p38MAPK, and p-Akt were blunted in CatS−/− VSMCs, with the exception of p-Erk (Figure 5A and 5B). And an increased HDAC6 activity in cell lysates of CatS+/+ VSMCs was also ablated by CatS−/− (Figure 5C). These results indicate that CatS regulates HDAC6 phosphorylation and activity in VSMCs. As shown in Figure 5D and 5E, CatS inhibitor reduced the PDGF-induced phosphorylation of HDAC6, p38MAPK, and Akt, but had no effect on that of p-Erk1/2. Likewise, the HDAC6 activity in cell lysates of CatS+/+ VSMCs was increased by PDGF-BB, and these changes were ablated by CatS inhibition (Figure 5F). We also found that p38MAPK and Akt inhibitor blocked the PDGF-induced phosphorylations of HDAC6 or eEF-2 (eukaryotic elongation factor-2; Figure VIIIC–VIIIE in the online-only Data Supplement), whereas Erk1/2 inhibitor alone had no effect (data not shown). These results indicate that CatS may regulate HDAC6 phosphorylation and activity through the p38MAPK/Akt signaling pathway in VSMCs. In addition, SMCs were transfected with a HDAC6 plasmid to dramatically increase HDAC6 expression, which resulted in enhanced levels of p-HDAC6 protein (Figure IX in the online-only Data Supplement).
Figure 5.
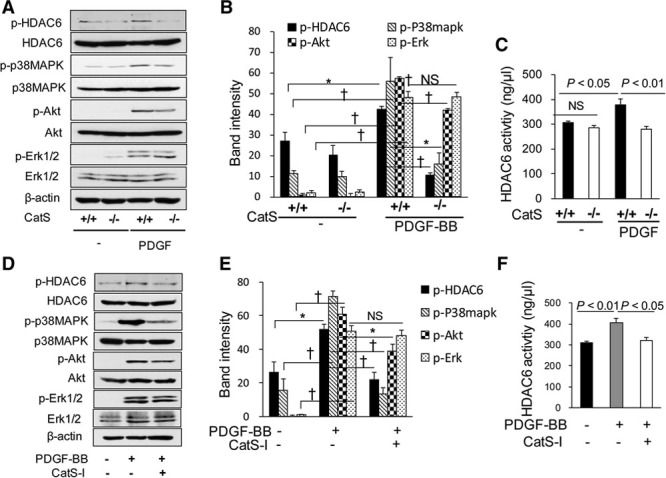
Cathepsin S (CatS) regulates histone deacetylase 6 (HDAC6) activity in vascular smooth muscle cells (VSMCs). Mouse aortic VSMCs were cultured in 10% FBS/DMEM medium and then subjected to serum-free medium for 12 h before the following treatment. Protein samples were isolated and used for a Western blotting analysis. Representative immunoblots (A) and quantitative data (B) show that the increased levels of p-HDAC6, p-p38MAPK, and p-Akt were blunted in CatS−/− VSMCs, with the exception of p-Erk. CatS−/− decreased HDAC6 activity in VSMCs in response to platelet-derived growth factor BB (PDGF-BB; C). Representative immunoblots (D) and quantitative data (E) show that the CatS inhibitor (CatS-I, 10 μM) reduced the PDGF-induced phosphorylation of HDAC6, p38MAPK, and Akt, but had no effect on that of p-Erk1/2. PDGF-BB–induced HDAC6 activity of CatS+/+ VSMCs was reduced by CatS inhibition (F). PDGF-BB, 20 ng/mL, for 10 minutes. Data are mean±SEM of 4 independent experiments, ANOVA and Bonferroni post hoc tests. ERK1/2 indicates extracellular signal-regulated protein kinases 1/2; p-HDAC6, phospho-histone deacetylase 6; and p-p38MAPK, phospho-p38 mitogen-activated protein kinase.
HDAC6 Plasmid Transfection Increased the In Vitro VSMC Proliferation, Migration, and Cell-Cycle Progression
To further determine the effect of HDAC6 on VSMC proliferation and migration, we enhanced the HDAC6 expression by transfecting HDAC6 plasmid into VSMCs in vitro. As anticipated, we observed increased proliferation and migration ability in VSMCs transfected with HDAC6 plasmid compared with those transfected with vector plasmids (Figure XA–XC in the online-only Data Supplement). HDAC6 transfection also enhanced the cell-cycle progression by increasing the proportion of cells in the S/G2/M stage (Figure XD and XE in the online-only Data Supplement).
Hypoxia Increased the HDAC6 Signaling and VSMC Proliferation and Migration In Vitro
Because we observed an increased expression of HIF-1α gene in injured arteries, which is a marker of hypoxia, we further investigated whether hypoxia plays a role in the CatS/p38MAPK/Akt/HDAC6 signaling pathway. As shown in Figure 6A and 6B, hypoxia increased the levels of PDGF-BB–induced p-HDAC6, p-p38MAPK, and p-Akt, indicating that hypoxic stress could enhance p38MAPK/Akt/HDAC6 signaling cascade activation in VSMCs. However, hypoxia had no effect on the levels of p-Erk1/2 protein (Figure 6C). Moreover, we found that hypoxia increased the proliferation and migration of VSMCs in response to PDGF-BB (Figure 6D–6F). In addition, CatS ablation diminished hypoxia-induced VSMC proliferation (0.48±0.6 versus 0.62±0.08; P<0.01) and migration (23.0±3.1 versus 47.2, cells; P<0.01).
Figure 6.
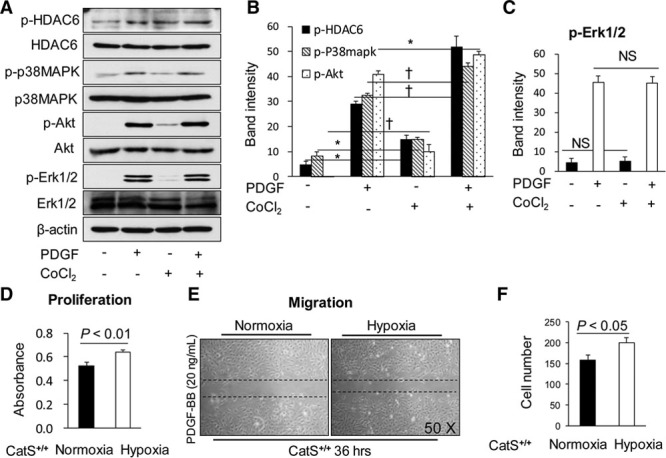
Hypoxia increased the histone deacetylase 6 (HDAC6) signaling and vascular smooth muscle cell (VSMC) proliferation and migration in vitro. Mouse VSMCs were cultured in 10% FBS/DMEM medium and then subjected to serum-free medium for 12 h. After starvation, cells were incubated with or without CoCl2 (600 μM) for 20 minutes and then stimulated with or without platelet-derived growth factor BB (PDGF-BB; 20 ng/mL) for 10 minutes. Protein samples were isolated and used for a Western blotting analysis as indicated. The representative image (A) and quantitative data (B) show that hypoxia increased the PDGF-BB–induced p-HDAC6, p-p38MAPK, and p-Akt levels, but had no effect on p-ERK1/2 signaling (C, n=3). Hypoxia increased VSMC proliferation (D, n=7). Representative images (E) and quantitative data (F) that hypoxia increased PDGF-BB–induced VSMC migration. Data are mean±SEM, Student unpaired t test, or ANOVA and Bonferroni post hoc tests. ERK1/2 indicates extracellular signal-regulated protein kinases 1/2; p-HDAC6, phospho-histone deacetylase 6; and p-p38MAPK, phospho-p38 mitogen-activated protein kinase.
TLR2 Was Required in HDAC6 Signaling and VSMC Events
To further investigate the interaction between TLR2 and HDAC6 signaling pathways, siTLR (short interfering RAN against toll-like receptor-2) was applied to use in the cellular experiments. Results indicated that TLR2 silencing mitigated HDAC6 as well as p38MAPK and Akt activations in cultured VSMCs in response to PDGF-BB (Figure XIA and XIB in the online-only Data Supplement). As anticipated, siTLR ameliorated cell proliferation and migration (Figure XIC–XIE in the online-only Data Supplement).
Discussion
Endovascular treatment–related maladaptive vascular remodeling represents the leading cause of restenosis and cardiovascular events.10 Identifying novel targets to suppress vascular negative remodeling will contribute to therapeutic strategies to preempt restenosis.10 The significant finding of our present study is that mice lacking the CatS gene are resistant to acute injury-induced intimal hyperplasia via the HDAC6-induced proliferation and migration of VSMCs. At the molecular level, CatS deletion retards injury-induced TLR2 gene and its downstream inflammatory genes and p38MAPK, Akt, and HDAC6 signaling activations. The pharmacological inhibition of HDAC6 activity also results in vascular protective actions via the reduction of VSMC proliferation. In vitro, the specific inhibition of CatS or HDAC6 attenuated VSMC migratory and proliferative abilities. We also observed that CatS regulates HDAC6 phosphorylation through the p38MAPK/Akt signaling pathway in cultured VSMCs. Moreover, TLR2 silencing mitigated the phosphorylations of HDAC6, p38MAPK, and Akt and VSMC functions. To the best of our knowledge, this is the first study to report that CatS controls injury-related vascular repair in mice via the TLR2-mediated p38MAPK/Akt-HDAC6 signaling pathway (Figure XII in the online-only Data Supplement).
Cathepsin cysteine proteases can degrade the basement membrane and surrounding extracellular matrix of artery walls.23 Cathepsin family members, such as CatS and CatK, have been demonstrated to play an important role in atherosclerosis-based proliferative diseases.10,24 Our research and previous studies by other groups have demonstrated increased expression and activity of CatS in balloon-injured arteries of rat12 or rabbit.4 In the present study, we also observed an increased expression of CatS in ligation-injured arteries of mice (Figure IA in the online-only Data Supplement). Interestingly, we observed that neointimal formation was reduced markedly in CatS−/− mice compared with wild-type mice. Intimal hyperplasia is believed to be the consequence of VSMC proliferation and migration from the media into the intima. In our present work, we observed that cell proliferative activity in the intima and media of injured arteries was decreased in the absence of CatS in vivo (Figure 1), and the pharmacological inhibition of CatS prevented the mitogen-induced VSMC proliferation and migration in vitro (Figure VI in the online-only Data Supplement). Moreover, the impaired VSMC migration from the arterial explants of CatS−/− reflects that the intrinsic ability of cells to migrate and degrade the local extracellular matrix proteins is controlled by CatS activity (Figure VII in the online-only Data Supplement). Collectively, these observations demonstrated that CatS controls injury-related vascular repair in mice by affecting VSMC migration and proliferation.
Recent evidence has shown that HDACs play a crucial role in the development of proliferative vascular diseases, including atherosclerosis and in-stent restenosis.13 Our present findings revealed that the level of phospho-HDAC6 protein was increased in injured arteries of CatS+/+ mice compared with noninjured arteries (Figure 2). The pharmacological inhibition of HDAC6 with tubastatin A markedly reduced the neointimal formation and neointimal VSMC proliferation in the ligation-injured arteries of CatS+/+ mice (Figure 4). Our in vitro study showed that tubastatin A reduced the mitogen-induced proliferation and migration of VSMCs and that the overexpression of HDAC6 exerted the opposite effect (Figure 3; Figure X in the online-only Data Supplement). These observations indicate that HDAC6 activity plays an important role in the process of intimal hyperplasia after vascular injury via the control of VSMC proliferation and migration. We further observed that the increased expression of p-HDAC6 protein in injured arteries was ablated in CatS−/− mice, and both the genetic ablation and pharmaceutical inhibition of CatS significantly suppressed the PDGF-induced HDAC6 phosphorylation and activities in cultured VSMCs (Figures 2 and 5). Thus, the present data suggest that CatS may control injury-related vascular repair via HDAC6-mediated VSMC migration and proliferation. It should be noted that the levels of p-HDAC6 but not the total HDAC6 expression changed in the injured lesions of CatS+/+ mice because neither HDAC6 mRNA nor protein changed from day 1 to day 4 after ligation in wild-type mice (Figure IIIB in the online-only Data Supplement), suggesting that HDAC6 activity plays the key role in vascular remodeling.
The engagement of TLRs on the cell surface by specific ligands leads to an increase in the expressions of proinflammatory cytokines and chemokines, such as MCP-1 (monocyte chemoattractant protein-1).10,22 Several lines of investigation demonstrated that TLR2 and TLR4 are required in the vascular SMC proliferation via MAPK activation.10,25 Previous studies reported that p38MAPK, Akt, and Erk1/2 signaling have critical roles in SMC proliferation and migration and in the process of neointimal formation.26 Our present observations show that CatS deletion reduced the levels of TLR2, p-p38MAPK, and p-Akt in the arterial lesions (Figure 2A, 2C, and 2D; Figure IIA and IIF in the online-only Data Supplement). Furthermore, both the genetic ablation and pharmaceutical inhibition of CatS significantly decreased the PDGF-BB–stimulated phosphorylation of p38MAPK and Akt but not that of Erk1/2 in cultured VSMCs (Figure 5A, 5B, 5D, and 5E), indicating that CatS controls vascular repair and that neointimal formation may act through p38MAPK and Akt signaling pathways. In addition, our present findings demonstrated that the inhibition of p38MAPK and Akt blocked the PDGF-induced phosphorylation of HDAC6 (Figure VIIIC–VIIIE in the online-only Data Supplement). HDAC6 inhibition has been shown to disrupt STAT3-related anti-inflammatory response in the antigen-presenting cells.27 A recent single study reported that PDGF regulated vascular SMC proliferation via STAT3 signaling pathway activation.28 Here, we have observed that TLR2 silencing mitigated VSMC proliferation and migration accompanied with the reduction of p-HDAC6, p-p38MAPK, and p-Akt levels (Figure XI in the online-only Data Supplement). Taken together, these results indicate that CatS controls injury-related vascular repair in mice via the TLR2-medated p38MAPK/Akt-HDAC6 signaling pathway. PDGF-BB has been known to be a key player in vascular remodeling and restenosis in response to mechanical stress. It should be noted that here the plasma PDGF-BB concentrations were lower in the CatS−/− mice than in the CatS+/+ mice at an early stage (day 4) after vascular injury, indicating that CatS deficiency–mediated vascular benefits may also be attributable, at least in part, to an attenuation of circulating PDGF-BB levels in a mouse carotid artery–injury model.
Recent studies have supported the existence of hypoxia in human and murine intima plaques, which augments plaque progression by stimulating plaque angiogenesis, altering glucose metabolism, promoting plaque proteolysis, inciting plaque inflammation, and increasing lipid accumulation in macrophage foam cells.29,30 In the present study, we observed that HIF-1α mRNA increased in the injured arteries, suggesting that ligation injury contributes to vascular local hypoxia (Figure IIG in the online-only Data Supplement). CatS deficiency had decreased levels of HIF-1α mRNA in injured arteries (Figure IIH in the online-only Data Supplement). The results of our in vitro study showed that hypoxia increased the levels of p-HDAC6, p-p38MAPK, and p-Akt in the PDGF-BB group (Figure 6A and 6B), indicating that hypoxia enhances the CatS/p38MAPK/Akt/HDAC6 signaling pathway in VSMCs. We also observed that hypoxia increased the proliferation and migration of VSMCs induced by PDGF-BB in vitro (Figure 6D–6F). These observations indicate that hypoxia may be involved in and may enhance the role of CatS in vascular repair. On the other hand, CatS ablation mitigates the levels of p-p38MAPK and p-HDAC6 (Figure 2C) as compared with the corresponding control, whereas it had increased levels of p-p38MAPK and p-HDAC6 proteins in cultured VSMCs in response to PDGF (Figure 5A and 5B). As known, it is too hard to completely reproduce in vivo conditions in one experiment. Although we have no direct evidence, this discrepancy might be because of the differences in the experimental conditions (in vivo and in vitro) and protein extractions (whole vessels or SMCs lysates).
In conclusion, our study demonstrated that CatS controls injury-related vascular repair via TLR2-mediated p38MAPK/Akt signaling activation and HDAC6-mediated VSMC migration and proliferation. Moreover, our present findings suggest that targeting CatS and HDAC6 represent an attractive therapeutic approach to treat vasculopathies that are involved in neointima formation and intima–media thickening.
Sources of Funding
This work was supported, in part, by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (no. 21590952) and The Scientific Research Fund of the Chinese Ministry of Education (no. 82160068 and 81560240).
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- CatS
- cathepsin S
- Erk1/2
- extracellular signal-regulated protein kinases 1/2
- HDAC6
- histone deacetylases 6
- HIF-1α
- hypoxia-inducible factor-1α
- PDGF-BB
- platelet-derived growth factor BB
- p-p38MAPK
- phospho-p38 mitogen-activated protein kinase
- TLR2
- toll-like receptor 2
- VSMC
- vascular smooth muscle cell
These authors contributed equally to this article.
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.115.307110/-/DC1.
Highlights
Cathepsin S gene and protein levels increased in mouse carotid vasculature to injuries.
Cathepsin S deficiency attenuated injury-related inflammatory actions and medial smooth muscle cellular events (including proliferation and migration) and mitigated vascular remodeling.
There is cross-interaction between toll-like receptor 2–mediated cathepsin S and histone deacetylase 6 signaling pathways during injury-related vascular repair.
Targeting the cross talk between cathepsin S and histone deacetylase 6 could provide a potential therapeutic route for the clinical treatment of proliferative vascular diseases that are involved in vascular remodeling and neointima hyperplasia.
References
- 1.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 2.Duran-Prado M, Morell M, Delgado-Maroto V, Castaño JP, Aneiros-Fernandez J, de Lecea L, Culler MD, Hernandez-Cortes P, O’Valle F, Delgado M. Cortistatin inhibits migration and proliferation of human vascular smooth muscle cells and decreases neointimal formation on carotid artery ligation. Circ Res. 2013;112:1444–1455. doi: 10.1161/CIRCRESAHA.112.300695. doi: 10.1161/CIRCRESAHA.112.300695. [DOI] [PubMed] [Google Scholar]
- 3.Lin SJ, Yen HT, Chen YH, Ku HH, Lin FY, Chen YL. Expression of interleukin-1 beta and interleukin-1 receptor antagonist in oxLDL-treated human aortic smooth muscle cells and in the neointima of cholesterol-fed endothelia-denuded rabbits. J Cell Biochem. 2003;88:836–847. doi: 10.1002/jcb.10431. doi: 10.1002/jcb.10431. [DOI] [PubMed] [Google Scholar]
- 4.Burns-Kurtis CL, Olzinski AR, Needle S, Fox JH, Capper EA, Kelly FM, McQueney MS, Romanic AM. Cathepsin S expression is up-regulated following balloon angioplasty in the hypercholesterolemic rabbit. Cardiovasc Res. 2004;62:610–620. doi: 10.1016/j.cardiores.2004.02.002. doi: 10.1016/j.cardiores.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Turk V, Turk B, Guncar G, Turk D, Kos J. Lysosomal cathepsins: structure, role in antigen processing and presentation, and cancer. Adv Enzyme Regul. 2002;42:285–303. doi: 10.1016/s0065-2571(01)00034-6. [DOI] [PubMed] [Google Scholar]
- 6.Reiser J, Adair B, Reinheckel T. Specialized roles for cysteine cathepsins in health and disease. J Clin Invest. 2010;120:3421–3431. doi: 10.1172/JCI42918. doi: 10.1172/JCI42918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng XW, Shi GP, Kuzuya M, Sasaki T, Okumura K, Murohara T. Role for cysteine protease cathepsins in heart disease: focus on biology and mechanisms with clinical implication. Circulation. 2012;125:1551–1562. doi: 10.1161/CIRCULATIONAHA.111.066712. doi: 10.1161/CIRCULATIONAHA.111.066712. [DOI] [PubMed] [Google Scholar]
- 8.Yang M, Zhang Y, Pan J, Sun J, Liu J, Libby P, Sukhova GK, Doria A, Katunuma N, Peroni OD, Guerre-Millo M, Kahn BB, Clement K, Shi GP. Cathepsin L activity controls adipogenesis and glucose tolerance. Nat Cell Biol. 2007;9:970–977. doi: 10.1038/ncb1623. doi: 10.1038/ncb1623. [DOI] [PubMed] [Google Scholar]
- 9.Jiang H, Cheng XW, Shi GP, et al. Cathepsin K-mediated Notch1 activation contributes to neovascularization in response to hypoxia. Nat Commun. 2014;5:3838. doi: 10.1038/ncomms4838. doi: 10.1038/ncomms4838. [DOI] [PubMed] [Google Scholar]
- 10.Hu L, Cheng XW, Song H, Inoue A, Jiang H, Li X, Shi GP, Kozawa E, Okumura K, Kuzuya M. Cathepsin K activity controls injury-related vascular repair in mice. Hypertension. 2014;63:607–615. doi: 10.1161/HYPERTENSIONAHA.113.02141. doi: 10.1161/HYPERTENSIONAHA.113.02141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest. 1998;102:576–583. doi: 10.1172/JCI181. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng XW, Kuzuya M, Sasaki T, Arakawa K, Kanda S, Sumi D, Koike T, Maeda K, Tamaya-Mori N, Shi GP, Saito N, Iguchi A. Increased expression of elastolytic cysteine proteases, cathepsins S and K, in the neointima of balloon-injured rat carotid arteries. Am J Pathol. 2004;164:243–251. doi: 10.1016/S0002-9440(10)63114-8. doi: 10.1016/S0002-9440(10)63114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Findeisen HM, Gizard F, Zhao Y, Qing H, Heywood EB, Jones KL, Cohn D, Bruemmer D. Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb Vasc Biol. 2011;31:851–860. doi: 10.1161/ATVBAHA.110.221952. doi: 10.1161/ATVBAHA.110.221952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou B, Margariti A, Zeng L, Xu Q. Role of histone deacetylases in vascular cell homeostasis and arteriosclerosis. Cardiovasc Res. 2011;90:413–420. doi: 10.1093/cvr/cvr003. doi: 10.1093/cvr/cvr003. [DOI] [PubMed] [Google Scholar]
- 15.Natarajan R. Drugs targeting epigenetic histone acetylation in vascular smooth muscle cells for restenosis and atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:725–727. doi: 10.1161/ATVBAHA.111.222976. doi: 10.1161/ATVBAHA.111.222976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan ZQ, Yao QP, Zhang ML, Qi YX, Guo ZY, Shen BR, Jiang ZL. Histone deacetylases modulate vascular smooth muscle cell migration induced by cyclic mechanical strain. J Biomech. 2009;42:945–948. doi: 10.1016/j.jbiomech.2009.01.012. doi: 10.1016/j.jbiomech.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 17.Usui T, Morita T, Okada M, Yamawaki H. Histone deacetylase 4 controls neointimal hyperplasia via stimulating proliferation and migration of vascular smooth muscle cells. Hypertension. 2014;63:397–403. doi: 10.1161/HYPERTENSIONAHA.113.01843. doi: 10.1161/HYPERTENSIONAHA.113.01843. [DOI] [PubMed] [Google Scholar]
- 18.Kaliszczak M, Trousil S, Åberg O, Perumal M, Nguyen QD, Aboagye EO. A novel small molecule hydroxamate preferentially inhibits HDAC6 activity and tumour growth. Br J Cancer. 2013;108:342–350. doi: 10.1038/bjc.2012.576. doi: 10.1038/bjc.2012.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valenzuela-Fernández A, Cabrero JR, Serrador JM, Sánchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18:291–297. doi: 10.1016/j.tcb.2008.04.003. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Cheng XW, Hu L, et al. Cathepsin S activity controls ischemia-induced neovascularization in mice. Int J Cardiol. 2015;183:198–208. doi: 10.1016/j.ijcard.2015.01.058. doi: 10.1016/j.ijcard.2015.01.058. [DOI] [PubMed] [Google Scholar]
- 21.Adams-Cioaba MA, Krupa JC, Xu C, Mort JS, Min J. Structural basis for the recognition and cleavage of histone H3 by cathepsin L. Nat Commun. 2011;2:197. doi: 10.1038/ncomms1204. doi: 10.1038/ncomms1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng XW, Song H, Sasaki T, Hu L, Inoue A, Bando YK, Shi GP, Kuzuya M, Okumura K, Murohara T. Angiotensin type 1 receptor blocker reduces intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Hypertension. 2011;57:981–989. doi: 10.1161/HYPERTENSIONAHA.110.168385. doi: 10.1161/HYPERTENSIONAHA.110.168385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng XW, Huang Z, Kuzuya M, Okumura K, Murohara T. Cysteine protease cathepsins in atherosclerosis-based vascular disease and its complications. Hypertension. 2011;58:978–986. doi: 10.1161/HYPERTENSIONAHA.111.180935. doi: 10.1161/HYPERTENSIONAHA.111.180935. [DOI] [PubMed] [Google Scholar]
- 24.Wu H, Cheng XW, Hu L, Hao CN, Hayashi M, Takeshita K, Hamrah MS, Shi GP, Kuzuya M, Murohara T. Renin inhibition reduces atherosclerotic plaque neovessel formation and regresses advanced atherosclerotic plaques. Atherosclerosis. 2014;237:739–747. doi: 10.1016/j.atherosclerosis.2014.10.098. doi: 10.1016/j.atherosclerosis.2014.10.098. [DOI] [PubMed] [Google Scholar]
- 25.Sasu S, LaVerda D, Qureshi N, Golenbock DT, Beasley D. Chlamydia pneumoniae and chlamydial heat shock protein 60 stimulate proliferation of human vascular smooth muscle cells via toll-like receptor 4 and p44/p42 mitogen-activated protein kinase activation. Circ Res. 2001;89:244–250. doi: 10.1161/hh1501.094184. [DOI] [PubMed] [Google Scholar]
- 26.Muto A, Fitzgerald TN, Pimiento JM, Maloney SP, Teso D, Paszkowiak JJ, Westvik TS, Kudo FA, Nishibe T, Dardik A. Smooth muscle cell signal transduction: implications of vascular biology for vascular surgeons. J Vasc Surg. 2007;45 Suppl A:A15–A24. doi: 10.1016/j.jvs.2007.02.06. doi: 10.1016/j.jvs.2007.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng F, Lienlaf M, Wang HW, et al. A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J Immunol. 2014;193:2850–2862. doi: 10.4049/jimmunol.1302778. doi: 10.4049/jimmunol.1302778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Q, Wang W, Li S, Nagarkatti P, Nagarkatti M, Windust A, Wang XL, Tang D, Cui T. American ginseng inhibits vascular smooth muscle cell proliferation via suppressing Jak/Stat pathway. J Ethnopharmacol. 2012;144:782–785. doi: 10.1016/j.jep.2012.09.046. doi: 10.1016/j.jep.2012.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parathath S, Mick SL, Feig JE, Joaquin V, Grauer L, Habiel DM, Gassmann M, Gardner LB, Fisher EA. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ Res. 2011;109:1141–1152. doi: 10.1161/CIRCRESAHA.111.246363. doi: 10.1161/CIRCRESAHA.111.246363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Libby P, Folco E. Tension in the plaque: hypoxia modulates metabolism in atheroma. Circ Res. 2011;109:1100–1102. doi: 10.1161/RES.0b013e31823bdb84. doi: 10.1161/RES.0b013e31823bdb84. [DOI] [PubMed] [Google Scholar]