

Abstract
All animals rely on their ability to sense and respond to their environment to survive. However, the suitability of a behavioral response is context-dependent, and must reflect both an animal’s life history and its present internal state. Based on the integration of these variables, an animal’s needs can be prioritized to optimize survival strategies. Nociceptive sensory systems detect harmful stimuli and allow for the initiation of protective behavioral responses. The polymodal ASH sensory neurons are the primary nociceptors in C. elegans. We show here that the guanylyl cyclase ODR-1 functions non-cell-autonomously to downregulate ASH-mediated aversive behaviors and that ectopic cGMP generation in ASH is sufficient to dampen ASH sensitivity. We define a gap junction neural network that regulates nociception and propose that decentralized regulation of ASH signaling can allow for rapid correlation between an animal’s internal state and its behavioral output, lending modulatory flexibility to this hard-wired nociceptive neural circuit.
Author Summary
Studying the logic of small neural circuits is an essential step toward understanding more complex circuits and, ultimately, the computational and integrative properties of whole nervous systems. With a compact nervous system (just 302 neurons) and a well-characterized behavioral repertoire, the small roundworm C. elegans serves as an excellent animal model to study circuit-level modulation of neuronal function. By employing a combination of genetic and behavioral approaches, we have identified a non-cell-autonomous role for the guanylyl cyclase ODR-1 in the regulation of nociceptive sensory behaviors, and we provide the first evidence for circuit-level modulation of neuronal activity by the second messenger cGMP. While revealing a new mechanism for the coordination and optimization of animal behavior, our work also supports a growing body of evidence that hard-wired neuronal circuitry can be dynamically repurposed in a context-dependent manner.
Introduction
Chemical stimuli, including odorants and tastants, can provide information about food availability and quality to affect appetitive behaviors across species. While all sensory circuits use specialized cells in the periphery to detect environmental stimuli, how the sensitivity of sensory neurons is tuned and how chemical information is processed and relayed through downstream interneurons remains largely unknown. As ultrastructural analyses of simple brains and small brain regions are providing connectome data [1–7], the challenge that lies ahead is in understanding the dynamic properties of circuitry usage. While mapped physical connections show the potential for information flow, the breadth of possibilities must also be reconciled with a circuit’s potential for neuromodulation as animals interact with a complex and changing environment. For example, studies in systems ranging from invertebrates to mammals have revealed that chemosensory responses and feeding behaviors are modulated by an animal’s nutritional state [8–16]. Furthermore, there is incredible complexity in the mechanisms by which nutritional status ultimately modulates chemosensory and feeding behaviors, reflecting the nervous system’s need to integrate information about what an animal has eaten, how much and when. For example, neuropeptides, neurotransmitters (e.g. serotonin and dopamine), hormones (e.g. insulin) and metabolites have all been shown to modulate chemosensory responses [8,12,15,17–21].
Chemical synapses allow neurons to communicate with each other through the vesicular release of neurotransmitters into synaptic clefts between the cells. These molecules then bind to receptors on the postsynaptic neurons. In contrast, gap junctions allow for direct cytoplasmic communication and electrical coupling between neurons. As such, gap junctions are often referred to as electrical synapses. Importantly, the presence of gap junctions in the nervous system allows for the establishment of even more complex circuits than can be generated by synaptic signaling alone.
Vertebrate gap junctions are formed through the association of transmembrane connexin proteins. Within one cell, six connexin proteins assemble to form one connexon hemichannel. Two hemichannels on neighboring cells then dock which each other, allowing homotypic (consisting of a single protein species on both cell membranes), heterotypic (consisting of two different homomeric connexons on the two cell membranes) or heteromeric (consisting of a mixture of protein species on both cell membranes) gap junctions to be made [22,23]. Gap junction communication allows for the transmission of action potentials [24,25], diffusion of metabolites and nutrients [26] and diffusion of second messengers, including Ca2+, IP3 and the cyclic nucleotides cAMP and cGMP [27–32]. The ability to pass such a diversity of molecules affords them the potential to repurpose hardwired circuitry to modify responses to diverse environmental stimuli.
The gap junction protein family consists of vertebrate connexins and invertebrate innexins (invertebrate analogues of the connexins) [33,34]. The C. elegans genome encodes 25 members of this protein family [35]. Although the functions of many C. elegans innexins remain unknown, the characterized cases have shown that these gap junction components are involved in diverse processes ranging from embryonic development and cell fate determination to adult neural functions [35–37].
C. elegans is an ideal model system in which to study the link between hardwired connectivity and the functional circuitry usage that drives animal behavior. The serial electron micrographs that showed the anatomical positioning of each of the 302 C. elegans hermaphrodite neurons also revealed all of the morphologically identifiable connections between neurons and between neurons and muscles [2,38–41]. The recently updated C. elegans wiring diagram shows a total of 6393 chemical synapses, 890 gap junctions and 1410 neuromuscular junctions [42]. It is through this interconnected neural network that C. elegans exploits a highly developed chemosensory system to detect olfactory and gustatory cues associated with food, danger and mating [43–45]. In general, animals move towards chemicals that indicate a favorable environment, such as a potential food source, and away from stimuli that suggest a harmful environment.
The 11 pairs of C. elegans head chemosensory neurons extend their ciliated ends to the tip of the animal’s nose, allowing for direct or indirect exposure to sensory stimuli in their environment [2,39,40]. Cellular laser ablation studies have been used to reveal the function of individual neuron pairs [43]. For example, the ASEs sense water-soluble attractants, while the AWA and AWC neurons detect volatile odorants that C. elegans are attracted to and chemotax towards. In addition, while the ASJ, ASI, ADF and ASG sensory neurons are primarily involved in regulating dauer formation, they do also play a role in other processes, including a minor role in chemotaxis. Conversely, the ASH, ADL, AWB and ASK chemosensory neurons detect aversive stimuli that animals avoid by initiating backward locomotion upon stimulus detection.
The two bilaterally symmetric ASH nociceptors are particularly important for the avoidance of noxious stimuli, as they are “polymodal.” This neuron pair responds to a broad range of aversive stimuli, including not only soluble chemicals (e.g. the bitter tastant quinine, heavy metals and SDS) and odorants (e.g. octanol), but also ions (e.g. Na+), osmotic stress and mechanosensory stimulation (nose touch) [46–54]. ASH activation elicits reversal and stimulus avoidance because these glutamatergic sensory neurons synapse onto command interneurons that drive backward locomotion via their connections with motor neurons. Thus, the nociceptive sensory system of C. elegans bears resemblance to its mammalian counterparts, in which noxious stimuli (including chemicals) are sensed predominantly by peripheral glutamatergic sensory neurons that synapse onto spinal dorsal horn neurons and, following further sensory processing in multiple regions of the brain, generate the perception of pain and aversive behavior [55–57].
We previously reported a role for the cGMP-dependent protein kinase EGL-4 in the modulation of ASH-evoked nociceptive behavioral responses [58]. C. elegans lacking EGL-4 function are hypersensitive in their response to a subset of ASH-detected stimuli; egl-4(lof) animals avoid dilute stimuli that wild-type animals do not respond to. Our data suggested that EGL-4 likely normally acts to dampen ASH sensitivity by phosphorylating and activating the GTPase activating proteins RGS-2 and RGS-3, which then downregulate G protein-coupled sensory signaling in the ASH nociceptors. Surprisingly, although EGL-4 requires cGMP binding to negatively regulate ASH sensitivity, no guanylyl cyclases are known to be expressed in ASH [59].
Herein we provide evidence that the C. elegans transmembrane guanylyl cyclase ODR-1 functions in a non-cell-autonomous manner to provide cGMP to regulate EGL-4 function in ASH. Like egl-4(lof) animals, odr-1(lof) animals are hypersensitive in their avoidance of a subset of ASH-detected stimuli. However, while EGL-4 functions directly in the ASHs, ODR-1 expression in the AWB, AWC and ASI head sensory neurons is sufficient to restore normal behavioral sensitivity to odr-1(lof) animals. We further provide evidence that the pool of cGMP produced by ODR-1 flows through a gap junction network from its site of production to the ASH nociceptors. Taken together, our data reveal a new way by which an animal’s nervous system can utilize information about the organism’s well-being to set the threshold of nociceptor sensitivity and coordinate behavioral responses that are appropriate for its internal state.
Results
ODR-1 Functions in Multiple Sensory Neurons to Dampen Quinine Sensitivity
We previously reported that animals lacking the function of the guanylyl cyclase ODR-1 are hypersensitive in their behavioral avoidance response to dilute concentrations of the bitter tastant quinine [58]. Significantly more odr-1 loss-of-function (lof) animals respond to dilute (1 mM) quinine than wild-type animals (Fig 1) [58]. The ASH sensory neurons are the main cells used to detect quinine in C. elegans, but the ASK neurons also contribute [52]. ODR-1 is not expressed in ASH, but is expressed in five other head sensory neurons—AWB, AWC, ASI, ASJ and ASK [60,61]. To determine in which cell(s) ODR-1 function is sufficient to dampen quinine sensitivity, we restored ODR-1 function in each neuron pair that it is natively expressed in, using the following cell-specific or -selective promoters: str-1p (AWB), ceh-36p3 (AWC), gpa-4p (ASI), trx-1p (ASJ) and srbc-66p (ASK) [62–66]. While expressing ODR-1 in ASJ or ASK had only a minimal effect on quinine sensitivity, individually expressing ODR-1 in either the AWB, AWC or ASI sensory neurons partially rescued the odr-1(lof) quinine hypersensitivity phenotype (Fig 1A). This suggested that ODR-1 function in more than one neuron pair regulates the quinine response. Co-injection of str-1p::odr-1, ceh36p3::odr-1 and gpa-4p::odr-1 to simultaneously express ODR-1 in the AWB, AWC and ASI sensory neurons of odr-1(lof) animals returned quinine response to the level seen when odr-1 was expressed under the control of its own promoter (odr-1p::odr-1), consistent with ODR-1 functioning in multiple neurons to regulate quinine sensitivity (Fig 1).
Fig 1. ODR-1 functions in adult sensory neurons.

(A) ODR-1 expression in the AWB, AWC and ASI sensory neurons is sufficient to rescue the behavioral hypersensitivity of odr-1(lof) animals. The str-1 [66], ceh-36p3 [65], gpa-4 [64], trx-1 [63] and srbc-66 [62] promoters were used to drive expression of wild-type odr-1 (genomic sequence) in odr-1(lof) animals. These promoters drive expression in the following cells: str-1 (AWB), ceh-36p3 (AWC), gpa-4 (ASI), trx-1 (ASJ and intestinal cells), srbc-66 (ASK). While more odr-1(lof) animal respond to 1 mM quinine than do wild-type animals, co-expressing odr-1 in the AWB, AWC and ASI sensory neurons returned response to wild-type levels (p > 0.1). (B) ODR-1 functions in adult animals to regulate behavioral sensitivity. Adult odr-1(lof) animals expressing odr-1 (genomic sequence) under the control of a heat shock inducible promoter (hsp) [68] were tested without heat shock (white bars) or 4 hours after heat shock treatment (grey bars). While odr-1(lof) animals have a hypersensitive response to dilute (1 mM) quinine, heat shock induced expression of odr-1 in adult odr-1(lof) animals abolished this hypersensitivity and returned quinine response to the degree seen in wild-type animals (p > 0.1 when comparing odr-1(lof) animals with heat shock treatment to wild-type animals either with or without heat shock). (C) The extracellular domain of ODR-1 is not required for regulation of quinine sensitivity. In odr-1(lof) animals, the odr-1 promoter was used to drive expression of either wild-type ODR-1 (genomic sequence), ODR-1 lacking its extracellular domain (ΔECD) or ODR-1 with a point mutation (E874A) that abolishes GTP binding in the catalytic domain. ODR-1(ΔECD) rescued the hypersensitivity of odr-1(lof) animals as well as wild-type ODR-1 (p > 0.1). Expression of ODR-1(E874A) had no effect on response sensitivity (p > 0.5 when compared to odr-1(lof) animals). The percentage of animals responding is shown. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Allele used: odr-1(n1936) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function. n.s. = not significant.
ODR-1 Function in Adult Animals Is Sufficient to Regulate Behavioral Sensitivity to Quinine
odr-1(lof) animals develop with altered membraneous structures at the distal segments of the AWB, but not ASI, dendritic cilia [67]; AWC cilia structure was not examined in this study. To assess when ODR-1 function is required to downregulate quinine sensitivity, the odr-1 gene was placed under the control of a heat shock inducible promoter [68] and introduced into odr-1(lof) animals. Induction of odr-1 expression by heat shock in adult animal stages returned the behavioral response to dilute quinine to wild-type levels when assayed four hours later (Fig 1B). Transgenic animals that were not heat shocked remained hypersensitive, similar to odr-1(lof) animals (Fig 1B). These results demonstrate that, even though cilia morphogenesis is likely an active process that continues through the late larval stages [67], odr-1 is only required in adult stages for normal behavioral sensitivity to dilute quinine. This heat shock-induced expression of odr-1 is also after developmental cell fate specification and neuronal connectivity is complete.
The Extracellular Domain of ODR-1 Is Not Required for Regulation of Quinine Sensitivity
ODR-1 is a receptor-type guanylyl cyclase with an extracellular domain (ECD) and an intracellular catalytic domain that processes GTP into cGMP [61]. To assess the contribution of the ECD to ODR-1 function in the regulation of quinine avoidance behavioral sensitivity, we expressed an ODR-1 construct lacking the extracellular domain (ΔECD) [61] under the control of its native promoter in odr-1(lof) animals (Fig 1C). Expression of the odr-1p::odr-1(ΔECD) construct restored quinine sensitivity to wild-type levels, similar to the expression of the wild-type odr-1 construct (odr-1p::odr-1) (Fig 1C). This suggests that the extracellular receptor region is not necessary for ODR-1 function in modulating quinine sensitivity.
To determine whether ODR-1’s ability to produce cGMP is necessary for regulation of quinine sensitivity, we expressed ODR-1 harboring a point mutation that abolishes GTP binding in the catalytic domain [61]. odr-1(lof) animals expressing odr-1p::odr-1(E874A) remained hypersensitive in their response to quinine (Fig 1C). This indicates an important role for cGMP in modulation of the quinine response and is consistent with our previous demonstration that the C. elegans cGMP-dependent protein kinase EGL-4 also regulates quinine response sensitivity [58].
The AWB, AWC and ASI Sensory Neurons Dampen Quinine Sensitivity
The above results suggested a modulatory role for the AWB, AWC and ASI neurons in quinine behavioral sensitivity. To further examine their contribution to the regulation of the quinine response, we genetically ablated these cells in wild-type animals, both as individual neuron pairs and in combination, using a reconstituted caspase approach [69,70]. Ablation of either the AWBs, AWCs or ASIs did not produce a marked hypersensitive phenotype (Fig 2A). While animals lacking two of the three neuron pairs displayed greater hypersensitivity than the single ablates, ablation of all three neuron pairs together resulted in the greatest degree of quinine hypersensitivity (Fig 2A). Taken together, our results reveal a role for the AWB, AWC and ASI sensory neurons in the negative regulation of quinine avoidance, and further suggest that ODR-1 function in these cells contributes to their modulatory role.
Fig 2. Neurotransmitter release from the AWB, AWC and ASI sensory neurons is not required to modulate quinine sensitivity.

(A) Genetic ablation of the AWB, AWC and ASI sensory neurons resulted in quinine hypersensitivity. The reconstituted caspase approach [69] was used to genetically ablate AWB, AWC [70] and ASI [70] either individually or in combination in otherwise wild-type animals. In each case, some hypersensitivity was observed (p < 0.05 for each when compared to wild-type). When all three neuron pairs were ablated simultaneously, the largest degree of behavioral hypersensitivity to dilute (1 mM) quinine was observed (p < 0.0001). (B) UNC-13-dependent synaptic signaling from AWB, AWC and ASI does not modulate quinine sensitivity. The str-1 (AWB) [66], ceh-36p3 (AWC) [65] and gpa-4 (ASI) [64] promoters were used to co-express a non-coding fragment of unc-13 in both the sense and antisense orientations of otherwise wild-type animals. RNAi knock-down of unc-13 in the AWB, AWC or ASI neurons, either individually or in combination, did not result in behavioral hypersensitivity to dilute (1 mM) quinine (p > 0.05 when compared to WT animals for all transgenes). The percentage of animals responding is shown. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Alleles used: odr-1(n1936) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function. n.s. = not significant. See also S1 Fig.
Neurotransmitter Release from AWB, AWC and ASI Is Not Required for the Modulation of Quinine Sensitivity
As the ASH nociceptors are the primary cells used to detect quinine [52], we sought to determine how the AWB, AWC and ASI sensory neurons might influence signaling through the ASH sensory circuit. One possibility could be via synaptic signaling between these neurons. UNC-13 protein is required for synaptic vesicle fusion and neurotransmitter release at synapses [71,72], and AWB and ASI are known to form direct synaptic connections onto ASH [2]. Because unc-13 null mutations are lethal, we used cell-specific RNAi [73] to knockdown unc-13 in AWB, AWC and ASI and block synaptic transmission from these neurons. We confirmed efficient unc-13 RNAi knockdown using chemosensory assays that require synaptic signaling from AWB and AWC for proper behavioral responses (S1 Fig). Animals in which unc-13 was simultaneously knocked down in the AWB, AWC and ASI sensory neurons did not show increased sensitivity to dilute quinine (Fig 2B), suggesting that vesicular synaptic transduction is not the mechanism by which these neurons influence the ASH-mediated response to quinine.
The INX-4 Innexin Gap Junction Component Is Required for Regulation of Quinine Sensitivity
A second way in which neurons can communicate with each other is via diffusion of ions and small metabolites through gap junctions. Studies in mammalian systems have suggested that cyclic nucleotides can also pass through gap junctions to affect cellular function. For example, cAMP movement between cells has been visualized following ectopic expression of gap junction components in human tissue culture [29,74,75]. cAMP movement through gap junctions also suppresses CD4+ T-cell function [76] and may alter gene expression in myelinating Schwann cells [77]. In addition, cGMP can pass through gap junctions in human cell culture, as well as between cultured mouse follicle cells and oocytes [29,30,32,78]. While 25 innexins are encoded by the C. elegans genome, the physiological roles of most are unknown [35–37]. To determine whether gap junction signaling can modulate quinine sensitivity, we assayed animals with loss-of-function alleles for 16 of the 25 innexins encoded by the C. elegans genome for response to dilute (1 mM) quinine (see Supplemental Materials and Methods). Two innexin mutants, inx-4(lof) and inx-20(lof), responded better than wild-type animals to dilute quinine (Fig 3A). While inx-20 expression has only been reported in the pharyngeal epithelium and the pm2 pharyngeal muscle cell, inx-4 expression was seen in several head and tail neurons, including the ASHs of early larvae and the ADFs of L1s [35]. Using an inx-4p::gfp reporter construct, we have also confirmed inx-4 expression in the ASH nociceptors of adult animals (S2 Fig).
Fig 3. Gap junctions regulate quinine sensitivity.

(A) Loss-of-function mutations in the innexin gap junction genes inx-4 and inx-20 resulted in behavioral hypersensitivity to 1 mM quinine (p < 0.001 for each when compared to wild-type animals). (B) The inx-4 [35], osm-10 (ASH, ASI, PHA and PHB) [48], srd-10 (ASH, PHA and PHB), srh-142 (ADF) [79] and odr-1 promoters were used to drive expression of wild-type inx-4 cDNA in inx-4(lof) animals. ASH-selective expression significantly dampened the inx-4(lof) hypersensitive response (p < 0.0001), while expression in the ODR-1-expressing neurons had no effect (p > 0.4). (C) INX-4 functions in adult animals to regulate behavioral sensitivity. Adult inx-4(lof) animals expressing inx-4 cDNA under the control of a heat shock inducible promoter (hsp) [68] were tested without heat shock (white bars) or 4 hours after heat shock treatment (grey bars). While inx-4(lof) animals had a hypersensitive response to dilute (1 mM) quinine, heat shock induced expression of inx-4 in adult inx-4(lof) animals dampened this hypersensitivity (p < 0.0001 when comparing inx-4(lof);hsp::inx-4 animals with heat shock treatment to inx-4(lof) animals either with or without heat shock). (D) INX-4 gap junctions are required for ODR-1 modulation of quinine sensitivity. Expression of only odr-1 or inx-4 in odr-1(lof);inx-4(lof) animals had no effect on quinine hypersensitivity (p > 0.2 when comparing rescue of either to odr-1(lof);inx-4(lof) animals). Simultaneous expression of inx-4 in ASH using the osm-10p promoter [48] and expression of odr-1 either behind its native promoter or using AWB, AWC and ASI cell-selective promoters (str-1 [66], ceh-36p3 [65], and gpa-4 [64], respectively), rescued hypersensitivity (p < 0.0001). The percentage of animals responding is shown. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Alleles used: inx-4(ok2373) loss-of-function, inx-20(ok426) loss-of-function and odr-1(n1936) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function. n.s. = not significant. See also S2 and S3 Figs.
To determine whether INX-4 function in the ASHs is sufficient to regulate quinine response, the ASH cell-selective promoters osm-10 [48] and srd-10 were used to restore INX-4 function and animals were assayed for response to 1 mM quinine. Expression in ASH using either promoter dampened the inx-4(lof) hypersensitive response, while simultaneous expression of INX-4 in ASH and ADF, using the osm-10 [48] and srh-142 [79] promoters, respectively, did not result in additional rescue (Fig 3B). Furthermore, consistent with the reported lack of inx-4 expression in ODR-1-expressing neurons [35], an odr-1p::inx-4 construct did not rescue the quinine hypersensitivity of inx-4(lof) animals (Fig 3B). These results suggest that the ASH nociceptors are the primary site for INX-4 function in regulating quinine sensitivity.
Since inx-4 is expressed from larval through adult stages, the inx-4 gene was placed under the control of a heat shock inducible promoter [68] and introduced into inx-4(lof) animals to determine when inx-4 function is required to modulate quinine sensitivity. Induction of inx-4 expression by heat shock in adult animal stages significantly dampened the behavioral hypersensitivity of inx-4(lof) animals to dilute quinine when assayed four hours later (Fig 3C). Transgenic animals that were not heat shocked remained hypersensitive, similar to inx-4(lof) animals (Fig 3C). These results demonstrate that, like ODR-1 (Fig 1B), INX-4 function in adult animal stages is sufficient to modulate behavioral sensitivity to dilute quinine.
ODR-1 and INX-4 Do Not Regulate ASH Sensitivity in General
We previously found that egl-4(lof) animals are hypersensitive in their response to distinct ASH-detected stimuli and, although EGL-4 functions in the ASH sensory neurons to regulate these behaviors, this cGMP-dependent protein kinase does not regulate ASH sensitivity in general [58]. For example, egl-4(lof) animals respond normally to the bitter tastant primaquine, the detergent SDS and the heavy metal copper [58], all of which are detected by the ASH nociceptors [49,51–53]. To determine whether loss of odr-1 or inx-4 increased overall sensitivity of ASH, or also selectively affected sensory signaling, we assayed odr-1(lof) and inx-4(lof) animals for their response to primaquine, SDS and copper. For each of these stimuli, odr-1(lof) and inx-4(lof) animals responded similarly to wild-type animals (S3 Fig), indicating that ODR-1 and INX-4 also do not regulate ASH signaling in general.
INX-4 Gap Junctions Are Required for ODR-1-Mediated Downregulation of Quinine Sensitivity
If INX-4 functions in the same pathway as ODR-1 to dampen quinine sensitivity, then odr-1(lof);inx-4(lof) double mutant animals should display a behavioral phenotype similar to odr-1(lof) animals and the hypersensitivity should not be additive. We found that the double mutant animals’ response to dilute (1 mM) quinine was indistinguishable from animals lacking only ODR-1 function (p > 0.2, Fig 3D), suggesting that they do function in the same regulatory pathway. Consistent with this observation, in the odr-1(lof);inx-4(lof) background, expression of only either odr-1 or inx-4 had no effect on the quinine hypersensitivity (Fig 3D). However, simultaneous expression of inx-4 in ASH and expression of odr-1 either behind its native promoter or using AWB, AWC and ASI cell-selective promoters, rescued hypersensitivity (Fig 3D).
cGMP Generation in ASH Is Sufficient to Dampen Behavioral Sensitivity to Quinine
No guanylyl cyclase has been found to be expressed in ASH [58,59] and the results described above suggest that cGMP generated at distant sites can dampen ASH response to sensory stimuli. To determine if the mere presence of cGMP in ASH is sufficient to dampen quinine sensitivity, the ASH cell-selective promoters osm-10 [48] and srb-6 [80] were used to express a blue light-inducible guanylyl cyclase (BlgC) [81] in the ASHs of animals lacking the blue-violet light receptor LITE-1 [82]. When assayed 10 minutes after a 30-second exposure to blue light, animals expressing BlgC in the ASH sensory neurons displayed a diminished response to both 5 mM (Fig 4A) and 10 mM quinine (S4 Fig), while transgenic animals that were not flashed with blue light displayed wild-type sensitivity. Although cAMP generation by BlgC was undetectable in E. coli, BlgC was shown to posses ~10% residual adenylyl cyclase activity in vitro [81]. To confirm that the dampened quinine response was due to production of cGMP, and not cAMP, we also assayed animals expressing a blue light-inducible adenylyl cyclase, BlaC [81]. Unlike BlgC, blue light-induction of BlaC to stimulate cAMP production did not alter animals’ response to quinine (Fig 4A and S4 Fig). Blue light induction of BlgC in the ASHs was also sufficient to significantly diminish the hypersensitivity of odr-1(lof) animals in response to 1 mM quinine (Fig 4B). Together, these results demonstrate that elevating cGMP levels in ASH is sufficient to dampen behavioral sensitivity to quinine.
Fig 4. cGMP generation in ASH dampens quinine sensitivity.

The ASH-selective osm-10 [48] and srb-6 [80] promoters were used to drive expression of a blue light-inducible guanylyl cyclase (BlgC) or blue light-inducible adenylyl cyclase (BlaC) [81]. osm-10p drives expression in ASH and weakly in ASI in the head, and in PHA and PHB in the tail. srb-6p drives expression in the ASH, ADL and ADF head sensory neurons, and in PHA and PHB in the tail. Adult animals expressing BlgC or BlaC were tested without blue light exposure (white bars) or after a 30 second exposure (grey bars). (A) While lite-1(lof) animals responded robustly to 5 mM quinine, similar to wild-type animals (p > 0.4), transgenic animals expressing BlgC displayed a significantly diminished response following blue light exposure (p < 0.0001 for each ASH-selective promoter). Transgenic animals expressing BlaC remained sensitive following blue light exposure (p > 0.05 when compared to wild-type animals). (B) While odr-1(lof) animals were hypersensitive in their response to 1 mM quinine, transgenic animals expressing BlgC displayed a significantly diminished response following blue light exposure (p < 0.0001). Transgenic animals expressing BlaC remained hypersensitive following blue light exposure (p > 0.5 when compared to odr-1(lof) animals). The percentage of animals responding is shown. The combined data of ≥ 2 independent lines and n ≥ 95 transgenic animals is shown. Alleles used: lite-1(ce314) loss-of-function, odr-1(n1936) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function. See also S4 Fig.
The ADF, AFD and AIA Neurons That Form Gap Junctions with ASH Modulate Quinine Sensitivity
Based on the C. elegans hermaphrodite wiring diagram (we referred to WormWiring.org for the most current wiring annotations based on the original electron micrograph series reported in [2]; Scott Emmons, personal communication), the ODR-1-expressing sensory neurons AWB, AWC and ASI do not form gap junction connections directly with ASH. However, these neurons are connected to ASH indirectly through a gap junction network via ADF (AWB, AWC), AFD (AWC) and AIA (AWB, AWC and ASI) (Fig 5A). The ADFs are chemosensory, the AFDs are thermosensory neurons, and the AIAs are interneurons [43,83]. ADF is also directly connected to both AFD and AIA by gap junctions (Fig 5A). These connections reveal a neuronal circuitry wherein ADF, AFD and AIA lay between the cGMP generating neurons (AWB, AWC and ASI) and ASH. We note that RMG and RIC also connect AWB to ASH, but their role in modulating the quinine response was not examined in this study. To determine whether ADF, AFD or AIA regulate the quinine response, we genetically ablated these cells in wild-type animals, both as individual neuron pairs and in combination, again using reconstituted caspases [69,84]. As shown in Fig 5B, loss of any one of the three neuron pairs resulted in significant behavioral hypersensitivity, as did simultaneous ablation of all three. In fact, the degree of quinine hypersensitivity seen in the ADF/AFD/AIA ablated animals was indistinguishable from that of the AWB/AWC/ASI animals (p > 0.5).
Fig 5. ADF, AFD and AIA neurons that form gap junctions with ASH modulate quinine sensitivity.
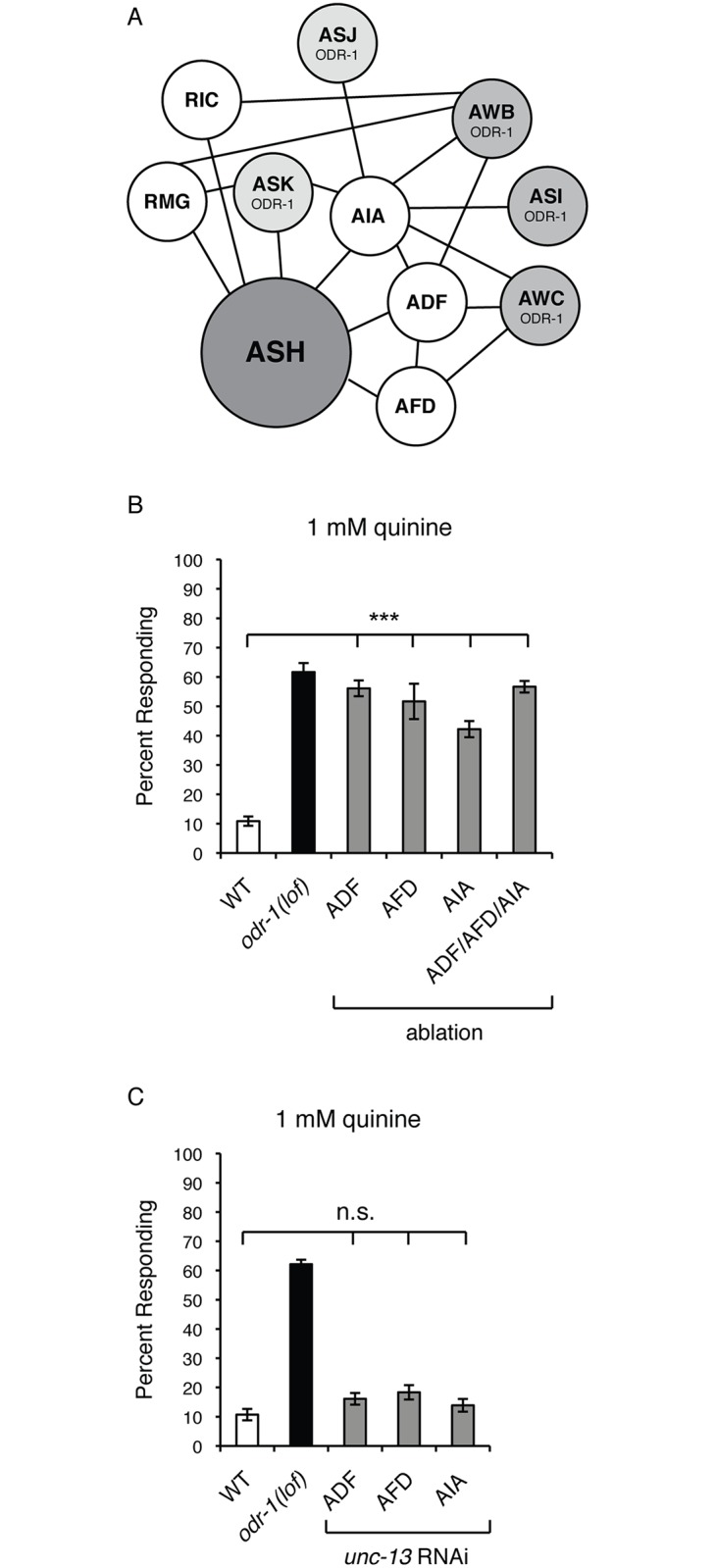
(A) A diagram of the gap junction network linking the AWB, AWC and ASI sensory neurons to ASH is shown. This diagram reflects the most current wiring annotations of the original electron micrograph series reported in [2], as curated at WormWiring.org. Black lines indicate gap junction connections between neurons. Only those neurons and gap junctions that would allow the ODR-1-expressing neurons to ultimately connect to ASH are shown. The head sensory neurons that express ODR-1 are shown. The ODR-1-expressing sensory neurons AWB, AWC and ASI do not form gap junction connections directly with ASH. However, these neurons are connected to ASH indirectly through a gap junction network via ADF (AWB, AWC), AFD (AWC), and AIA (AWC, AWB and ASI) [2]. (B) Genetic ablation of the ADF, AFD and AIA neurons resulted in quinine hypersensitivity. The reconstituted caspase approach [69] was used to genetically ablate ADF, AFD and AIA either individually or in combination in otherwise wild-type animals. Loss of any of the three neuron pairs individually, or all three neuron pairs simultaneously, resulted in significant behavioral hypersensitivity to dilute (1 mM) quinine (p < 0.0001 for each when compared to wild-type animals). (C) UNC-13-dependent synaptic signaling from ADF, AFD and AIA does not modulate quinine sensitivity. The srh-142p (ADF) [79], gcy-8p (AFD) [121] and gcy-28dp (AIA) [122] promoters were used to co-express a non-coding fragment of unc-13 in both the sense and antisense orientations in otherwise wild-type animals. RNAi knock-down of unc-13 in the ADF, AFD or AIA neurons did not result in behavioral hypersensitivity to dilute (1 mM) quinine (p > 0.05 when compared to wild-type animals for each transgene). The percentage of animals responding is shown. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Allele used: odr-1(n1936) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function. n.s. = not significant.
We noted that ADF and AIA do also chemically synapse onto ASH [2] (and WormWiring.org). To confirm that vesicular synaptic signaling from these neurons does not underlie their ability to decrease ASH sensitivity, we again utilized cell-specific RNAi [73] to knockdown unc-13 in ADF and AIA to block synaptic transmission from these neurons. Animals in which unc-13 was knocked down in either the ADF or AIA neurons did not show increased sensitivity to dilute quinine (Fig 5C). Even though AFD does not synapse onto ASH, we also confirmed that unc-13 RNAi in this neuron pair did not affect quinine sensitivity (Fig 5C). Taken together, our data suggest that gap junction-mediated communication between AWB/AWC/ASI and ASH, via ADF/AFD/AIA can regulate ASH sensitivity and an animal’s response to environmental stimuli.
No guanylyl cyclases are known to be expressed in the ADF sensory neurons [59]. Therefore, we next sought to determine whether ectopic cGMP generation in these cells, which lay between the ODR-1-expressing neurons and the ASHs in the gap junction network (Fig 5A), would be sufficient to dampen quinine sensitivity. The ADF-specific srh-142 promoter [79] was used to drive expression of BlgC [81] in lite-1(lof) or inx-4(lof);lite-1(lof) animals. After exposure to blue light, lite-1(lof) animals expressing BlgC in the ADFs displayed a diminished response to 5 mM (Fig 6) and 10 mM (S5 Fig) quine, while transgenic animals that were not exposed to blue light displayed wild-type sensitivity. Conversely, inx-4(lof);lite-1(lof) transgenic animals expressing BlgC in the ADFs did not display a diminished response following blue light exposure (Fig 6 and S5 Fig). Consistent with the results described above (Fig 4A), blue light-induction of BlaC to stimulate cAMP production in the ADFs did not alter animals’ response to quinine (Fig 6 and S5 Fig). Together, these data demonstrate that elevating cGMP levels in the ADFs is sufficient to dampen behavioral sensitivity to quinine, and that function of the INX-4 gap junction component is required for this effect.
Fig 6. cGMP generation in ADF dampens quinine sensitivity.

The ADF-specific srh-142 promoter [79] was used to drive expression of a blue light-inducible guanylyl cyclase (BlgC) or blue light-inducible adenylyl cyclase (BlaC) [81] in lite-1(lof) or inx-4(lof);lite-1(lof) animals. srh-142p drives expression only in the two ADF head sensory neurons. Adult lite-1(lof) or inx-4(lof);lite-1(lof) animals expressing BlgC or BlaC were tested without blue light exposure (white bars) or after a 30 second exposure and 30 second recovery (grey bars). While lite-1(lof) animals responded robustly to 5 mM quinine, similar to WT animals (p > 0.1), transgenic animals expressing BlgC in the ADFs displayed a significantly diminished response following blue light exposure (p < 0.0001). Transgenic animals expressing BlaC did not display a diminished response following blue light exposure (p > 0.2 when compared to wild-type animals). inx-4(lof);lite-1(lof) transgenic animals expressing either BlgC or BlaC did not display a diminished response following blue light exposure (p > 0.7 when compared to wild-type animals). The percentage of animals responding is shown. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Alleles used: lite-1(ce314) loss-of-function and inx-4(ok2373) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function. See also S5 Fig.
Discussion
The sheer number of possible circuit outcomes revealed by anatomical wiring diagrams means that we cannot possibly predict how information might flow through a circuit based on physical connections alone. Even in the relatively simple nervous system of C. elegans, the function of most of the connections, or which connections will be preferentially used under different circumstances, is not known [85]. For example, neuropeptide signaling regulates a sensory context-dependent switch in the composition of a C. elegans salt sensory circuit [86]. In this case, one of the AWC olfactory sensory neurons is recruited to function as an interneuron during response to high salt concentrations [86]. It was also recently reported that, in a reciprocal inhibition circuit that fine-tunes copper (a heavy metal) avoidance, the ADF sensory neurons can be activated by neuropeptides to act as interneurons downstream of the ASI sensory neurons. The ADFs then dampen ASH sensitivity via serotonin release [87]. In addition, the C. elegans nose touch circuit appears to utilize electrical synapses to mediate both excitatory and inhibitory interactions between neurons [88]. Adding another layer of complexity, the internal state of an animal, such as its nutritional status and degree of satiety, can influence its sensitivity to environmental stimuli [58,89–93], and may even alter which neurons participate in stimulus detection [89].
Sensory systems in particular may be subject to extensive modulation [8,15]. For example, feeding state and food availability can alter gustatory and olfactory responses in diverse species, where the complexity of the nervous systems ranges from just hundreds of neurons to billions of neurons. To better understand the neuromodulatory mechanisms that regulate chemosensation, we focused on one component of the regulatory pathway that controls C. elegans response to aversive sensory stimuli, the guanylyl cyclase ODR-1. As previously reported, the two AWC olfactory neurons require ODR-1 function to mediate chemotaxis towards attractive odorants that they detect, while the AWBs require ODR-1 for 2-nonanone avoidance [61]. In addition, ODR-1 plays a role within the AWCs to regulate adaption in response to prolonged odor exposure [61]. Here, we describe a new role for ODR-1 in modulating animals’ avoidance response to the bitter tastant quinine in a non-cell-autonomous manner. This is, to our knowledge, the first in vivo demonstration of a guanylyl cyclase functioning to modulate the activity of another cell.
odr-1(lof) animals are hypersensitive in their response to dilute concentrations of the bitter tastant quinine, and ODR-1 function in three distinct pairs of sensory neurons (the AWBs, AWCs and ASIs) appears to contribute to the modulation of ASH-mediated avoidance (Fig 1). However, the evolutionary advantage of this sort of decentralized regulation of sensory signaling is not immediately clear. One possibility could be to prevent overstimulation of ASH, which is the main nociceptor in C. elegans. Nociceptors in general have high thresholds of activation, understandably to prevent organisms from unnecessarily reacting to minute or low-risk stimuli [94]. An additive modulatory role for AWB, AWC and ASI may help to assure that the threshold of ASH sensitivity to noxious stimuli remains high, consistent with the observed behavioral hypersensitivity we observed upon ablation of these three neurons pairs (Fig 2).
Another potential benefit of decentralized ASH regulation is that by utilizing multiple sensory neurons to regulate ASH function, the integration of diverse sets of environmental information can maximize the appropriateness of an animal’s response to its surroundings. For example, in addition to detecting the aversive odorants 100% octanol and 2-nonanone [66,89], which may indicate the presence of fungi or pathogenic bacteria [95–97], AWB also mediates lawn avoidance upon encountering the pathogenic bacteria Serratia marcescens [97]. All of these roles for AWB could help C. elegans avoid environments that might be harmful to them. Conversely, scents detected by AWC (benzaldehyde, butanone, isoamyl alcohol, 2,3-pentanedione, and 2,4,5-trimethylthiazole) [98,99] are primarily produced in nature by plants (e.g. fruits, nuts and coffee) and microorganisms. Therefore, these natural odorants signal potential sources of nutritive bacteria and it has been shown that C. elegans will populate areas of decaying plant matter [100]. Finally, if developing larvae encounter harsh environmental conditions, including elevated population density and/or a limited food supply, they can enter dauer arrest at the second molt [101]. This transition is repressed by the ASI, ADF and ASG neurons, which detect the dauer pheromone that serves as an indicator of population density. A high density of C. elegans in a given area could signify low or dwindling food availability there, which could in turn result in poor nutritional status. Thus, the AWBs, AWCs and ASIs detect distinct sets of environmental stimuli that can all provide the animal with information about potential food quality and availability.
Following the initial detection of an environmental stimulus, the AWB, AWC or ASI sensory neurons may then subsequently modulate ASH sensitivity to indirectly optimize nociceptive responses in a context-dependent manner. For example, when animals are well-fed, they are more sensitive to aversive stimuli, including quinine (Fig 7), than they are upon food deprivation [58,89–93]. This may reflect the need of animals to reprioritize their behaviors to balance the need to avoid potentially dangerous situations with the need to find food. If starving, minimized aversive responses may maximize entry into new environments to increase the likelihood of encountering new food sources.
Fig 7. Response to quinine is modulated by feeding status.

The response of wild-type animals to dilute quinine diminishes upon food deprivation (p < 0.00001 for 2.5 mM when comparing animals assayed “off food” versus “on food”). The percentage of animals responding is shown. The combined data of n ≥ 60 animals is shown for each concentration. Animals were assayed 10–20 minutes after transfer to plates with or without food (no bacterial lawn). conc = concentration. n.s. = not significant. WT = the N2 wild-type strain.
We speculate that food somehow suppresses ODR-1 activity in the upstream AWB, AWC and/or ASI sensory neurons, while removal of food allows for ODR-1 activation and cGMP accumulation (Fig 8A). ODR-1 is most similar to transmembrane guanylyl cyclases, which are often regulated by extracellular peptides [102]. However, we found that the extracellular domain is not required for ODR-1 function in modulating ASH sensitivity (Fig 1). In addition, while soluble (cytoplasmic) guanylyl cyclases are generally activated by nitric oxide [103], C. elegans lack a nitric oxide synthase. Thus, the mechanistic link between an animal’s feeding status and ODR-1 activity is unclear. Interestingly, calcium levels rise in the AWCs upon withdrawal of either odorant or bacterial-conditioned media [104]. An attractive possibility is that this increase in calcium could directly or indirectly activate ODR-1 activity in the AWCs, which would provide a link between food withdrawal, cGMP generation in this neuron pair and the downstream dampening of ASH-mediated responses. Alternatively, ODR-1 may be constitutively active and phosphodiesterase activity may be regulated by an animal’s feeding status to adjust the pool of available cGMP.
Fig 8. Model for ODR-1 modulation of ASH-mediated nociceptive signaling.

(A) Our working model is that the transmembrane guanylyl cyclase ODR-1 functions in the AWB, AWC and ASI sensory neurons to decrease C. elegans behavioral sensitivity to the bitter tastant quinine (and possibly the volatile odorant octanol). Removal of food leads to cGMP accumulation, at least in the AWCs, likely by direct/indirect activation of ODR-1 in these neurons. Based on the re-annotated wiring diagram (WormWiring.org), we propose that cGMP then flows via gap junction connections from the site of its production in the ODR-1-expressing AWB/AWC/ASI sensory neurons, through ADF, AFD and AIA, to the ASH nociceptors. Once in ASH, cGMP activates the cGMP-dependent protein kinase EGL-4, which likely directly phosphorylates the regulator of G protein signaling proteins RGS-2 and RGS-3, stimulating their activity [58]. RGS-2 and RGS-3 downregulate Gα proteins that signal downstream of G protein-coupled receptors (GPCRs) that are activated by ligands such as quinine and octanol. When animals are well-fed, cGMP levels are low and there is only minimal inhibition of G protein-coupled signaling in ASH. Upon food remove, cGMP influx into ASH activates EGL-4, resulting in diminished nociceptive behavioral sensitivity to a subset of ASH-detected aversive stimuli. Decentralized modulation of ASH sensitivity may allow an animal to integrate multiple environmental cues with its internal state to maximize the appropriateness of its response to its surroundings. (B) The same diagram depicted in Fig 5A is shown here, but with only those gap junction connections originally reported in White et al. [2] included. (C) The left column shows a list of the neurons reported by White et al. [2] to make gap junctions with ASH, while the right column lists the neurons currently annotated at WormWiring.org to make gap junctions with ASH. The neurons shown in bold are those that are common to both lists.
Collectively, our data support a model wherein, upon food removal, the ODR-1-expressing neurons AWB, AWC and ASI provide a pool of cGMP that flows through a gap junction network from the site of its production in these sensory neurons, through ADF, AFD and AIA, to the ASH nociceptors (Fig 8A). However, the gap junctions reported in the wiring diagram first published by White et al. [2] do not provide a straightforward explanation for our experimental observations (Fig 8B). For example, White et al. [2] did not report gap junctions between ASH and ADF, AFD or AIA (Fig 8C). Therefore, these neurons were not predicted to connect the ODR-1-expressing neurons (AWB, AWC and ASI) to ASH via a gap junction circuit (Fig 8B) [2]. For our study, we referred to the re-annotated wiring diagram, which is based on the original electron micrograph series [2] and curated at WormWiring.org (Scott Emmons, personal communication) (Fig 5A). If the presence of the newly annotated gap junctions is not corroborated by future analysis, then alternate pathways/circuits for the non-cell-autonomous role of ODR-1 in the regulation of ASH-mediated nociceptive sensory behaviors must be considered.
INX-4 function in the ASHs is sufficient to rescue the quinine hypersensitivity of inx-4(lof) animals (Fig 3), suggesting that INX-4 expressed in ASH likely forms heterotypic gap junctions with an unidentified innexin(s) in ADF/AFD/AIA. We note that the reported expression patterns for most of the innexins were determined using only short upstream regulatory sequences to drive GFP expression [35]. Therefore, it is likely that the full expression pattern of each innexin is not yet known. For example, while the 706 bp upstream promoter for inx-19 (also known as nsy-5) drove GFP expression in the AVBs and AVKs [35], a 5.8 kb inx-19 promoter did not express in these neurons but instead was seen to drive GFP expression in 18 other neuron pairs (including AWB, AWC, ASI, ADF, AFD and ASH) [105]. Thus, further analysis is needed to correlate endogenous innexin expression with the anatomically defined gap junctions present throughout the C. elegans nervous system. While our combination of cell-specific rescue and ablation experiments strongly supports a role for the proposed gap junction network in the modulation of ASH activity, it is possible that extracellular cGMP could also enter ASH through an INX-4 hemichannel.
Delayed delivery of cGMP to the ASHs, due to flux through such a network, may help to optimize an animal’s navigation through an unpredictable environment [106]. Additionally, since C. elegans lack a complex central nervous system with which to simultaneously process several sets of sensory information, a gap junction network may allow for the efficient integration of diverse signals representing the environmental landscape with an animal’s experiences and internal state. This may allow for a more sophisticated modulation of behavior than could be achieved by expressing a guanylyl cyclase(s) directly in the ASHs themselves, especially given that the ASHs detect multiple forms of aversive stimuli [46–54]. Consistent with this possibility, we found that odr-1(lof) animals are hypersensitive in their response to some (e.g. quinine), but not all (primaquine, SDS, copper), ASH-detected stimuli (Fig 1, S3 Fig).
ODR-1 is not the only guanylyl cyclase that appears to downregulate ASH-mediated behavioral responses. We previously reported that animals lacking GCY-27, GCY-33 or GCY-34 function are also hypersensitive in their avoidance of dilute quinine [58]. GCY-27 is expressed in the ASJ, ASK and ASI head sensory neurons, although its role in these cells has not been characterized [59]. GCY-33 and GCY-34 contribute to the detection of oxygen levels by the BAG neurons and the URX, AQR, and PQR neurons, respectively [107–112]. Oxygen detection has been hypothesized to be important for C. elegans because aerobic bacteria, which utilize oxygen for metabolic processes, serve as a food source for animals in their native soil environment. Therefore, C. elegans may correlate low ambient oxygen levels with locations having nutritive potential. Thus, guanylyl cyclase function in neurons besides AWB/AWC/ASI could also serve to modulate ASH sensitivity based on food availability cues. Future studies focused on GCY-27, GCY-33 and GCY-34 are needed to shed light on the mechanism by which these guanylyl cyclases ultimately influence ASH-mediated behavioral responses. If they regulate ASH function non-cell-autonomously, in a manner similar to ODR-1, this would allow for the integration of an even greater array of sensory information to generate animal behavior that is optimized based on environment and experience.
Gap junctions have been classically described for their role in electrical coupling of neurons [113,114]. However, it is now appreciated that a variety of ions and small molecules can pass through these channels [24–32]. In mammalian systems, changes in gap junction protein expression have been documented in central nervous system pathologies [115,116], and studies in cultured cells have suggested possible opposing contributions for gap junctions. For example, while they may play a neuroprotective role in brain ischemia, gap junctions appear to contribute to the propagation of excitatory activity in epilepsy [115–117]. With a compact nervous system and a well-characterized behavioral repertoire, C. elegans serves as an excellent animal model to study the in vivo roles of gap junctions and their contribution to circuit-level modulation of neuronal function.
Materials and Methods
C. elegans Culture
Strains were maintained under standard conditions on NGM agar plates seeded with OP50 E. coli bacteria [118]. For a list of strains used in this study, see Supplemental Information.
Behavioral Assays
Well-fed young adults were used for analysis, and all behavioral assays were performed on at least three separate days, in parallel with controls. Response to the soluble aversive tastants was scored as the percentage of animals that initiated backward locomotion within 4 seconds of encountering a drop of the tastant placed on the agar plate in front of a forward moving animal [51,52,119]. Tastants were dissolved in M13, pH 7.4 [120]. For soluble tastant avoidance assays, animals were tested 30 minutes after transfer to NGM plates lacking bacteria (“off food”). Response to octanol was scored as the amount of time it took an animal to initiate backward locomotion when presented with a hair dipped in octanol [48,80]. The nonanone avoidance assays were performed essentially as described [66] with the following modifications: round 100x15 mm plates were used with 14 ml of agar and animals were incubated on the nonanone plates for two hours. The Avoidance Index (A.I.) was calculated as the number of animals in sectors A and B, minus the number of animals in sectors E and F, divided by the total number of animals (not including animals that did not move from their original placement) [66]. The benzaldehyde chemotaxis assays were performed as previously described [60]. After one hour, the Chemotaxis Index (C.I.) was calculated as the number of animals that had accumulated at the attractant, minus the number of animals at the control, divided by the total number of animals [60]. For heat shock experiments, animals were raised to young adulthood and then shifted to 33°C for two hours. They were allowed to recover for four hours at 20°C prior to assaying. All data is presented as ± standard error of the mean (SEM). The Student’s two-tailed t-Test and one-way Anova with Tukey’s Honestly Significant Difference (HSD) were used for statistical analyses. In all figures, * denotes p < 0.05, ** denotes p < 0.001, and *** denotes p < 0.0001. n.s. denotes p ≥ 0.05.
Supporting Information
(A) The loss of UNC-13-dependent synaptic signaling from the AWB head sensory neurons resulted in failure to avoid the aversive odorant 2-nonanone, which is detected by the AWBs [66]. The str-1 (AWB) [66] promoter was used to co-express a non-coding fragment of unc-13 in both the sense and antisense orientations of otherwise wild-type animals. RNAi knock-down of unc-13 in the AWB neurons resulted in diminished avoidance of a 1:10 dilution of 2-nonanone (p < 0.0001 when compared to wild-type animals). (B) Loss of UNC-13-dependent synaptic signaling from the AWC head sensory neurons resulted in failure to chemotax towards benzadehyde, which is detected by the AWCs [60]. The ceh-36p3 (AWC) [65], gpa-4 (ASI) [64] and str-1p (AWB) [66] promoters were used to co-express a non-coding fragment of unc-13 in both the sense and antisense orientations of otherwise wild-type animals. RNAi knock-down of unc-13 in the AWC neurons or in the AWB, AWC or ASI neurons in combination abolished chemotaxis to a 1:200 dilution of benzaldehyde. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Avoidance index = ((A + B)–(E + F)) ÷ total number of animals on the assay plate [66]. Chemotaxis index = (number of animals at odorant − number of animals at control) ÷ total number of animals on the assay plate [60]. Alleles used: odr-1(n1936) loss-of-function and inx-4(ok2373) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function.
(TIF)
The inx-4 promoter [35] was used to drive expression of GFP (inx-4p::gfp). Transgenic animals were incubated with the lipophilic dye DiD (shown in red), which is taken up by the ASH sensory neurons and was used to visualize the neuronal cell body. As previously reported [35], inx-4 expression was seen in the ASH sensory neurons of larval-stage animals, including the L2 stage (top panel). In addition, inx-4 expression was observed in the ASHs of adult animals (bottom panel).
(TIF)
C. elegans respond to aversive stimuli in addition to quinine. (A-C) odr-1(lof) and inx-4(lof) animals responded similarly to wild-type animals to the bitter tastant primaquine, the detergent SDS and the heavy metal copper, across a range of concentrations (p > 0.2 for each concentration). The percentage of animals responding is shown. n ≥ 60 for each. All tastants were dissolved in M13 buffer, pH 7.4. Alleles used: inx-4(ok2373) loss-of-function and odr-1(n1936) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function.
(TIF)
The ASH-selective osm-10 [48] and srb-6 [80] promoters were used to drive expression of a blue light-inducible guanylyl cyclase (BlgC) or blue light-inducible adenylyl cyclase (BlaC) [81] in lite-1(lof) animals. osm-10p drives expression in ASH and weakly in ASI in the head, and in PHA and PHB in the tail. srb-6p drives expression in the ASH, ADL and ADF head sensory neurons, and in PHA and PHB in the tail. Adult lite-1(lof) animals expressing BlgC or BlaC were tested without blue light exposure (white bars) or after a 30 second exposure (grey bars). While lite-1(lof) animals responded robustly to 5 mM quinine, similar to WT animals (p > 0.2), transgenic animals expressing BlgC displayed a significantly diminished response following blue light exposure (p < 0.0001 for each ASH-selective promoter). Transgenic animals expressing BlaC remained sensitive following blue light exposure (p > 0.1 when compared to wild-type animals). The percentage of animals responding is shown. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Allele used: lite-1(ce314) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function.
(TIF)
The ADF-specific srh-142 promoter [79] was used to drive expression of a blue light-inducible guanylyl cyclase (BlgC) or blue light-inducible adenylyl cyclase (BlaC) [81] in lite-1(lof) or inx-4(lof);lite-1(lof) animals. Adult lite-1(lof) or inx-4(lof);lite-1(lof) animals expressing BlgC or BlaC were tested without blue light exposure (white bars) or after a 30 second exposure and 30 second recovery (grey bars). While lite-1(lof) animals responded robustly to 10 mM quinine, similar to WT animals (p > 0.1), transgenic animals expressing BlgC in the ADFs displayed a significantly diminished response following blue light exposure (p < 0.001). Transgenic animals expressing BlaC did not display a diminished response following blue light exposure (p > 0.1 when compared to wild-type animals). inx-4(lof);lite-1(lof) transgenic animals expressing either BlgC or BlaC did not display a diminished response following blue light exposure (p > 0.5 when compared to wild-type animals). The percentage of animals responding is shown. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Alleles used: lite-1(ce314) loss-of-function and inx-4(ok2373) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function.
(TIF)
(DOCX)
(DOCX)
Acknowledgments
We thank Cori Bargmann, Alexander Gottschalk, Gal Haspel, Keith Nehrke, Doug Portman and Andy Samuelson for valuable discussions, and Cori Bargmann and Doug Portman for feedback on this manuscript. We are grateful to Cori Bargmann, Mark Gomelsky, Keith Nehrke, Atsunori Oshima and Piali Sengupta for reagents. We thank Paolo Bazzicalupo for sharing information about the srd-10 promoter and its expression pattern, Clark Driscoll for generating pFG233, Bi-Tzen Juang for generating ceh-36p3::mCherry, and Chao He and Damien O’Halloran for technical advice for the nonanone avoidance assay. Some strains used in this study were obtained from the Caenorhabditis Genetics Center, which is funded in part by the National Institutes of Health—National Center for Research Resources.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the National Science Foundation (MCB-0917896 and IOS-1351649, DMF). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kleinfeld D, Bharioke A, Blinder P, Bock DD, Briggman KL, et al. (2011) Large-scale automated histology in the pursuit of connectomes. J Neurosci 31: 16125–16138. 10.1523/JNEUROSCI.4077-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White JG, Southgate E, Thomson JN, Brenner S (1986) The structure of the nervous system of the nematode Caenorhabditis elegans. Phil Trans R Soc Lond 314: 1–340. [DOI] [PubMed] [Google Scholar]
- 3.Seung HS (2009) Reading the book of memory: sparse sampling versus dense mapping of connectomes. Neuron 62: 17–29. 10.1016/j.neuron.2009.03.020 [DOI] [PubMed] [Google Scholar]
- 4.Meinertzhagen IA, Lee CH (2012) The genetic analysis of functional connectomics in Drosophila. Adv Genet 80: 99–151. 10.1016/B978-0-12-404742-6.00003-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson JR, Jones BW, Watt CB, Shaw MV, Yang JH, et al. (2011) Exploring the retinal connectome. Mol Vis 17: 355–379. [PMC free article] [PubMed] [Google Scholar]
- 6.Kucyi A, Davis KD (2015) The dynamic pain connectome. Trends Neurosci 38: 86–95. 10.1016/j.tins.2014.11.006 [DOI] [PubMed] [Google Scholar]
- 7.Soden ME, Gore BB, Zweifel LS (2014) Defining functional gene-circuit interfaces in the mouse nervous system. Genes Brain Behav 13: 2–12. 10.1111/gbb.12082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sengupta P (2013) The belly rules the nose: feeding state-dependent modulation of peripheral chemosensory responses. Curr Opin Neurobiol 23: 68–75. 10.1016/j.conb.2012.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Critchley HD, Rolls ET (1996) Hunger and satiety modify the responses of olfactory and visual neurons in the primate orbitofrontal cortex. J Neurophysiol 75: 1673–1686. [DOI] [PubMed] [Google Scholar]
- 10.Magni P, Dozio E, Ruscica M, Celotti F, Masini MA, et al. (2009) Feeding behavior in mammals including humans. Ann N Y Acad Sci 1163: 221–232. 10.1111/j.1749-6632.2008.03627.x [DOI] [PubMed] [Google Scholar]
- 11.Savigner A, Duchamp-Viret P, Grosmaitre X, Chaput M, Garcia S, et al. (2009) Modulation of spontaneous and odorant-evoked activity of rat olfactory sensory neurons by two anorectic peptides, insulin and leptin. J Neurophysiol 101: 2898–2906. 10.1152/jn.91169.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niki M, Yoshida R, Takai S, Ninomiya Y (2010) Gustatory signaling in the periphery: detection, transmission, and modulation of taste information. Biol Pharm Bull 33: 1772–1777. [DOI] [PubMed] [Google Scholar]
- 13.Dietrich MO, Horvath TL (2009) Feeding signals and brain circuitry. Eur J Neurosci 30: 1688–1696. 10.1111/j.1460-9568.2009.06963.x [DOI] [PubMed] [Google Scholar]
- 14.Shin YK, Egan JM (2010) Roles of hormones in taste signaling. Results Probl Cell Differ 52: 115–137. 10.1007/978-3-642-14426-4_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komuniecki R, Hapiak V, Harris G, Bamber B (2014) Context-dependent modulation reconfigures interactive sensory-mediated microcircuits in Caenorhabditis elegans. Curr Opin Neurobiol 29: 17–24. 10.1016/j.conb.2014.04.006 [DOI] [PubMed] [Google Scholar]
- 16.Ryan DA, Miller RM, Lee K, Neal SJ, Fagan KA, et al. (2014) Sex, age, and hunger regulate behavioral prioritization through dynamic modulation of chemoreceptor expression. Curr Biol 24: 2509–2517. 10.1016/j.cub.2014.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palouzier-Paulignan B, Lacroix MC, Aime P, Baly C, Caillol M, et al. (2012) Olfaction under metabolic influences. Chem Senses 37: 769–797. 10.1093/chemse/bjs059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelley AE, Bakshi VP, Haber SN, Steininger TL, Will MJ, et al. (2002) Opioid modulation of taste hedonics within the ventral striatum. Physiol Behav 76: 365–377. [DOI] [PubMed] [Google Scholar]
- 19.Breunig E, Czesnik D, Piscitelli F, Di Marzo V, Manzini I, et al. (2010) Endocannabinoid modulation in the olfactory epithelium. Results Probl Cell Differ 52: 139–145. 10.1007/978-3-642-14426-4_11 [DOI] [PubMed] [Google Scholar]
- 20.Jyotaki M, Shigemura N, Ninomiya Y (2010) Modulation of sweet taste sensitivity by orexigenic and anorexigenic factors. Endocr J 57: 467–475. [DOI] [PubMed] [Google Scholar]
- 21.Kaji I, Karaki S, Kuwahara A (2014) Taste sensing in the colon. Curr Pharm Des 20: 2766–2774. [DOI] [PubMed] [Google Scholar]
- 22.Phelan P, Starich TA (2001) Innexins get into the gap. Bioessays 23: 388–396. [DOI] [PubMed] [Google Scholar]
- 23.Sohl G, Maxeiner S, Willecke K (2005) Expression and functions of neuronal gap junctions. Nat Rev Neurosci 6: 191–200. [DOI] [PubMed] [Google Scholar]
- 24.Simon AM (1999) Gap junctions: more roles and new structural data. Trends Cell Biol 9: 169–170. [DOI] [PubMed] [Google Scholar]
- 25.Kirchhoff S, Nelles E, Hagendorff A, Kruger O, Traub O, et al. (1998) Reduced cardiac conduction velocity and predisposition to arrhythmias in connexin40-deficient mice. Curr Biol 8: 299–302. [DOI] [PubMed] [Google Scholar]
- 26.Goldberg GS, Lampe PD, Nicholson BJ (1999) Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nat Cell Biol 1: 457–459. [DOI] [PubMed] [Google Scholar]
- 27.Saez JC, Connor JA, Spray DC, Bennett MV (1989) Hepatocyte gap junctions are permeable to the second messenger, inositol 1,4,5-trisphosphate, and to calcium ions. Proc Natl Acad Sci U S A 86: 2708–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson E, Albertini DF (1976) Gap junctions between the oocyte and companion follicle cells in the mammalian ovary. J Cell Biol 71: 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mao GK, Li JX, Bian FH, Han YY, Guo M, et al. (2013) Gap junction -mediated cAMP movement between oocytes and somatic cells. Front Biosci (Elite Ed) 5: 755–767. [DOI] [PubMed] [Google Scholar]
- 30.Norris RP, Ratzan WJ, Freudzon M, Mehlmann LM, Krall J, et al. (2009) Cyclic GMP from the surrounding somatic cells regulates cyclic AMP and meiosis in the mouse oocyte. Development 136: 1869–1878. 10.1242/dev.035238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaccari S, Weeks JL 2nd, Hsieh M, Menniti FS, Conti M (2009) Cyclic GMP signaling is involved in the luteinizing hormone-dependent meiotic maturation of mouse oocytes. Biol Reprod 81: 595–604. 10.1095/biolreprod.109.077768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shuhaibar LC, Egbert JR, Norris RP, Lampe PD, Nikolaev VO, et al. (2015) Intercellular signaling via cyclic GMP diffusion through gap junctions restarts meiosis in mouse ovarian follicles. Proc Natl Acad Sci U S A 112: 5527–5532. 10.1073/pnas.1423598112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yen MR, Saier MH Jr. (2007) Gap junctional proteins of animals: the innexin/pannexin superfamily. Prog Biophys Mol Biol 94: 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phelan P, Bacon JP, Davies JA, Stebbings LA, Todman MG, et al. (1998) Innexins: a family of invertebrate gap-junction proteins. Trends Genet 14: 348–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altun ZF, Chen B, Wang ZW, Hall DH (2009) High resolution map of Caenorhabditis elegans gap junction proteins. Dev Dyn 238: 1936–1950. 10.1002/dvdy.22025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phelan P (2005) Innexins: members of an evolutionarily conserved family of gap-junction proteins. Biochim Biophys Acta 1711: 225–245. [DOI] [PubMed] [Google Scholar]
- 37.Simonsen KT, Moerman DG, Naus CC (2014) Gap junctions in C. elegans. Front Physiol 5: 40 10.3389/fphys.2014.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Albertson DG, Thomson JN (1976) The pharynx of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci 275: 299–325. [DOI] [PubMed] [Google Scholar]
- 39.Ward S, Thomson N, White JG, Brenner S (1975) Electron microscopical reconstitution of the anterior sensory anatomy of the nematode Caenorhabditis elegans. J Comp Neurol 160: 313–337. [DOI] [PubMed] [Google Scholar]
- 40.Ware RW, Clark D, Crossland K, Russell RL (1975) The nerve ring of the nematode Caenorhabditis elegans: sensory input and motor output. J Comp Neurol 162: 71–110. [Google Scholar]
- 41.Hall DH, Russell RL (1991) The posterior nervous system of the nematode Caenorhabditis elegans: serial reconstruction of identified neurons and complete pattern of synaptic interactions. J Neurosci 11: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varshney LR, Chen BL, Paniagua E, Hall DH, Chklovskii DB (2011) Structural properties of the Caenorhabditis elegans neuronal network. PLoS Comput Biol 7: e1001066 10.1371/journal.pcbi.1001066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bargmann CI Chemosensation in C. elegans. In: Community TCeR, editor. WormBook: WormBook. [Google Scholar]
- 44.Barr MM, Garcia LR Male mating behavior. In: Community TCeR, editor. WormBook: WormBook. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Bono M, Maricq AV (2005) Neuronal substrates of complex behaviors in C. elegans. Annu Rev Neurosci 28: 451–501. [DOI] [PubMed] [Google Scholar]
- 46.Bargmann CI, Thomas JH, Horvitz HR (1990) Chemosensory cell function in the behavior and development of Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol 55: 529–538. [DOI] [PubMed] [Google Scholar]
- 47.Kaplan JM, Horvitz HR (1993) A dual mechanosensory and chemosensory neuron in Caenorhabditis elegans. Proc Natl Acad Sci U S A 90: 2227–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hart AC, Kass J, Shapiro JE, Kaplan JM (1999) Distinct signaling pathways mediate touch and osmosensory responses in a polymodal sensory neuron. J Neurosci 19: 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sambongi Y, Nagae T, Liu Y, Yoshimizu T, Takeda K, et al. (1999) Sensing of cadmium and copper ions by externally exposed ADL, ASE, and ASH neurons elicits avoidance response in Caenorhabditis elegans. Neuroreport 10: 753–757. [DOI] [PubMed] [Google Scholar]
- 50.Troemel ER (1999) Chemosensory signaling in C. elegans. Bioessays 21: 1011–1020. [DOI] [PubMed] [Google Scholar]
- 51.Hilliard MA, Bargmann CI, Bazzicalupo P (2002) C. elegans responds to chemical repellents by integrating sensory inputs from the head and the tail. Curr Biol 12: 730–734. [DOI] [PubMed] [Google Scholar]
- 52.Hilliard MA, Bergamasco C, Arbucci S, Plasterk RH, Bazzicalupo P (2004) Worms taste bitter: ASH neurons, QUI-1, GPA-3 and ODR-3 mediate quinine avoidance in Caenorhabditis elegans. Embo J 23: 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hilliard MA, Apicella AJ, Kerr R, Suzuki H, Bazzicalupo P, et al. (2005) In vivo imaging of C. elegans ASH neurons: cellular response and adaptation to chemical repellents. Embo J 24: 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chatzigeorgiou M, Bang S, Hwang SW, Schafer WR (2013) tmc-1 encodes a sodium-sensitive channel required for salt chemosensation in C. elegans. Nature 494: 95–99. 10.1038/nature11845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Basbaum AI, Bautista DM, Scherrer G, Julius D (2009) Cellular and molecular mechanisms of pain. Cell 139: 267–284. 10.1016/j.cell.2009.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luo DS, Huang J, Dong YL, Wu ZY, Wei YY, et al. (2014) Connections between EM2- and SP-containing terminals and GABAergic neurons in the mouse spinal dorsal horn. Neurol Sci 35: 1421–1427. 10.1007/s10072-014-1774-9 [DOI] [PubMed] [Google Scholar]
- 57.Pezet S, McMahon SB (2006) Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci 29: 507–538. [DOI] [PubMed] [Google Scholar]
- 58.Krzyzanowski MC, Brueggemann C, Ezak MJ, Wood JF, Michaels KL, et al. (2013) The C. elegans cGMP-Dependent Protein Kinase EGL-4 Regulates Nociceptive Behavioral Sensitivity. PLoS Genet 9: e1003619 10.1371/journal.pgen.1003619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ortiz CO, Etchberger JF, Posy SL, Frokjaer-Jensen C, Lockery S, et al. (2006) Searching for neuronal left/right asymmetry: genomewide analysis of nematode receptor-type guanylyl cyclases. Genetics 173: 131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bargmann CI, Hartwieg E, Horvitz HR (1993) Odorant-selective genes and neurons mediate olfaction in C. elegans. Cell 74: 515–527. [DOI] [PubMed] [Google Scholar]
- 61.L'Etoile ND, Bargmann CI (2000) Olfaction and odor discrimination are mediated by the C. elegans guanylyl cyclase ODR-1. Neuron 25: 575–586. [DOI] [PubMed] [Google Scholar]
- 62.Kim K, Sato K, Shibuya M, Zeiger DM, Butcher RA, et al. (2009) Two chemoreceptors mediate developmental effects of dauer pheromone in C. elegans. Science 326: 994–998. 10.1126/science.1176331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miranda-Vizuete A, Fierro Gonzalez JC, Gahmon G, Burghoorn J, Navas P, et al. (2006) Lifespan decrease in a Caenorhabditis elegans mutant lacking TRX-1, a thioredoxin expressed in ASJ sensory neurons. FEBS Lett 580: 484–490. [DOI] [PubMed] [Google Scholar]
- 64.Jansen G, Thijssen KL, Werner P, van der Horst M, Hazendonk E, et al. (1999) The complete family of genes encoding G proteins of Caenorhabditis elegans. Nat Genet 21: 414–419. [DOI] [PubMed] [Google Scholar]
- 65.Etchberger JF, Lorch A, Sleumer MC, Zapf R, Jones SJ, et al. (2007) The molecular signature and cis-regulatory architecture of a C. elegans gustatory neuron. Genes Dev 21: 1653–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Troemel ER, Kimmel BE, Bargmann CI (1997) Reprogramming chemotaxis responses: sensory neurons define olfactory preferences in C. elegans. Cell 91: 161–169. [DOI] [PubMed] [Google Scholar]
- 67.Mukhopadhyay S, Lu Y, Shaham S, Sengupta P (2008) Sensory signaling-dependent remodeling of olfactory cilia architecture in C. elegans. Dev Cell 14: 762–774. 10.1016/j.devcel.2008.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stringham EG, Dixon DK, Jones D, Candido EP (1992) Temporal and spatial expression patterns of the small heat shock (hsp16) genes in transgenic Caenorhabditis elegans. Mol Biol Cell 3: 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chelur DS, Chalfie M (2007) Targeted cell killing by reconstituted caspases. Proc Natl Acad Sci U S A 104: 2283–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beverly M, Anbil S, Sengupta P (2011) Degeneracy and neuromodulation among thermosensory neurons contribute to robust thermosensory behaviors in Caenorhabditis elegans. J Neurosci 31: 11718–11727. 10.1523/JNEUROSCI.1098-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Richmond JE, Davis WS, Jorgensen EM (1999) UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci 2: 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tokumaru H, Augustine GJ (1999) UNC-13 and neurotransmitter release. Nat Neurosci 2: 929–930. [DOI] [PubMed] [Google Scholar]
- 73.Esposito G, Di Schiavi E, Bergamasco C, Bazzicalupo P (2007) Efficient and cell specific knock-down of gene function in targeted C. elegans neurons. Gene 395: 170–176. [DOI] [PubMed] [Google Scholar]
- 74.Bedner P, Niessen H, Odermatt B, Kretz M, Willecke K, et al. (2006) Selective permeability of different connexin channels to the second messenger cyclic AMP. J Biol Chem 281: 6673–6681. [DOI] [PubMed] [Google Scholar]
- 75.Kanaporis G, Mese G, Valiuniene L, White TW, Brink PR, et al. (2008) Gap junction channels exhibit connexin-specific permeability to cyclic nucleotides. J Gen Physiol 131: 293–305. 10.1085/jgp.200709934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bodor J, Bopp T, Vaeth M, Klein M, Serfling E, et al. (2012) Cyclic AMP underpins suppression by regulatory T cells. Eur J Immunol 42: 1375–1384. 10.1002/eji.201141578 [DOI] [PubMed] [Google Scholar]
- 77.Oh S, Ri Y, Bennett MV, Trexler EB, Verselis VK, et al. (1997) Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron 19: 927–938. [DOI] [PubMed] [Google Scholar]
- 78.Norris RP, Freudzon M, Mehlmann LM, Cowan AE, Simon AM, et al. (2008) Luteinizing hormone causes MAP kinase-dependent phosphorylation and closure of connexin 43 gap junctions in mouse ovarian follicles: one of two paths to meiotic resumption. Development 135: 3229–3238. 10.1242/dev.025494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sagasti A, Hobert O, Troemel ER, Ruvkun G, Bargmann CI (1999) Alternative olfactory neuron fates are specified by the LIM homeobox gene lim-4. Genes Dev 13: 1794–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Troemel ER, Chou JH, Dwyer ND, Colbert HA, Bargmann CI (1995) Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans. Cell 83: 207–218. [DOI] [PubMed] [Google Scholar]
- 81.Ryu MH, Moskvin OV, Siltberg-Liberles J, Gomelsky M (2010) Natural and engineered photoactivated nucleotidyl cyclases for optogenetic applications. J Biol Chem 285: 41501–41508. 10.1074/jbc.M110.177600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Edwards SL, Charlie NK, Milfort MC, Brown BS, Gravlin CN, et al. (2008) A novel molecular solution for ultraviolet light detection in Caenorhabditis elegans. PLoS Biol 6: e198 10.1371/journal.pbio.0060198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hobert O Specification of the nervous system. In: Community TCeR, editor. WormBook: WormBook. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Glauser DA, Chen WC, Agin R, Macinnis BL, Hellman AB, et al. (2011) Heat avoidance is regulated by transient receptor potential (TRP) channels and a neuropeptide signaling pathway in Caenorhabditis elegans. Genetics 188: 91–103. 10.1534/genetics.111.127100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bargmann CI, Marder E (2013) From the connectome to brain function. Nat Methods 10: 483–490. [DOI] [PubMed] [Google Scholar]
- 86.Leinwand SG, Chalasani SH (2013) Neuropeptide signaling remodels chemosensory circuit composition in Caenorhabditis elegans. Nat Neurosci 16: 1461–1467. 10.1038/nn.3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guo M, Wu TH, Song YX, Ge MH, Su CM, et al. (2015) Reciprocal inhibition between sensory ASH and ASI neurons modulates nociception and avoidance in Caenorhabditis elegans. Nat Commun 6: 5655 10.1038/ncomms6655 [DOI] [PubMed] [Google Scholar]
- 88.Rabinowitch I, Chatzigeorgiou M, Schafer WR (2013) A gap junction circuit enhances processing of coincident mechanosensory inputs. Curr Biol 23: 963–967. 10.1016/j.cub.2013.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chao MY, Komatsu H, Fukuto HS, Dionne HM, Hart AC (2004) Feeding status and serotonin rapidly and reversibly modulate a Caenorhabditis elegans chemosensory circuit. Proc Natl Acad Sci U S A 101: 15512–15517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ezcurra M, Tanizawa Y, Swoboda P, Schafer WR (2011) Food sensitizes C. elegans avoidance behaviours through acute dopamine signalling. EMBO J 30: 1110–1122. 10.1038/emboj.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ferkey DM, Hyde R, Haspel G, Dionne HM, Hess HA, et al. (2007) C. elegans G protein regulator RGS-3 controls sensitivity to sensory stimuli. Neuron 53: 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wragg RT, Hapiak V, Miller SB, Harris GP, Gray J, et al. (2007) Tyramine and octopamine independently inhibit serotonin-stimulated aversive behaviors in Caenorhabditis elegans through two novel amine receptors. J Neurosci 27: 13402–13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harris GP, Hapiak VM, Wragg RT, Miller SB, Hughes LJ, et al. (2009) Three distinct amine receptors operating at different levels within the locomotory circuit are each essential for the serotonergic modulation of chemosensation in Caenorhabditis elegans. J Neurosci 29: 1446–1456. 10.1523/JNEUROSCI.4585-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fong A, Schug SA (2014) Pathophysiology of pain: a practical primer. Plast Reconstr Surg 134: 8S–14S. 10.1097/PRS.0000000000000682 [DOI] [PubMed] [Google Scholar]
- 95.Kaminski E, Stawicki S, Wasowicz E (1974) Volatile Flavor Compounds Produced by Molds of Aspergillus, Penicillium, and Fungi imperfecti. Appl Microbiol 27: 1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sharpell FH (1985) Microbial flavors and fragrances In: Moo-Young M, editor. Comprehensive Biotechnology: The principles, applications, and regulations of biotechnology in industry, agriculture, and medicine. 1st ed ed. Oxford, [Oxfordshire]; New York: Pergamon Press; pp. 965981. [Google Scholar]
- 97.Pradel E, Zhang Y, Pujol N, Matsuyama T, Bargmann CI, et al. (2007) Detection and avoidance of a natural product from the pathogenic bacterium Serratia marcescens by Caenorhabditis elegans. Proc Natl Acad Sci U S A 104: 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bargmann CI (1993) Genetic and cellular analysis of behavior in C. elegans. Annu Rev Neurosci 16: 47–71. [DOI] [PubMed] [Google Scholar]
- 99.Chou JH, Bargmann CI, Sengupta P (2001) The Caenorhabditis elegans odr-2 gene encodes a novel Ly-6-related protein required for olfaction. Genetics 157: 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Felix MA, Duveau F (2012) Population dynamics and habitat sharing of natural populations of Caenorhabditis elegans and C. briggsae. BMC Biol 10: 59 10.1186/1741-7007-10-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hu PJ Dauer. In: Community TCeR, editor. WormBook: WormBook. [Google Scholar]
- 102.Sharma RK (2010) Membrane guanylate cyclase is a beautiful signal transduction machine: overview. Mol Cell Biochem 334: 3–36. 10.1007/s11010-009-0336-6 [DOI] [PubMed] [Google Scholar]
- 103.Russwurm M, Russwurm C, Koesling D, Mergia E (2013) NO/cGMP: the past, the present, and the future. Methods Mol Biol 1020: 1–16. 10.1007/978-1-62703-459-3_1 [DOI] [PubMed] [Google Scholar]
- 104.Chalasani SH, Chronis N, Tsunozaki M, Gray JM, Ramot D, et al. (2007) Dissecting a circuit for olfactory behaviour in Caenorhabditis elegans. Nature 450: 63–70. [DOI] [PubMed] [Google Scholar]
- 105.Chuang CF, Vanhoven MK, Fetter RD, Verselis VK, Bargmann CI (2007) An innexin-dependent cell network establishes left-right neuronal asymmetry in C. elegans. Cell 129: 787–799. [DOI] [PubMed] [Google Scholar]
- 106.Milton JG (2011) The delayed and noisy nervous system: implications for neural control. J Neural Eng 8: 065005 10.1088/1741-2560/8/6/065005 [DOI] [PubMed] [Google Scholar]
- 107.Zimmer M, Gray JM, Pokala N, Chang AJ, Karow DS, et al. (2009) Neurons detect increases and decreases in oxygen levels using distinct guanylate cyclases. Neuron 61: 865–879. 10.1016/j.neuron.2009.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wedel BJ, Garbers DL (1997) New insights on the functions of the guanylyl cyclase receptors. FEBS Lett 410: 29–33. [DOI] [PubMed] [Google Scholar]
- 109.Derbyshire ER, Winter MB, Ibrahim M, Deng S, Spiro TG, et al. (2011) Probing domain interactions in soluble guanylate cyclase. Biochemistry 50: 4281–4290. 10.1021/bi200341b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cheung BH, Cohen M, Rogers C, Albayram O, de Bono M (2005) Experience-dependent modulation of C. elegans behavior by ambient oxygen. Curr Biol 15: 905–917. [DOI] [PubMed] [Google Scholar]
- 111.Chang AJ, Chronis N, Karow DS, Marletta MA, Bargmann CI (2006) A distributed chemosensory circuit for oxygen preference in C. elegans. PLoS Biol 4: e274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Qin H, Zhai Z, Powell-Coffman JA (2006) The Caenorhabditis elegans AHR-1 transcription complex controls expression of soluble guanylate cyclase genes in the URX neurons and regulates aggregation behavior. Dev Biol 298: 606–615. [DOI] [PubMed] [Google Scholar]
- 113.Cheung G, Chever O, Rouach N (2014) Connexons and pannexons: newcomers in neurophysiology. Front Cell Neurosci 8: 348 10.3389/fncel.2014.00348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Palacios-Prado N, Huetteroth W, Pereda AE (2014) Hemichannel composition and electrical synaptic transmission: molecular diversity and its implications for electrical rectification. Front Cell Neurosci 8: 324 10.3389/fncel.2014.00324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rouach N, Avignone E, Meme W, Koulakoff A, Venance L, et al. (2002) Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell 94: 457–475. [DOI] [PubMed] [Google Scholar]
- 116.Nakase T, Naus CC (2004) Gap junctions and neurological disorders of the central nervous system. Biochim Biophys Acta 1662: 149–158. [DOI] [PubMed] [Google Scholar]
- 117.Mylvaganam S, Ramani M, Krawczyk M, Carlen PL (2014) Roles of gap junctions, connexins, and pannexins in epilepsy. Front Physiol 5: 172 10.3389/fphys.2014.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fukuto HS, Ferkey DM, Apicella AJ, Lans H, Sharmeen T, et al. (2004) G protein-coupled receptor kinase function is essential for chemosensation in C. elegans. Neuron 42: 581–593. [DOI] [PubMed] [Google Scholar]
- 120.Wood WB (1988) The Nematode Caenorhabditis elegans; researchers tcoCe, editor. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory. [Google Scholar]
- 121.Yu S, Avery L, Baude E, Garbers DL (1997) Guanylyl cyclase expression in specific sensory neurons: a new family of chemosensory receptors. Proc Natl Acad Sci U S A 94: 3384–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shinkai Y, Yamamoto Y, Fujiwara M, Tabata T, Murayama T, et al. (2011) Behavioral choice between conflicting alternatives is regulated by a receptor guanylyl cyclase, GCY-28, and a receptor tyrosine kinase, SCD-2, in AIA interneurons of Caenorhabditis elegans. J Neurosci 31: 3007–3015. 10.1523/JNEUROSCI.4691-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) The loss of UNC-13-dependent synaptic signaling from the AWB head sensory neurons resulted in failure to avoid the aversive odorant 2-nonanone, which is detected by the AWBs [66]. The str-1 (AWB) [66] promoter was used to co-express a non-coding fragment of unc-13 in both the sense and antisense orientations of otherwise wild-type animals. RNAi knock-down of unc-13 in the AWB neurons resulted in diminished avoidance of a 1:10 dilution of 2-nonanone (p < 0.0001 when compared to wild-type animals). (B) Loss of UNC-13-dependent synaptic signaling from the AWC head sensory neurons resulted in failure to chemotax towards benzadehyde, which is detected by the AWCs [60]. The ceh-36p3 (AWC) [65], gpa-4 (ASI) [64] and str-1p (AWB) [66] promoters were used to co-express a non-coding fragment of unc-13 in both the sense and antisense orientations of otherwise wild-type animals. RNAi knock-down of unc-13 in the AWC neurons or in the AWB, AWC or ASI neurons in combination abolished chemotaxis to a 1:200 dilution of benzaldehyde. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Avoidance index = ((A + B)–(E + F)) ÷ total number of animals on the assay plate [66]. Chemotaxis index = (number of animals at odorant − number of animals at control) ÷ total number of animals on the assay plate [60]. Alleles used: odr-1(n1936) loss-of-function and inx-4(ok2373) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function.
(TIF)
The inx-4 promoter [35] was used to drive expression of GFP (inx-4p::gfp). Transgenic animals were incubated with the lipophilic dye DiD (shown in red), which is taken up by the ASH sensory neurons and was used to visualize the neuronal cell body. As previously reported [35], inx-4 expression was seen in the ASH sensory neurons of larval-stage animals, including the L2 stage (top panel). In addition, inx-4 expression was observed in the ASHs of adult animals (bottom panel).
(TIF)
C. elegans respond to aversive stimuli in addition to quinine. (A-C) odr-1(lof) and inx-4(lof) animals responded similarly to wild-type animals to the bitter tastant primaquine, the detergent SDS and the heavy metal copper, across a range of concentrations (p > 0.2 for each concentration). The percentage of animals responding is shown. n ≥ 60 for each. All tastants were dissolved in M13 buffer, pH 7.4. Alleles used: inx-4(ok2373) loss-of-function and odr-1(n1936) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function.
(TIF)
The ASH-selective osm-10 [48] and srb-6 [80] promoters were used to drive expression of a blue light-inducible guanylyl cyclase (BlgC) or blue light-inducible adenylyl cyclase (BlaC) [81] in lite-1(lof) animals. osm-10p drives expression in ASH and weakly in ASI in the head, and in PHA and PHB in the tail. srb-6p drives expression in the ASH, ADL and ADF head sensory neurons, and in PHA and PHB in the tail. Adult lite-1(lof) animals expressing BlgC or BlaC were tested without blue light exposure (white bars) or after a 30 second exposure (grey bars). While lite-1(lof) animals responded robustly to 5 mM quinine, similar to WT animals (p > 0.2), transgenic animals expressing BlgC displayed a significantly diminished response following blue light exposure (p < 0.0001 for each ASH-selective promoter). Transgenic animals expressing BlaC remained sensitive following blue light exposure (p > 0.1 when compared to wild-type animals). The percentage of animals responding is shown. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Allele used: lite-1(ce314) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function.
(TIF)
The ADF-specific srh-142 promoter [79] was used to drive expression of a blue light-inducible guanylyl cyclase (BlgC) or blue light-inducible adenylyl cyclase (BlaC) [81] in lite-1(lof) or inx-4(lof);lite-1(lof) animals. Adult lite-1(lof) or inx-4(lof);lite-1(lof) animals expressing BlgC or BlaC were tested without blue light exposure (white bars) or after a 30 second exposure and 30 second recovery (grey bars). While lite-1(lof) animals responded robustly to 10 mM quinine, similar to WT animals (p > 0.1), transgenic animals expressing BlgC in the ADFs displayed a significantly diminished response following blue light exposure (p < 0.001). Transgenic animals expressing BlaC did not display a diminished response following blue light exposure (p > 0.1 when compared to wild-type animals). inx-4(lof);lite-1(lof) transgenic animals expressing either BlgC or BlaC did not display a diminished response following blue light exposure (p > 0.5 when compared to wild-type animals). The percentage of animals responding is shown. The combined data of ≥ 3 independent lines and n ≥ 120 transgenic animals is shown in each panel. Alleles used: lite-1(ce314) loss-of-function and inx-4(ok2373) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function.
(TIF)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.