

Abstract
Introduction
Non-small cell lung cancer (NSCLC) patients with ROS1 chromosomal rearrangements benefit from treatment with the ROS1 inhibitor crizotinib. Limited data exist on the spectrum of resistance mechanisms in ROS1+ NSCLC. To delineate mechanisms of acquired resistance, we analyzed tumor samples from biopsies of progressing tumor lesions on crizotinib.
Methods
An activating mutation in the KIT proto-oncogene receptor tyrosine kinase (KIT) (p.D816G) was identified by SNaPshot sequencing in a tumor sample from a patient with ROS1-positive NSCLC identified by fluorescence in situ hybridization whose disease progressed after initial response to crizotinib. In vitro studies included evaluation of KIT mRNA expression by qRT-PCR, transduction of Ba/F3 cells and NSCLC cell lines with lentiviral plasmids modified by site-directed mutagenesis, immunoblotting and MTS cellular proliferation assays.
Results
KITD816G is an activating mutation that induces autophosphorylation and cell proliferation. Expression of the mutant KITD816G receptor in ROS1+ NSCLC cell lines led to constitutively activated KIT as measured by phosphorylation of the KIT receptor. Expression of the KITD816G rendered the HCC78 and CUTO2 cell lines resistant to crizotinib and only dual inhibition of ROS1 and KIT with crizotinib and ponatinib could resensitize the cells to proliferation inhibition. The oncogenic switch observed in ROS1+ cell lines was not immediate and required pharmacologic inactivation of ROS1.
Conclusions
Activation of KIT by a gain-of-function, somatic mutation is a novel mechanism of resistance to crizotinib in ROS1 rearranged NSCLC. This bypass-signaling pathway serves as a ROS1-independent mechanism of resistance, similarly to previously identified EGFR or RAS signaling pathways, and can potentially be targeted by KIT inhibitors.
Introduction
Chromosomal rearrangements or deletions involving the ROS proto-oncogene 1, receptor tyrosine kinase (ROS1) generate fusion genes that are expressed as chimeric oncoproteins.1 ROS1 fusions occur in approximately 1-2% of non-small cell lung cancers (NSCLC), but have also been identified in multiple other tumors.1-3 Treatment of patients with NSCLC and other malignancies using the ROS1 inhibitor crizotinib often generates tumor regression that is durable.4-5 Despite this initial success, disease progression inevitably ensues. Although there are emerging data on acquired drug-resistance in ROS1+ cancers, little remains known about the full spectrum of mechanisms of acquired drug resistance in this disease. Resistance to crizotinib can occur through the clonal outgrowth of cells harboring kinase domain mutations that decrease sensitivity to crizotinib.6 In vitro data suggest that structurally unique kinase inhibitors may be able to overcome drug resistance mutations in ROS1.7 Bypass signaling via either EGFR or RAS has also been described and may be amenable to single agent or combinatorial targeted therapy to overcome drug resistance in these cases.8,9
In an ongoing effort to understand the mechanisms by which oncogene-driven cancers become resistant to kinase inhibitors, we have performed directed analysis of tumor samples from NSCLC patients treated with oncogene-targeted therapy.10 We describe here a novel mechanism of drug resistance, an activating KIT mutation, in a ROS1+ patient with acquired resistance to crizotinib.
Materials and Methods
Patient Samples
Written informed consent was obtained from the patient prior to tumor sample analyses.
Fluorescence in situ Hybridization
Break-apart ROS1 FISH analysis was designed and performed as previously described in the Molecular Pathology Shared Resource of the University of Colorado Cancer Center.2 KIT FISH was performed using the Vysis LSI KIT SpectrumRed and CEP4 SpectrumAqua (4p11-q11 alpha satellite DNA) probes (Abbott Molecular).
KIT and ROS1 DNA Sequencing
The post-treatment sample was manually microdissected to enrich for tumor, followed by DNA extraction (Qiagen, Inc.). Molecular testing of samples was performed using a multiplex single nucleotide base extension assay (SNaPshot®, Life Technologies, Carlsbad, CA), designed to detect > 60 point mutations in 15 genes targets, including in EGFR, KRAS, BRAF, KIT and others as previously described.11 SNaPshot was performed in the Molecular Pathology Shared Resource of the University of Colorado Cancer Center.
KIT mRNA expression was determined by RT-PCR. RNA was extracted from both pre- and post-progression tumor samples followed by reverse transcription and PCR amplification as previously described (KIT primers available upon request).2 Exons 35-43 of the ROS1 gene were amplified from genomic DNA and sequenced as previously described (ROS1 primers available upon request).10
Cell Lines and Reagents
HCC78 and CUTO2 cell lines have been previously described and were cultured in RPMI1640 supplemented with 10% FBS.2,8 Ba/F3 cells and Ba/F3 cells expressing the SDC4-ROS1 cDNA have previously been described.2 Crizotinib and ponatinib were obtained from Selleck Chemicals (Boston, MA).
Lentiviral Expression Constructs and Transduction
Lentiviral plasmids expressing wild type KIT and KIT D816V were a kind gift from Dr. Ronnstrand.12 Site-directed mutagenesis of the c.2447A>G (encoding the D816G substitution) was accomplished using the Quik Change II kit (Life Technologies, primers available upon request). The Genbank accession number used for KIT is NM_000222. Lentiviral transduction of HCC78 and CUTO2 cells and subsequent puromycin selection was performed as previously described.2
Immunoblotting and Antibodies
Immunoblotting was performed as previously described.2 Antibodies for pKIT Y703 (D12E12), Y719 (3391), total KIT (Ab81), pROS1 Y2274 (3078), total ROS1 (D4D6), pSHP-2 Y542(3751), total SHP-2 (D50F2), pAKT S437 (4058), total AKT (2920), pERK1/2 T202/Y204 (D13.14.4E), and total ERK1/2 (L34F12) were purchased from Cell Signaling (Danvers, MA). 4G10 antibody and anti-GAPDH (MAB274) antibodies were obtained from Miilipore (Bedford, MA). Anti-mouse and anti-rabbit secondary IR anitibodies were purchased from LiCor Biosciences (Lincoln, Nebraska)
Cellular Proliferation
Cell proliferation assays using Ba/F3 cells expressing the indicated cDNA constructs in the absence of drug were performed by manual cell counting at each time point. Cell proliferation assays to measure drug inhibition were performed using MTS assays as previously described.2
Results
Case report of a ROS1+ NSCLC patient with acquired resistance to crizotinib
A 38-year-old woman with a smoking history of 15 pack years initially presented with severe dyspnea, cough and weight loss and she was ultimately diagnosed with stage IV lung adenocarcinoma with papillary features.13 Break-apart fluorescence in situ hybridization (FISH) analysis revealed a ROS1 gene rearrangement and the patient was treated with crizotinib (Fig. 1A). A partial response was achieved and was accompanied by symptomatic improvement (Figure 2A,B). Disease progression in the chest was noted after approximately 15 months on crizotinib therapy and a biopsy of the growing tumor was obtained by CT-guided biopsy (Figure 2C).
Figure 1. FISH analyses for ROS1 and KIT in patient tumor samples.
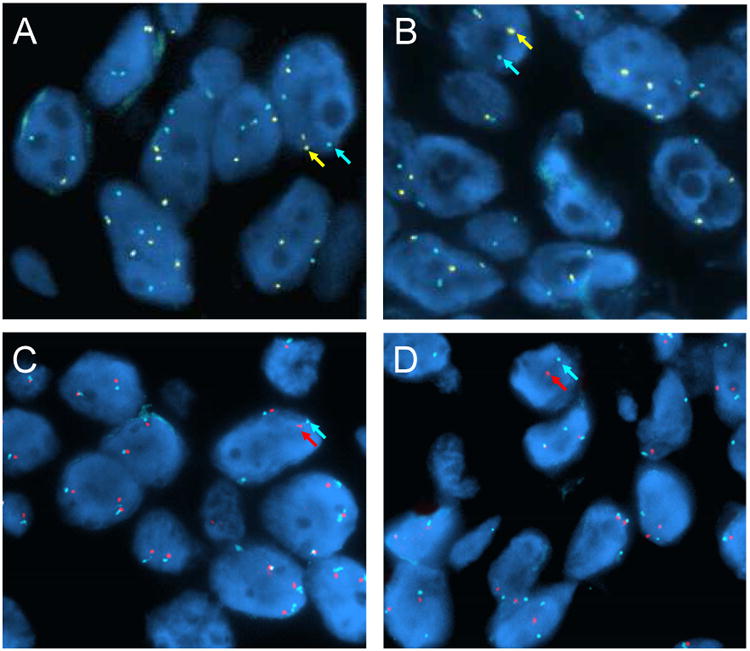
FISH analysis was performed on the patient's tumor samples before crizotinib treatment (A,C) and at the time of progression on crizotinib (B,D). ROS1 break apart FISH demonstrates no gain or loss of ROS1 fusion gene copies in the post-crizotinib sample (B) compared to the pre-crizotinib sample (A). 5′ ROS1 probes are indicated in yellow and 3′ ROS1 probes are indicated in aqua. Normal, unrearranged copies are indicated by white arrows. KIT FISH demonstrates no evidence of KIT gene amplification in the post-crizotinib sample (D) compared to the pre-treatment sample (C). The KIT probe is indicated in red and the centromere enumeration probes from chromosome 4 (CEP4) in aqua.
Figure 2. Crizotinib-resistance in a ROS1 NSCLC patient.
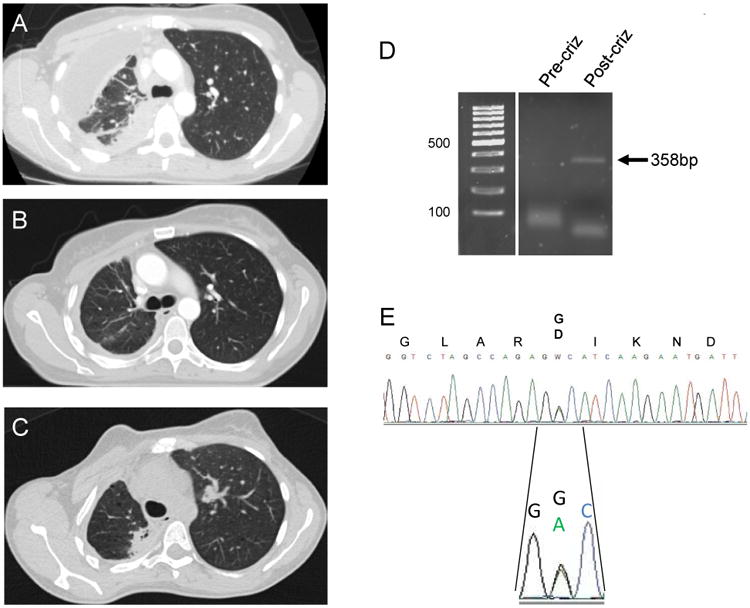
Computed tomography (CT) scans demonstrating pre-treatment disease (A), response to therapy on crizotinib (B) and disease-progression on crizotinib (C) in a ROS1+ NSCLC patient. (D) Reverse transcriptase polymerase-chain (RT-PCR) reaction of a region of the KIT kinase domain in pre- and post-crizotinib treatment tumor biopsies. The correct-sized PCR product is indicated by an arrow and is only observed in the post-crizotinib tumor sample. (E) Sanger DNA sequencing of the RT-PCR product observed in (D), demonstrating the presence of the mutation c.2447A>G encoding the KIT p.D816G amino acid substitution. Expression of both the wildtype and mutant KIT mRNA was observed.
Analysis of tumor specimens
We performed ROS1 break-apart FISH analysis on the crizotinib-resistant tumor specimen to determine whether there was any gain or loss of the ROS1 fusion gene as previously observed for ALK and ROS1 drug resistance in patient samples and/or in vitro.8,10 No change in ROS1 gene fusion copy number was observed (Fig. 1A,B). DNA sequencing of the exons encoding the ROS1 kinase domain did not demonstrate the presence of any mutations (not shown). Analysis of the tumor sample for other activating mutations in proto-oncogenes using a multi-analyte targeted assay demonstrated the presence of a KIT gene mutation encoding the amino acid substitution, p.D816G (not shown). No other gene mutations were identified using this targeted assay. Analysis of the pre-crizotinib tumor biopsy demonstrated wild-type KIT sequence and also did not detect any other mutations (not shown). RT-PCR analysis failed to demonstrate mRNA expression of KIT in the pre-crizotinib specimen, but demonstrated expression in the progression sample (Figure 2D). Sequence analysis of this transcript in the post-progression sample re-identified the KIT p.D816G mutation (Figure 2E). KIT gene amplification has previously been shown to be a mechanism of crizotinib resistance in ALK+ patients.14 We therefore determined KIT gene copy number in this sample and in three other ROS1+, crizotinib tumor samples. No evidence of KIT gene amplification was found in any of the four samples tested (Tab. 1). One other sample had sufficient tissue for KIT mutation analysis and demonstrated a wildtype sequence. None of the samples tested had evidence of a ROS1 kinase domain mutation.
Table 1. Molecular analysis of re-biopsy samples of evaluable ROS1+ patients.
| Patient number | Age | Sex | Smoking status (py) | Histology | Possible Mechanisms of Resistance | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| ROS1 Kinase Domain | KIT mutation | KIT FISH | |||||
| 1 | 38 | M | 0 | Large cell carcinoma | NA | NA | Not amplified |
| 2 | 65 | M | 0 | Adenocarcinoma | WTa | WTb | Not amplified |
| 3 | 37 | F | 15 | Adenocarcinoma | WTa | KIT p.D816Gb | Not amplified |
| 4 | 41 | M | 0 | Adenocarcinoma | NA | NA | Not amplified |
direct sequencing
SNaPshot® analysis
FISH, fluorescence in situ hybridization
NA, not available due to insufficient tissue
Modeling of KITD816G in ROS1+ models
Although other substitutions at this position, KITD816V/Y/F, have been shown to induce constitutive activation of the KIT kinase domain, KITD816G has not been previously evaluated to determine whether it is activating.15 We therefore first introduced a cDNA expressing the KITD816G or the KITWT as a control into 293T cells. Immunoblot analysis demonstrated increased KIT autophosphorylation for the KITD816G, compared to wild-type KIT (KITWT) (Fig. 3A). KITD816G has been demonstrated as a secondary resistance mutation identified in gastrointestinal stromal tumors (GIST) that harbor a primary KIT activating mutation following treatment with imatinib.16 In vitro data suggests that ponatinib has more potency against KITD816G compared to other commonly used KIT inhibitors.17 We therefore evaluated whether treatment with ponatinib could inhibit activity of the KITD816G kinase activity in these cells. Treatment of 293T cells expressing KITD816G with ponatinib shows inhibition of KIT phosphorylation (Fig. 3A). Notably, ponatinib demonstrates greater inhibition of phosphorylation of KITD816G than KITD816V. Expression of KITD816G in Ba/F3 cells induced IL-3 independent growth similar to WT KIT supplemented with SCF or KITD816V, a known activating mutation (Figure 3B). Ba/F3 cells expressing KITD816G or KITD816V were resistant to crizotinib in comparison to Ba/F3 cells expressing the SDC4-ROS1 fusion gene and showed similar sensitivity to control Ba/F3 cells supplemented with IL-3 (Figure 3C). Conversely, Ba/F3 cells expressing KITD816G or KITD816V were sensitive to ponatinib in comparison to Ba/F3 cells expressing SDC4-ROS1, which had a similar IC50 to control Ba/F3 cells (Figure 3D).
Figure 3. KITD816G is an activating mutation and can be inhibited by ponatinib.
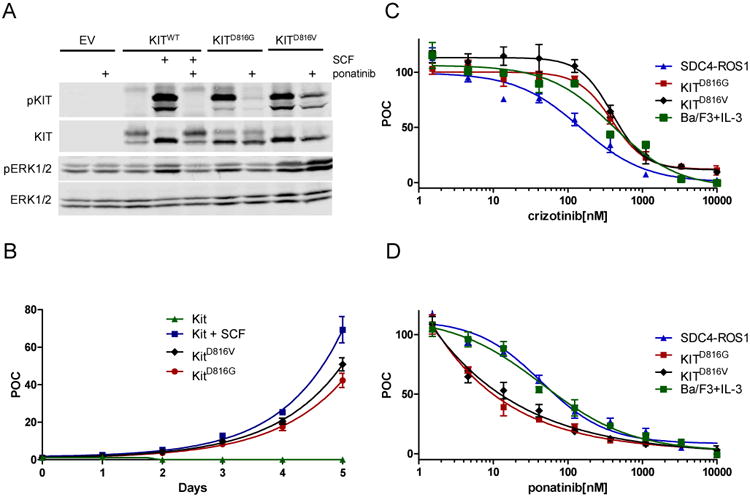
(A) KITWT, KITD816G, KITD816V or an empty vector construct was expressed in 293T cells by transient transfection. Cells were treated with 100 ng/ml stem cell factor (SCF) for 20 minutes and/or 500 nM ponatinib for 2 hours where indicated. Protein expression and phosphorylation were measured by western blot. KITD816G is autophosphorylated in the absence of its cognate ligand SCF and can be inhibited by ponatinib. KITD816V is also autophosphorylated but is inhibited to a lesser degree by ponatinib. (B) Cellular proliferation of Ba/F3 cells expressing WT KIT with or without 100ng SCF, KITD816G or KITD816V mutation in the absence of supplemental IL-3 was measured by cell counting at the indicated intervals. Cell number was plotted as percent of control (POC) relative to day 0 counts. WT KIT with SCF, KITD816G and KITD816V induced cell proliferation, whereas WT KIT in the absence of SCF failed to support cellular proliferation. (C) Cell proliferation assays were performed on Ba/F3 cells expressing SDC4-ROS1, KITD816G, KITD816V, or parental BA/F3 cells supplemented with IL-3 in the presence of the indicated doses of crizotinib (C) or ponatinib (D). Cells were assayed for cell proliferation using an MTS assay (n = 3, error bars represent ± S.E.M.). (C) Ba/F3-SDC4-ROS1 cells, but not KITD816G, KITD816V, or BA/F3 parental cells, are sensitive to crizotinib. The half-maximal inhibitory concentration (IC50) of crizotinib was calculated for each cell line as follows: Ba/F3-SDC4-ROS1, 112.3 ± 26.7 nM; Ba/F3--KITD816G, 391.2 nM ± 4.7 nM; Ba/F3-KITD816V, 393.8 ± 6.1 nM; Ba/F3 parental + IL-3, 444.5 ± 15.9 nM. (D) Ba/F3-KITD816G and -KITD816V cells, but not SDC4-ROS1 or BA/F3 parental cells, are sensitive to ponatinib. The IC50 of ponatinib was calculated for each cell line as follows: Ba/F3-Ba/F3-KITD816G, 9.9 ± 2.5 nM; Ba/F3--KITD816V, 12.3 nM ± 2.3 nM; Ba/F3-SDC4-ROS1, 73.4 ± 12.2 nM; Ba/F3 parental + IL-3, 68.3 ± 13.1 nM.
We introduced cDNAs encoding KITWT, the KITD816G observed in the patient sample or another activating KIT mutation, D816V (KITD816V), or an empty vector into two ROS1+ NSCLC cell lines, HCC78 and CUTO2,2,8 and measured cell proliferation in the presence of crizotinib. KITWT had no effect on proliferation compared to the empty vector control in either ROS1 cell line, but stimulation with the cognate ligand, stem cell factor (SCF) induced partial resistance to crizotinib (not shown). Introduction of KITD816V (not shown) or KITD816G induced substantial resistance to crizotinib in both CUTO2 and HCC78 (Figure 4A,B).
Figure 4. KITD816G induces resistance to crizotinib in ROS1+ cell line models.
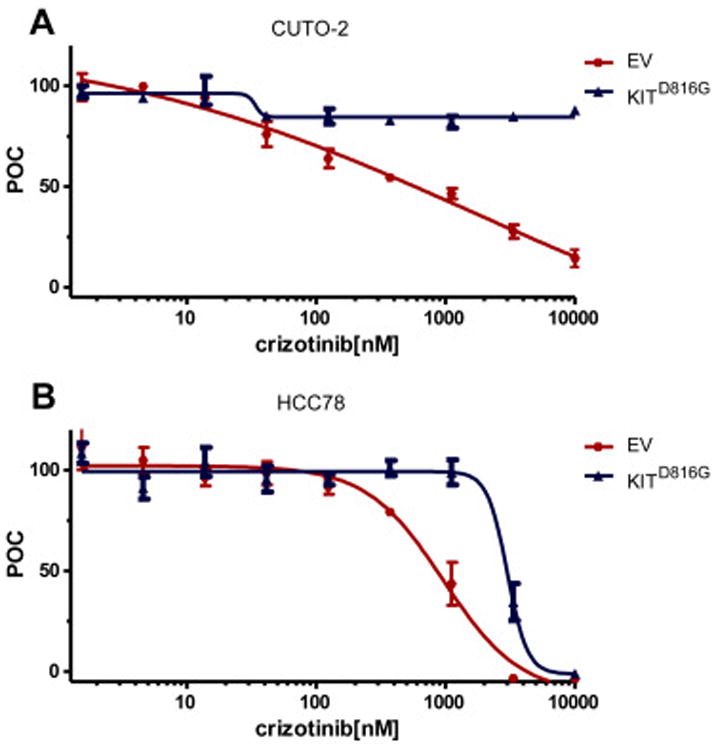
Human NSCLC cell lines CUTO2 (A) and HCC78 (B) were transduced with lentivirus expressing KITD816G (blue) or an empty vector control (red). Cells were treated with the indicated dose range of crizotinib and assayed for cell proliferation using an MTS assay (n = 3, error bars represent ± S.E.M.). The IC50 was calculated for each cell line as follows: CUTO2 EV, 598 ± 110 nM; CUTO2 KITD816G, not reached at 10μM, HCC78 EV, 872 ±138 nM; HCC78 KITD816G, 3222 ±138 nM.
We next interrogated ROS1 and KIT signaling in CUTO2 and HCC78 cells expressing the KITD816G activating mutation. Treatment of the CUTO2 KITD816G cells with the ROS1 inhibitor crizotinib resulted in reduction of phosphorylated ROS1 and its adaptor SHP2 similar to CUTO2 cells transduced with the empty vector (Fig 5. A,B). ROS1 inhibition by crizotinib inhibits ERK1/2 activation at 24 hours, suggesting that ROS1 was still the primary oncogene driver at this timepoint (Fig. 5A). Ponatinib monotherapy inhibits phospho-KIT but has no effect on AKT or ERK1/2 activation. Dual inhibition of ROS1 and KIT was required to maximally inhibit AKT and ERK1/2 at both 24 and 72 hours, suggesting that either oncogene was sufficient to activate these signaling pathways in CUTO2 cells.
Figure 5. Analysis of signaling in ROS1+ cells harboring KITD816G.
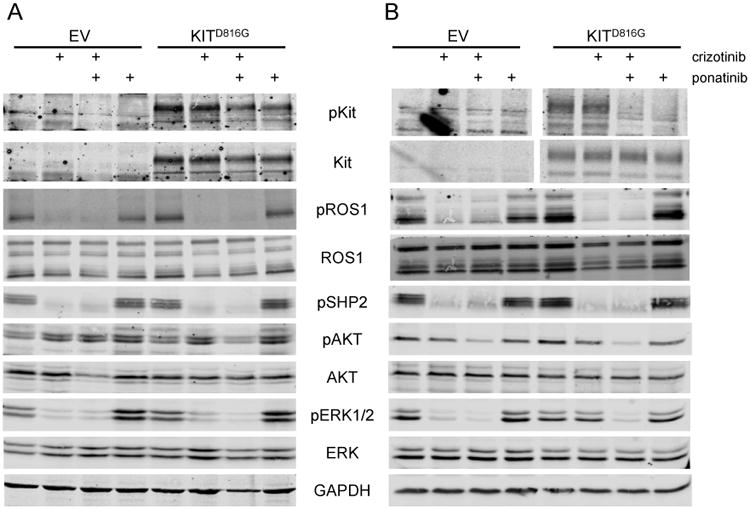
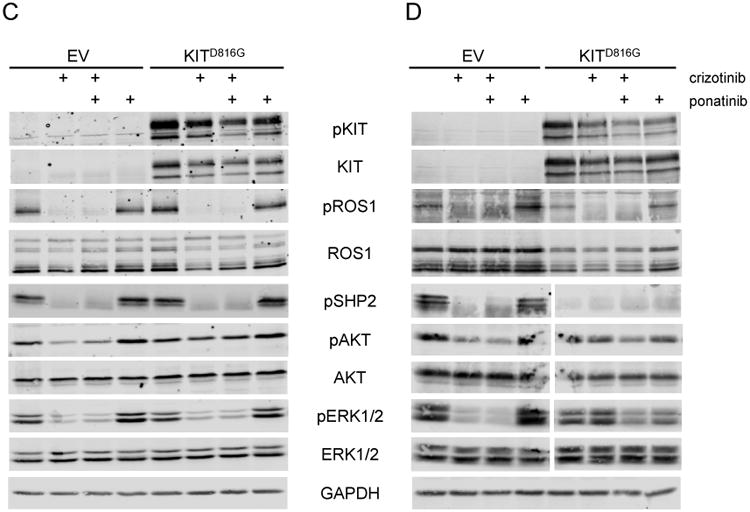
Human NSCLC cell lines CUTO2 (A,B) and HCC78 (C,D) transduced with lentivirus expressing KITD816G or an empty vector (EV) control. CUTO2 cells were treated with 500nM crizotinib for 24 hours (A) or 72 hours (B) followed by drug washout for 2 hours. HCC78 cells were treated with 1 μM crizotinib for 24 hours (C) or 72 hours (D) followed by drug washout for 2 hours. Cells were then treated with crizotinib or 500 nM ponatinib for 2 hours as indicated and protein expression and phosphorylation was measured for the indicated proteins.
We performed similar experiments in the HCC78 cell line. Notably, in HCC78 cells expressing KITD816G, ROS1 was still primarily regulating ERK1/2 activation at 24 hours following crizotinib treatment (Figure 5C). At 72 hours, however, ERK1/2 activation was no longer inhibited by crizotinib alone, but was inhibited by ponatinib. Ponatinib treatment alone inhibited ERK1/2 activation at 72 hours and was similar to combination therapy with ponatinib and crizotinib at 72 hours (Fig. 5D). Furthermore, the ROS1 adaptor SHP2 was no longer phosphorylated at 72 hours in the HCC78 KITD816G cells in the absence of crizotinib (Fig. 5D), whereas pSHP2 was observed in the CUTO2 KITD816G cells at 72 hours (Fig. 5C). These data suggest that KIT activation cannot immediately replace the function of ROS1 in HCC78 cells and requires prolonged suppression of ROS1 before the oncogenic switch from ROS1 to KIT can occur.
To determine whether KIT inhibition could overcome KIT-mediated crizotinib resistance, we performed cell proliferation experiments in ROS1+ cells expressing empty vector or the KITD816G construct. In ROS1+ cells expressing KITD816G addition of ponatinib resensitized cells to crizotinib (Fig 6).
Figure 6. Combination ROS1 and KIT inhibition overcomes crizotinib-resistance mediated by KITD816G.

Human NSCLC cell lines CUTO2 (A) and HCC78 (B) transduced with lentivirus expressing KITD816G and were treated with the indicated dose range of crizotinib alone (blue) or crizotinib plus a fixed dose ponatinib at 500nM (red). Cells were assayed for cell proliferation using an MTS assay (n = 3, error bars represent ± S.E.M.). The IC50 was calculated for each cell line as follows: CUTO2-KITD816G crizotinib only, not reached at 1000nM; CUTO2-KITD816G crizotinib plus ponatinib, 489nM ± 29 nM; HCC78-KITD816G crizotinib only, >3000nM; HCC78-KITD816G crizotinib plus ponatinib, 271 ± 12 nM.
Discussion
Here we describe a novel mechanism of crizotinib resistance in a ROS1+ NSCLC patient. The KITD816G mutation identified in the genomic DNA of the crizotinib-resistant ROS1+ tumor sample and confirmed by sequencing of the mRNA by RT-PCR was deemed a somatic mutation that was acquired following drug treatment with crizotinib as it was not present in the pre-treatment sample. We demonstrate that the KITD816G mutation, which has previously been identified as a secondary resistance mutation in GIST, is itself oncogenic by showing that it can promote autophosphorylation and proliferation in cells. We demonstrate that an activating mutation in KIT can drive resistance in vitro using two different ROS1+ cell lines and demonstrate that this cellular resistance can be overcome by the addition of a KIT inhibitor to crizotinib. This mechanism of resistance adds to a growing list of acquired crizotinib-resistance observed in patients or cell line models. KITD816G serves as a bypass signaling mechanism of resistance similar to that described for RAS and EGFR because it occurs independently of ROS1.8,9 KIT gene amplification has been observed as a mechanism of bypass signaling in ALK+ lung cancer treated with crizotinib.14 Although KIT kinase domain mutations are a common mechanism of acquired resistance to imatinib in tumors harboring KIT activating mutations,18 this is the first example of a KIT activating mutation acting as a bypass signaling resistance mechanism to a different oncogene.
This bypass mechanism of resistance is in contrast to ROS1 resistance mutations that maintain ROS1 resistance despite the presence of a ROS1 inhibitor.6,7 This distinction between oncogene-dependent and –independent resistance is important because it may dictate whether a more potent inhibitor can be used to overcome resistance (in the case of a resistance mutation) or whether combination therapy will be needed (in the case of bypass signaling mechanism).19 The degree of drug resistance induced by KITD816G was notably different between the two ROS1 cell lines suggesting that the cellular context may an important role beyond the class of oncogene. Although both ROS1+ cell lines expressing KITD816G require dual ROS1 and KIT inhibition to restore drug sensitivity, dissection of cell signaling following ROS1 inhibition suggests two different reasons for the necessity of dual inhibition. In CUTO2 cells both ROS1 and KIT oncogenic signaling remain active and can engage downstream signaling such as MAPK. In HCC78 cells, crizotinib inhibition induces a change in dependence from ROS1 to KIT over a period of time. Notably, following 72 hours of crizotinib treatment followed by drug washout, ROS1 phosphorylation and signaling via SHP2 is re-engaged in CUTO2 cells, but not in HCC78 cells and correlates with a switch in dependence to KIT. This kinase inhibitor-induced reprogramming of cells has been described previously for the BCR-ABL oncogene.20 Notably, the KIT gene expression was only observed in the post-crizotinib patient tumor sample, but not in the pre-treatment tumor sample. This emergence of KIT gene expression could occur either by clonal selection or derepression of KIT gene expression following ROS1 inhibition.
KIT activating mutations, such as KITD816G, can be detected in many standard clinical assays used both at tumor diagnosis and following acquisition of drug-resistance. Although our results demonstrate that ponatinib can overcome KIT-mediated resistance in vitro, it remains unknown whether ponatinib can overcome this or other KIT activation mutations at the D816 position in patients. Detection of this mutation may allow enrollment of patients with ROS1+ cancer (or other oncogenes), onto clinical trials of KIT inhibitors, however it is likely that dual inhibition of both KIT and ROS1 would need to be maintained.
Acknowledgments
This work was made possible by generous support from the Christine J. Burge Endowment for Lung Cancer Research at the University of Colorado Cancer Center, the Burge family, and the Miramont Cares foundation. Drs. Doebele, Varella-Garcia, and Aisner acknowledge funding from the University of Colorado Lung Cancer SPORE (P50 CA058187); Drs. Varella-Garcia, and Aisner also acknowledge support from the University of Colorado Cancer Center Support Grant (P30 CA046934).
Disclosure: RD has received honoraria from Pfizer, Novartis, Roche, Clovis, Boehringer-Ingelheim and Tesaro. ATL has received licensing fees from Abbott Molecular and GVKbio. JJ has received honoraria from Roche, Boehringer-Ingelheim and AstraZeneca; research grant from GSK. RCD has received honoraria from Pfizer, OxOnc, Loxo Oncology, Array BioPharma, Ariad, Clovis and AstraZeneca; research grants from Pfizer, Mirati Therapeutics, Loxo Oncology and Abbott Molecular; Licensing fees from Abbott Molecular, Ariad Pharmaceuticals, Chugai, Blueprint Medicines, and GVKbio.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davies KD, Doebele RC. Molecular pathways: ROS1 fusion proteins in cancer. Clin Cancer Res. 2013;19:4040–4045. doi: 10.1158/1078-0432.CCR-12-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies KD, Le AT, Theodoro MF, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. 2012;18:4570–4079. doi: 10.1158/1078-0432.CCR-12-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le AT, Doebele RC. The democratization of the oncogene. Cancer Discov. 2014;4:870–872. doi: 10.1158/2159-8290.CD-14-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw AT, Ou SA, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–1971. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lovly CM, Gupta A, Lipson D, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4:889–895. doi: 10.1158/2159-8290.CD-14-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awad MM, Engelman JA, Shaw AT. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368:2395–2401. doi: 10.1056/NEJMoa1215530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davare MA, Saborowski A, Eide CA, et al. Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc Natl Acad Sci USA. 2013:19519–19524. doi: 10.1073/pnas.1319583110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies KD, Mahale S, Astling DP, et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS One. 2013;8:e82236. doi: 10.1371/journal.pone.0082236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cargnelutti M, Corso S, Pergolizzi M, et al. Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget. 2015;6:5182–5194. doi: 10.18632/oncotarget.3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dias-Santagata D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2:146–158. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pedersen M, Ronnstrand L, Sun J. The c-Kit/D816V mutation eliminates the differences in signal transduction and biological responses between two isoforms of c-Kit. Cell Signal. 2009;21:413–418. doi: 10.1016/j.cellsig.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 13.Dziadziuszko K, Szurowska E, Pienkowska J, et al. Milliary brain metastases in a patient with ROS1-rearranged lung adenocarcinoma: a case report. J Thorac Oncol. 2014;9:e34–e36. doi: 10.1097/JTO.0000000000000091. [DOI] [PubMed] [Google Scholar]
- 14.Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Longley BJ, Longley BJ, Jr, Metcalfe DD, et al. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci U S A. 1999;96:1609–14. doi: 10.1073/pnas.96.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–9. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 17.Garner AP, Gozgit JR, Anjum R, et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin Cancer Res. 2014;20:5745–5755. doi: 10.1158/1078-0432.CCR-14-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–5359. doi: 10.1200/JCO.2007.15.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camidge DR, Doebele RC. Treating ALK-positive lung cancer – early successes and future challenges. Nat Rev Clin Oncol. 2012;9:268–277. doi: 10.1038/nrclinonc.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asmussen J, Lasater EA, Tajon C, et al. MEK-dependent negative feedback underlies BCR-ABL-mediated oncogene addiction. Cancer Discov. 2014;4:200–215. doi: 10.1158/2159-8290.CD-13-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]