

Tau protein can transfer between neurons transneuronally and trans-synaptically, which is thought to explain the progressive spread of tauopathy observed in the brain of patients with Alzheimer’s Disease. Here we show that physiological tau released from donor cells can transfer to recipient cells via the medium, suggesting that at least one mechanism by which tau can transfer is via the extracellular space. Neuronal activity has been shown to regulate tau secretion, but its effect on tau pathology is unknown. Using optogenetic and chemogenetic approaches, we found that increased neuronal activity stimulates the release of tau in vitro, and enhances tau pathology in vivo. These data have implications for disease pathogenesis and therapeutic strategies for Alzheimer’s disease and other tauopathies.
Misfolded, hyperphosphorylated forms of the microtubule stabilizing protein tau accumulate as neurofibrillary tangles in Alzheimer’s disease (AD). In the earliest stages of the disease, tau is found in the somatodendritic compartment of neurons located in the transentorhinal cortex (EC)1. Tau pathology spreads medially and worsens in the EC; it then accumulates within limbic areas, followed by neocortical areas in a pattern that suggests spread along neuroanatomical connections1. Various mouse models have been created to replicate the progressive spread of pathology including those using regional promoters2–4, inoculation models5–9 and viral models10,11 and it is now well established that tau has the capability to transfer between cells both in vivo and in vitro8,12. Two hypotheses have been proposed to explain the propagation of tau pathology. First, intracellular tau could transfer directly between cells through physical connections such as tunneling nanotubes, as shown in vitro for misfolded proteins such as prion13. Alternatively, tau could be released into the extracellular space, either as free tau or within vesicles such as exosomes14–16 or ectosomes17. Data showing that tau is present in the cerebrospinal fluid (CSF) and interstitial fluid (ISF) of tau transgenic mouse lines18–20 and the CSF and ISF of AD brain21 together with data showing tau is released into the medium of cultured neurons22 support the second hypothesis. Although there is strong evidence that tau is released from cells, the competence of extracellular tau generated in vitro, by neurons, to contribute to cell to cell propagation has not been tested. Using physiologically relevant culture systems, including human iPSCs, we have shown that tau released from donor cells into the medium can be internalized by recipient cells where it can stimulate the generation of more aberrant tau. Furthermore, using a tripartite microfluidic system, we have shown that the aberrant tau generated in recipients can be passed to subsequent recipient cells indicating true propagation.
The mechanism by which tau can be released from neurons is unknown, however its release can be stimulated by enhanced neuronal activity both in vitro22 and in vivo23. As cells within the AD13 brain could be hyperexcitable24–28, the impact of enhanced neuronal activity on tauopathy is of significant interest. Using sophisticated optogenetic and chemogenetic manipulations we have demonstrated that increasing neuronal activity can not only enhance release and transfer of tau in vitro, but also exacerbate tau pathology in vivo.
Results
Endogenously generated tau transfers from cell to cell
To examine whether physiologically relevant, human tau (hTau) can transfer between neurons, we cultured neurons derived from a transgenic mouse line expressing hTau P301L (line rTg4510, “donor”)29 with neurons from a MAPT−/−:GFP (tau knockout-GFP line (KO, “recipient”)30 (Fig. 1a). After 2 weeks, hTau accumulated in the cytosol and neurites of recipient cells grown on plates (Fig. 1b, cell C) and in microfluidic chambers (MFs, Fig. 1c, arrows). To determine whether highly aggregated forms of tau have a greater propensity for neuron-to-neuron transfer, we compared the effects of two types of seeds on tau transfer; clone 9 seeds that have been shown to induce the formation of robust aggregates of tau, or clone 1 seeds that do not induce the formation of aggregates of tau8. In this experiment, the tau that is expressed in donor cells was fluorescently tagged (repeat domain RD P301S YFP) so that it could be directly visualized within the recipients. Lysate containing clone 1 seeds did not readily induce any obvious aggregates in donor cells even after extensive culturing (20 days in vitro). In contrast, when lysate containing clone 9 seeds was added, YFP-tagged endogenous tau began forming perinuclear aggregates in cells after 5 days in vitro (DIV). The number of cells forming tau aggregates increased dramatically after 20 DIV, demonstrating that template induced tau aggregation is time-dependent (Fig. 2a). To assess whether induced tau aggregates were transferred to recipients, seeded donor cells were co-cultured with mCherry-expressing recipient neurons. YFP-tau from clone 9 seeded cells, but not clone 1 seeded cells readily accumulated in the cytoplasm of recipient cells (Fig. 2b, cell A, B; Fig. 2c). Controls included neurons expressing YFP but no tau, co-cultured with neurons expressing mCherry. In the absence of tau, no transfer of YFP or mCherry was seen (data not shown).
Figure 1.
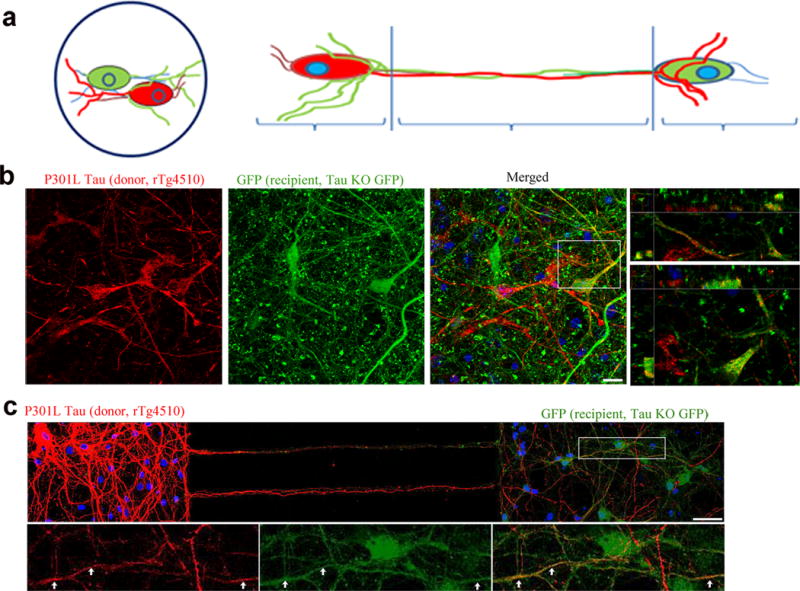
Endogenously generated human tau can transfer from cell to cell. (a) Cartoon model showing co-cultures of neurons on coverslips and in microfluidic chamber devices. Co-cultures of mutant hTau-expressing (red, from rTg4510 line, cell A) and recipient KO neurons (green, from KO line, cells B, C). (b) hTau in recipient KO cell (cell C, yellow) on coverslip and (c) in MFs (yellow, arrows). Scale bars, 10 and 50 μm, respectively. Images are representative of four neuronal cultures.
Figure 2.
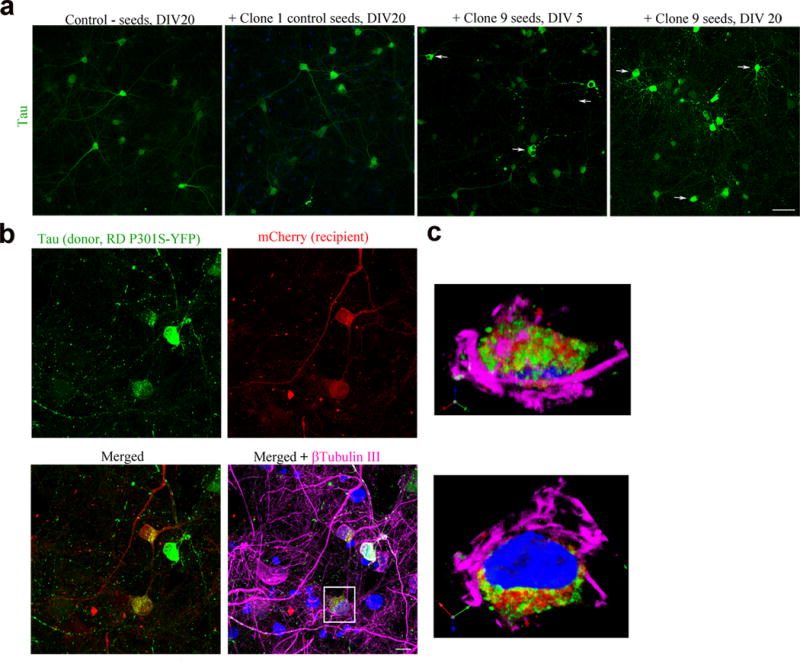
Endogenously generated human tau aggregates can transfer from cell to cell. (a) Tau repeat domain-expressing neurons (hTau RD P301S-YFP, green) do not readily form aggregates when exposed to PBS or clone 1 lysate after 20 DIV. Clone 9 lysate seeded tau cells start forming aggregates (arrow) at 5 DIV and increase after 20 DIV. Scale bar, 50 μm. (b) Seeded tau RD aggregates (green) transferred to recipient cells (mCherry, red) (cell A, B, yellow). Scale bar, 10 μm. (c) Enlarged orthogonal view of cell A showing transferred tau in the cytoplasm of the cell. Images are representative of four neuronal cultures.
To test whether aggregate formation can be induced in downstream cells, we turned to a bi- and tripartite microfluidic chamber device (Figs. 3a,d). The chambers allow axons from one compartment to grow through microchannels into an adjacent compartment while isolating cell bodies and dendrites within one compartment. Importantly, the fluidically isolated microenvironment in each chamber allows for the delivery of agents such as seeds to one cell population without crossing over into the next compartment9,31. RD YFP neurons were grown in each chamber. Unseeded tau expressing cells did not form visible aggregates (Fig. 3b); however, adding clone 9 seeded lysate to cells in the first compartment (population 1) induced endogenous YFP-tagged tau to form aggregates (Fig. 3c). Endogenous tau in neurons grown in the second compartment (population 2) formed aggregates after 12–15 DIV in response to aggregated tau being formed and transferred from population 1 (Fig. 3c). To confirm true cell-to-cell tau propagation, we utilized tripartite MFs and added seed-containing lysate only to neurons in the first compartment (population 1). We then examined whether tau aggregates could propagate not only to the second compartment (population 2), but also to the third compartment (population 3) (Fig. 3d). After 15–18 DIV, tau aggregates were found in some population 3 neurons (Fig. 3e). The misfolded conformation of these aggregates was confirmed by staining with X-34, a fluorescent congo red derivative that binds to neurofibrillary tangles (Fig. 3f)32.
Figure 3.
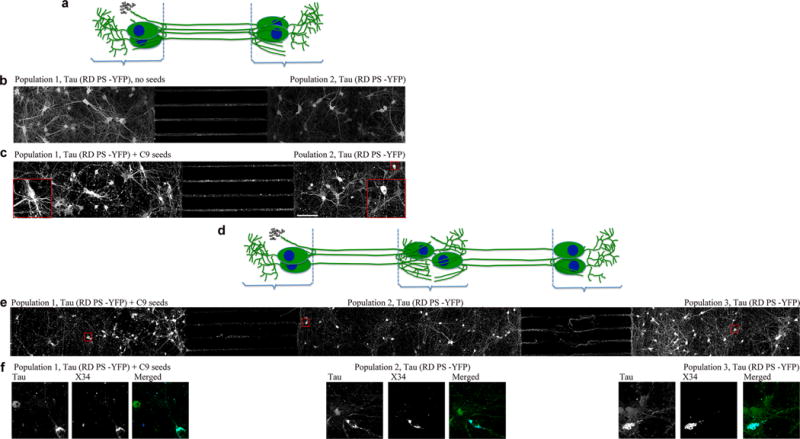
Seed-induced tau pathology propagates from cell to cell. (a) Cartoon model showing co-cultures in microfluidic devices with two chambers. (b) Unseeded tau-expressing cells (population 1, 2) grown in bipartite chamber device do not form visible aggregates. (c) Seeding population 1 with clone 9 lysate triggered endogenous tau to form aggregates in population 1 and population 2. Images are representative of three cultures. (d) Cartoon model showing co-cultures in microfluidic devices with three chambers. (e) Pathology propagated from population 1 cells to population 3 cells grown in tripartite chambers. Scale bars, 100 μm. Enlarged images show tau aggregate-containing neurons in population 1, 2 and 3. Images are representative of two cultures. (f) Tau aggregate-containing neurons from populations 1, 2, and 3 stained with X-34.
Tau transfers to recipients via the extracellular medium
We next examined whether tau transfer from donor neurons to recipient neurons requires cell-to-cell contact, or if tau released into the extracellular fluid is sufficient. Lysate and conditioned medium (CM) from primary neurons overexpressing tau, or neurons infected with tau virus was collected, tau was immunoprecipitated using a human tau specific antibody and analyzed by western blot analysis. Tau was detected in the media of both cell models (Fig. 4a). Actin was present in lysates but absent in media fraction demonstrating that the media was not contaminated with cell debris. Next, we incubated wild-type (wt) recipient neurons with CM from hTau-expressing donor cells for the following incubation periods: 1 hour, 6 hours, 12 hours, 1 day or 6 days. Recipient neurons accumulated tau within hours and had a significant amount of tau accumulation by 6 days (Fig. 4b). Immunofluorescence staining revealed that the internalized tau initially accumulated in discrete puncta in the cytosol and dendrites of recipient neurons after 2 days (Fig. 4c) but it later appeared more diffusely distributed in cell bodies and neurites (Fig. 4c). To confirm that tau was released into the medium from cells expressing it at physiological levels, we examined induced pluripotent stem cells (iPSC) that were differentiated into cortical neurons by dual SMAD inhibition followed by an extended period of in vitro corticogenesis (Figs. 4d,e)33. In vitro corticogenesis using this method has been shown to take up to 90 days, producing all classes of glutamatergic projection neurons that make mature, functional synapses that are capable of firing repetitive action potentials34. Immunoblotting of whole cell lysates at time points throughout differentiation (Fig. 4d) showed a single band that was immunoreactive for tau at all time points from day 20 onwards. Comparison with a recombinant tau ladder confirmed this was the 0N3R tau isoform, consistent with previous reports that iPSC-neurons mainly express fetal tau isoform and not the adult isoform ratio of equivalent 3R:4R35. Cortical progenitor rosettes positive for the early forebrain marker Pax6 and the proliferation marker Ki67 were observed at day 20 of differentiation (Fig. 4e). At this time point, tau was observed in a subset of cells but it did not colocalize with Ki67, suggesting that tau expression is restricted to post-mitotic neurons. By day 100 of differentiation, the majority of cultured cells showed stereotypic neuronal morphology such as high levels of tau expression and extended neurites. To assess whether tau could be internalized, we transferred CM from iPSCs to mouse primary wt neurons. After six days, human tau was observed in the neurites of the recipient cells with a similar distribution pattern to the tau generated by the transgenic mouse neurons (Fig. 4f). This result is consistent with a recent finding showing tau from mouse and human brain extracts, and the ISF from the rTg4510 line are internalized by recipients20.
Figure 4.
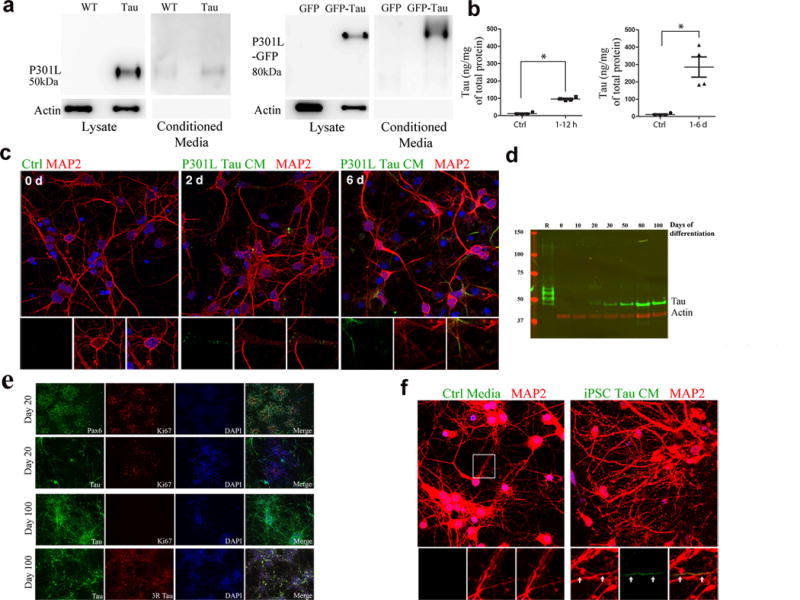
Tau from mouse primary neurons and human iPSCs can transfer via the extracellular medium (a) Lysate from rTg4510 primary neurons and P301L-GFP transduced neurons, and conditioned media from the same cells, labeled with TauC (full-length blots shown in Supplementary Figure 4). Actin shows equal amounts of protein loaded. (Full-length blots shown in Supplementary Figure 4.) (b) ELISA using tau-specific antibodies showing uptake of tau from media by tau-KO cells. Compared to controls (n = 4 cultures), tau was significantly higher in neurons after 1–6 hours of incubation with tau-conditioned media (n = 6, z = −2.558, adjusted P = 0.021) (left), and also after 1–6 days of incubation (n = 4, z = −2.309, adjusted P = 0.042). (c) Neurons (wild-type), incubated with rTg4510 conditioned media for 2 and 6 days (d), labeled with anti-tau antibody (green) and anti-MAP2 (red). Insets show tau in cell bodies and neurites. (d) Total tau in human iPSC neuron lysates. Recombinant tau ladder (R) separates in the following order of decreasing molecular weight: 2N4R, 2N3R, 1N4R, 1N3R, 0N4R, 0N3R. The single tau band corresponds to the 0N3R isoform. (e) iPSC cultures immunolabeled with the early forebrain marker Pax6 and tau, which was only expressed in post-mitotic neurons. By day 100 of differentiation, the majority of the cells in culture were immunoreactive for total and 3R tau. Scale bar, 100. (f) Neurons (wild-type) treated for 6 d with tau conditioned media collected from iPSC neurons, stained with antibodies (anti-tau, green, anti-MAP2, red) and DAPI (blue). Enlarged insets show tau (green) accumulating inside neurites (yellow). Scale bar, 10 μm.
Neuronal activity stimulates tau release and spread
Increased neuronal activity has been shown to stimulate tau release in wild type and human tau-overexpressing transgenic mouse models in vitro22 and in vivo23, however, it is not known if stimulation leads to release of tau from human neurons with physiological levels of tau. To examine this, we measured the amount of tau released into media from human iPSC neurons, as well as murine tau expressing neurons (wild type) and neurons overexpressing mutant human tau (rTg4510 line), before, and after treatment with picrotoxin, a non-competitive channel blocker for the GABA receptor chloride channels36 (Fig. 5a). In all three systems, stimulation with picrotoxin increased tau in the media by 150% – 210% compared to tau levels in the media prior to the addition of picrotoxin, or compared to media from neurons treated with buffer alone, without causing cell death (Fig. 5b). To examine whether increasing neuronal activity leads to enhanced tau release, we used an optogenetic approach in one of our culture models to directly stimulate neuronal action potentials37. Cells expressing channelrhodopsin (ChR2) can be depolarized by exposure to blue light at 473 nm wavelength. Primary neurons from a wt mouse were transfected with fluorescently tagged hTau and ChR2 expressing viral vectors (Figs. 5c,d). Media was removed and analyzed for human tau levels before, and after light stimulation. Whole cell recording was performed simultaneously to confirm activation (Figs. 5e,f). After neurons were stimulated for 30 min (0.1 mW mm−2, 2 s intervals), tau in the media increased approximately 250% (Fig. 5g), similar to the amount of tau released from neurons following picrotoxin treatment (Fig. 5a). Optogenetic stimulation did not result in cell death as shown by patch clamp recording of individual cells (Figs. 5e,f) and more globally, by lactate dehydrogenase (LDH) assessment (Fig. 5h). To test whether stimulating neuronal activity can enhance cell-to-cell transfer of tau in addition to tau release, we added picrotoxin to co-cultures of neurons that made hTau aggregates (RD YFP with C9 seeded lysate) or mCherry (Fig. 6), and quantified the percent of mCherry cells containing YFP tau aggregates. After two weeks of co-culturing, neurons were fixed and examined using confocal microscopy (Fig. 6a). Quantification of IF images demonstrated that stimulating neuronal activity increased the percentage of cells that had taken up tau from approximately 20% of mCherry cells to 45% of mCherry cells (Fig. 6b).
Figure 5.
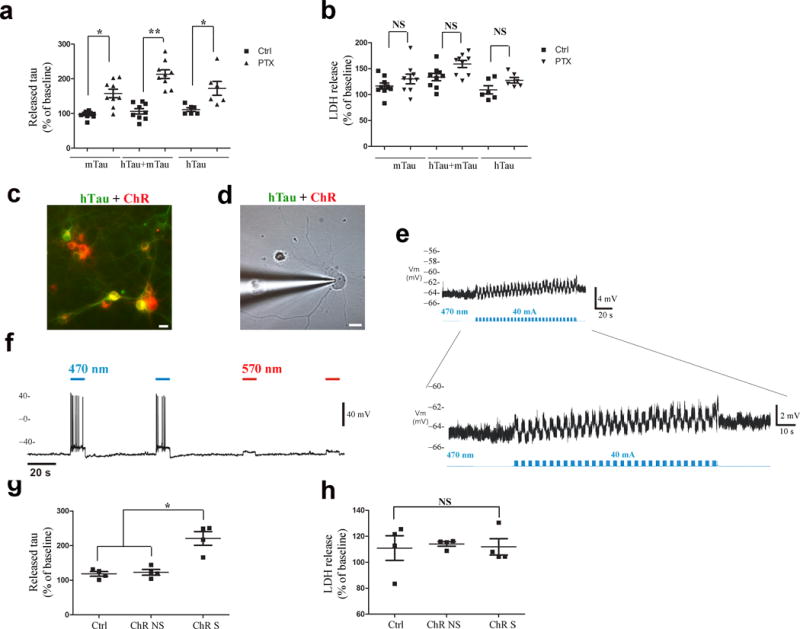
Tau release is enhanced by stimulating neuronal activity. (a) Sandwich ELISA showed that tau is released into the media from cultured cells (mTau, n = 9 cultures per treatment group; hTau + mTau, n = 9 cultures per treatment group; hTau, human and iPSC neurons, n = 6 cultures per treatment group), when treated with picrotoxin for 30 min. Raw data was normalized to baseline levels for each cell population, and plotted as the percent change. A significant difference was observed between treated and untreated groups for each model: mTau t(9.167) = −4.75, Bonferroni adjusted P =0.003; hTau + mTau t(16) = −7.25, adjusted P < 0.0001; hTau t(10) = −2.95, adjusted P = 0.044. (b) No significant difference was observed between treated and untreated groups in LDH release. Group n’s were as above. P values from adjusted t-tests = 0.73, 0.061, and 0.233. (c) Transduced neurons co-express both tau (green) and ChR2 (red). (d) Tau and ChR2-expressing neuron was patch-recorded and (e) exhibited ~5 mV depolarization of membrane potential (RMP) with light stimulation at 30 Hz. (f) Light at 470 nm, but not 570 nm caused a 12mV depolarization in ChR2-cells. (g) Significant difference in tau released after stimulation (30 min) (n = 4 cultures per condition) (Kruskal-Wallis χ2(2) = 7.42, P = 0.024) with significantly higher tau in stimulated cells compared to non-stimulated (adjusted P = 0.036) and control neurons (adjusted P = 0.021). (h) LDH release measured before and after treatment did not differ significantly χ2(2) = 0.731, P = 0.694 (n = 4 cultures per condition).
Figure 6.
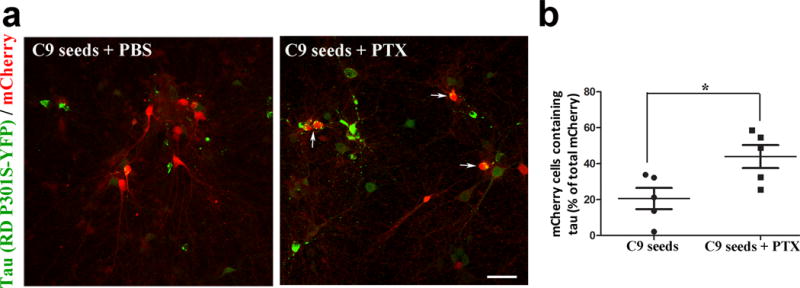
Transfer of tau from cell to cell is enhanced by stimulating neuronal activity. (a) Images showing tau protein (green) from tau-expressing donor cells in recipient mCherry cells (red) (merged in yellow) seeded with clone 9 lysate. Data shows cells without, and with picrotoxin (+PTX) stimulation. Scale bar, 50 μm. (b) Tau transfer, as determined by the number of mCherry cells containing tau, as a percentage of the total number of mCherry cells, was significantly higher in the stimulated group (n = 5) compared to the unstimulated group (n = 5); t(8)= −2.68, P = 0.028.
Enhanced neuronal activity accelerates tauopathy in vivo
Tau has been shown to accumulate in the ISF following administration of picrotoxin in vivo23, however it is not known if enhancing neuronal activity accelerates tauopathy. To address this important question, we used two approaches to increase neuronal activity in two different tau mouse models. Using an optogenetic approach, ChR2 was expressed in neurons under the CamKII promoter37 for 10 days and optical fibers were implanted in both left and right hippocampi of the rTg4510 line, but only the left hippocampus was stimulated with blue light (470 nm). Tau pathology in the left (stimulated) hemisphere was compared to the right (non stimulated) hemisphere (Fig. 7a). In vivo recordings confirmed that blue light induced an increase in hippocampal activity (Fig. 7b). Neuronal firing in response to blue light ON was instantaneous with minimal time lag (<10 ms) and the firing rate approximately doubled during stimulation. When the blue light was OFF, the firing rate returned to normal. The EEG and spike firing pattern did not show any epileptic-like phenotype and seizures were not observed (data not shown). Recording from the right hemisphere showed that ChR2-controlled firing did not lead to firing in the non-stimulated hemisphere (data not shown). After 20 days of stimulation, mice were sacrificed within 75 mins of the last stimulation, and the brain tissue was processed for immunolabeling of the activity-induced immediate early gene, c-Fos, which was readily observed in the stimulated hippocampus (Fig. 7c). When immunolabeled with anti-tau antibodies, we observed that the stimulated hippocampus had accumulated more human tau (labeled with TauY9 and CP27 antibodies) in the cell bodies. This was particularly evident in the posterior and anterior planes, especially in CA1 and CA3 pyramidal cells, (Figs. 7d,e, insets) which was consistent with the placement of the optical fiber. Enhanced tau pathology was accompanied by hippocampal cell layer atrophy (Fig. 7e) that was more evident in brain tissues stained using Nissl (Fig. 7f,h). Optogenetic stimulation using the same parameters did not result in detectible atrophy in mice without tauopathy (a MAPT−/− line (Figs. 7g,h) suggesting the stimulation itself was not causing the atrophy. To ensure that enhanced tauopathy was not due to enhanced expression of the transgene, we performed quantitative RT-PCR on both stimulated and non-stimulated hippocampal tissue from mice stimulated for 20 d. While there was variable human tau expression between mice, we did not detect differences in tau expression between hemispheres of rTg4510 mice (Supplementary Fig. 1).
Figure 7.
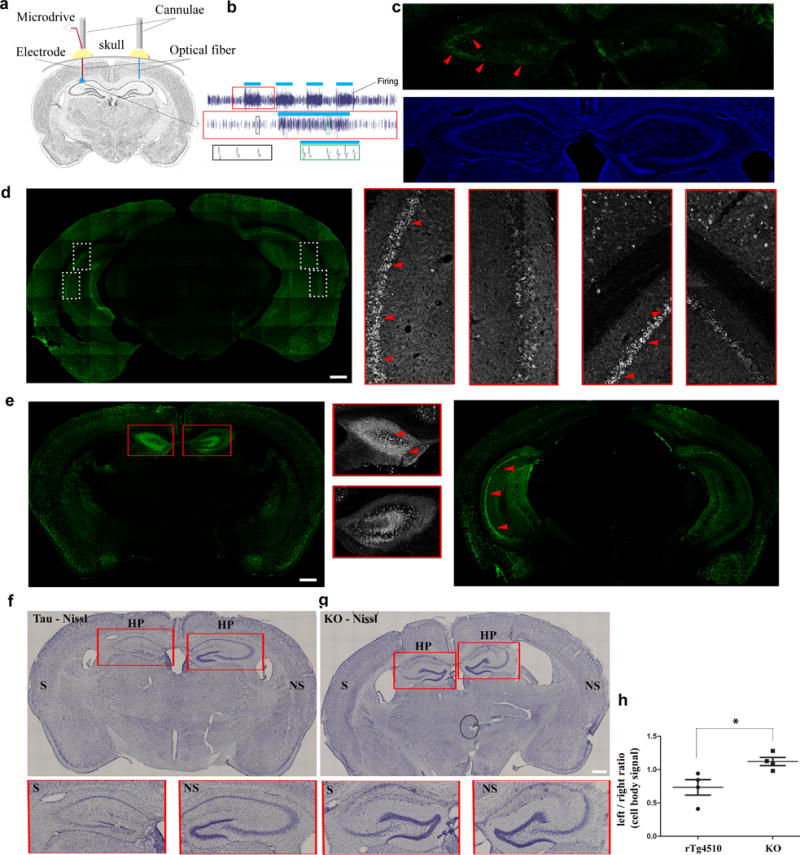
Increased neuronal activity induced optogenetically exacerbates tau pathology in the hippocampus. (a) In vivo optogenetic stimulation: Cartoon shows the experimental setup. Tau expressing mice (rTg4510) were injected with ChR2 expressing vector and implanted with optical fibers in both hemispheres; in some mice a recording electrode was also inserted in one or both hemispheres. (b) Extracellular recordings show that pulses of blue light (blue bars) increased the firing activity consistently. The same neurons could be recorded longitudinally demonstrating that chronic, repetitive stimulation did not impact cell viability. Red boxed inset: zoom-in of stimulation #15. Bottom row: example waveforms from stimulated (green inset) vs. unstimulated (black inset) neurons. (c) Representative image of c-Fos immunostaining (d) Brain tissues of stimulated mice labeled with anti-tau antibody (TauY9, green) Scale bar, 500 μm. n = 2 mice. White boxed inset: enlargements of CA1 and 3. (e) Posterior and anterior tissues labeled with anti-tau antibody (CP27, green) shows more tau pathology (arrows, red) in the stimulated side. Red boxed inset: enlargements of anterior hippocampus. (f, g) Nissl stain of brain tissues of stimulated tau mouse and tau-KO (KO-GFP line) shows reduced cell body staining, especially in the hippocampus. Scale bar, 500 μm. n = 4 mice for each line. (h) Assessment of Nissl stain signal of hippocampal pyramidal cell layers shows significant reduction in the stimulated hemisphere of the tau mice (n = 4) but not the tau-KO mice (n = 4 mice, z = 2.31, P = 0.021). S = stimulated hemisphere, NS = non-stimulated hemisphere.
To confirm our results using a different stimulation approach, in a different mouse line, we used the Designer Receptor Exclusively Activated by Designer Drug (DREADD)38 chemogenetic approach to transiently stimulate neurons in the entorhinal cortex (EC) of the EC-Tau mouse line2. The clozapine-n-oxide (CNO)-responsive DREADD (CaMKIIa-hM3D(Gq)-mCherry) was expressed in the left EC and a receptor that was unresponsive to CNO (CaMKIIa-ChR-mCherry) was expressed in the right EC as a control. The DREADD was expressed in the MEC for at least 10 days before stimulation, and viral expression remained stable for at least 6 weeks (Fig. 8a). CNO administration enhanced neuronal activity in the MEC approximately 20 min post injection without causing seizures, as shown by in vivo electrophysiology (data not shown), and enhanced c-Fos labeling in the stimulated EC compared to the non-stimulated EC (Fig. 8b). After 6 weeks of CNO administration, we observed increased accumulation of somatodendritic tau (antibody MC1) in neurons in the stimulated EC as compared to the non-stimulated EC of the same mouse (Fig. 8c, outlined, arrows, insets, low laser power images). Stimulation for 2 weeks also showed enhanced tauopathy, but to a lesser degree (Supplementary Fig. 2a). Images taken using higher laser power demonstrated that the increase in somatodendritic tau was most evident in the EC region where DREADD virus was expressed whereas tau pathology in other regions (for example the granule cells of the dentate gyrus) did not show any obvious enhancement (Fig. 8c). A similar enhancement of tau pathology was observed using a second human specific tau antibody, CP27 (Supplementary Fig. 3b, arrows). Non-specific staining was not observed when the secondary antibody was used in the absence of the primary antibody (data not shown).
Figure 8.
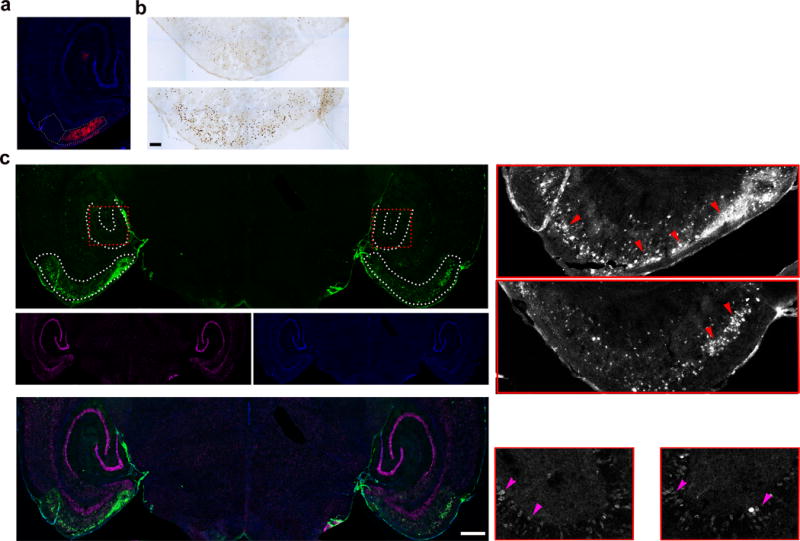
Increased neuronal activity induced chemogenetically accelerates tau pathology in the EC. (a) CaMKIIa-hM3D(Gq)-mCherry (red) was injected and expressed in the MEC of the left hemisphere of EC-Tau mice. (b) Representative image of c-Fos immunostaining. Scale bar, 100 μm. (c) Representative image of brain tissues of mice stimulated for six weeks labeled with an antibody against pathological human tau (MC1, green), anti-NeuN (magenta) and DAPI (blue). Inset: Enlarged images from the entorhinal cortex (EC) and the dentate gyrus (DG) of stimulated and non-stimulated hemispheres. Red arrows, somatodendritic tau in neurons of the EC. Magenta arrows, tau in granule cells of the DG. DAPI (blue). Scale bar, 500 μm, n = 3 mice. S = stimulated hemisphere. NS = non-stimulated hemisphere.
Discussion
Using a variety of in vitro cell models, we have shown that 1) neuron-derived full-length tau can transfer to recipients and that aggregated tau induced by seeding transfers efficiently, 2) that neuron-derived tau can not only be internalized by recipient neurons, but it can undergo transcellular propagation to distant cells, and 3) that neuron-derived tau (including tau from human iPSCs) can transfer to recipients via the extracellular space. Several studies have shown internalization of exogenously-added tau8,9,12,39,40 into recipient cells, while more recent studies show that endogenously generated tau can also be internalized, including tau that was released into the extracellular space either in conditioned medium from mutant tau expressing SH-SY5Y cells41 or the ISF from rTg4510 mice23. In our study we found that non-mutant tau from wt mice and from non-mutant human iPSCs can also be released and internalized. Tau in these cells does not form overt, morphologically distinct aggregates even when cultured for long periods of time (over 20 days, data not shown), suggesting that the majority of tau released from these neurons is likely to be soluble which is consistent with a study that showed that soluble tau is released into the medium of cells22, the ISF of transgenic and wt mice18,23, and the ISF of an anti-aggregating mouse model42. Whether the soluble tau from wt neurons or iPSCs is monomeric or oligomeric is unknown. Exogenously added monomeric tau can be internalized41 but in our experience9, it does not accumulate to any degree. It is possible, especially for neurons from the rTg4510 line, that small, early stage misfolded tau oligomers have formed in the cultures that are not readily discernable by morphology, nor by dyes such as X-34 that recognize more aggregated beta-sheet structures and that these oligomers are released into the media and taken up by recipient neurons. What is clear from our seeding experiments however is that higher order aggregated forms of tau can accumulate in recipients more readily. A recent study23 has examined the uptake of tau from brain extract and has shown that a low abundance, higher molecular weight, phosphorylated form of tau is internalized and propagates. The same study showed that both high and low molecular weight tau is present in the ISF from rTg4510 mice, and while tau in the ISF was shown to be internalized by recipients, it is not known if it was the same type of tau that was internalized from the lysate. In our experience, tau released from cells making aggregates promotes propagation much more robustly than tau from neurons making soluble tau. Taking advantage of this, we have now shown that tau aggregates generated within one population of neurons can induce tau misfolding (X-34 positive aggregates) in a fluidically isolated, second- and third population of neurons cultured in microfluidic chambers.
The fact that tau can be transferred between cells via the media suggests that structures such as tunneling nanotubes are not required, at least not for in vitro transfer (in vivo transfer mechanisms are not known). We do not yet know whether tau in the media is vesicle bound or free. Exogenously added free (recombinant) tau12,39,43 including oligomers9 has been shown to be internalized by bulk endocytosis, and uptake can be mediated by binding to heparan sulfate proteoglycans43. Exogenously added tau in cell or brain extract can also be internalized by recipient cells8,44,45, but it is not known whether it is free or vesicle-bound in these preparations. A small proportion of tau released from cells (for example, into the ISF of wt tau overexpressing rodents17) has been identified in vesicles (ectosomes and to a lesser extent exosomes). Tau has only been found in ectosomes, and not exosomes when it is expressed at physiological levels17. Importantly, using wt neurons and iPSCs we have shown that tau release and internalization can occur without overexpressing tau. How internalized tau accesses endogenous tau to enable templating is currently unknown but the creation of more physiologically relevant models of tau release, internalization and propagation such as those described here will allow us to address these questions better. Taken together, our data show that endogenously produced tau aggregates are released and can induce subsequent misfolding and seed formation in downstream neurons, resulting in cell-to-cell propagation that could explain the widespread distribution of tau pathology in AD.
One of the observations from transgenic mice overexpressing the amyloid precursor protein (APP) is that elevated amyloid beta (Aβ) is somehow associated with hyperexcitability at the cellular level24–28. The observation that cellular hyperexcitability can stimulate the release of tau in vitro22 and in vivo23 suggests that in humans, Aβ-induced hyperexcitability within regions of the brain that are vulnerable to early tauopathy, such as the hippocampal formation, could lead to enhanced release of pathogenic tau and accelerated propagation of tauopathy through circuits. In support of this, we have shown that optogenetically stimulated tau mice undergo robust worsening of pathology (accumulation of cell body tau) in the stimulated hippocampus. Of note, the worsened pathology appears to be neurotoxic as it correlated with exacerbated hippocampal cell layer atrophy. The EC-tau mouse model, when stimulated chemogenetically, demonstrated worsening tauopathy in the stimulated entorhinal cortex. Additional pathology in cell populations that are synaptically connected with the EC such as the dentate gyrus granule cell layer was not obvious. However, in the EC-tau line, the accumulation of tau in the granule cell layer is a slow process, which takes more than 18 months to become apparent2. In our experiments, the EC was stimulated for a maximum of 6 weeks, starting at an age when pathology was mild. More time may be needed to effectively induce tau propagation into the granule cells of the dentate gyrus, or the mice may need to be at a more advanced stage when first stimulated for it to become apparent within the relatively short timeframe. In general, these data have several implications. Firstly, they may have relevance for AD where therapeutic approaches to dampen down excitability46–48 may retard the spread of tau pathology, which, if performed during the earliest Braak stages, could prevent the onset of cognitive decline. Second, these results may explain the observation of tauopathy associated with epileptic seizures, although the link between brain injury and tauopathy obscures the relationship49. Lastly, there may be negative implications for stimulation therapies such as deep brain stimulation (DBS), or transcranial magnetic stimulation (TMS) that are currently in clinical trials for AD patients.
Methods
Transgenic mice
Protocols and procedures were approved by the Committee on the Ethics of Animal Experiments of Columbia University and according to Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Mouse models used in this study included: rTg4510 (CAMKII:hTau-P301L, embryonic day 15–16 for culture, 2–4 months for in vivo studies, parental lines were a gift of K. Hsiao22, MAPT−/− GFP (Tau-KO-GFP) (embryonic day 15–16, parental line was a gift of K. Tucker)30, Tau-KO (Jackson Labs), and EC-Tau (neuropsin-tTA, hTau-P301L line r4510, 12–14 months, parental lines were a gift of M. Mayford and K. Hsiao)2. Both male and female mice were used for each in vivo experiment. Strains of mice were C129/FVB F1 (rTg4510), C57blk6 (hTau, tau KO).
Neuronal culture
Primary neuronal cultures were prepared and maintained as previously described9. For co-cultures, hippocampal and cortical neurons were isolated from male and female rTg4510 and Tau-KO-GFP mouse lines, plated into separate reservoirs of the MFs or together at 1:1 ratio on coverslips. For co-cultures of transduced neurons, male and female wild type rat primary neurons were plated in MFs and then transduced at 3–5 DIV with either mCherry (1:1000) or tau-GFP (hTau-P301L-GFP or GFP alone, 2 × 109 particles), or RD P301S YFP (1:10) vectors. Amounts of tau protein generated in cultured neurons was as follows: P301L GFP transduced neurons: 9.28 ± 0.675 ug mg (tau per total protein) (n = 3 cultures); rTg4510 neurons: 7.84 ± 1.97 ug mg (tau per total protein) (n = 12 cultures); WT neurons: 2.81 ± 1.14 ug mg (tau total protein) (n = 12 cultures); iPSC neurons: 6.05 ± 1.07 ug mg (tau per total protein) (n = 12 cultures). For seeding experiment, 2.5 μg of clone 1, or clone 9 lysates prepared as previously described were added to neurons that were transduced with RD P301S YFP at DIV 6–8, and cultured for an additional 9–10 days.
Neural Induction
iPSC were differentiated into cortical neurons. Briefly, iPSC were converted to neural epithelium using the dual SMAD inhibitors dorsomorphin and SB431452, followed by an extended period of in vitro neurogenesis to generate cortical glutamatergic neurons. Neurons were maintained in 1:1 mixture of N-2 medium and B-27 medium33. Medium was changed every 48 h. Neuron-conditioned media were prepared by collecting media after 48 h, centrifuging at 3,000 × g for 10 min to remove cell debris and stored at −80 °C.
Immunoprecipitation
Neurons were transduced with either tau-GFP or GFP (75,000 neurons/well, tau-P301L-GFP, 2 × 109 particles, GFP, 2 × 109 particles). Media from wild-type (wt), GFP expressing, or tau expressing cells was collected every 3–4 days from mature neurons (tau or tau-GFP, 14–21 DIV) and centrifuged at 3,000 × g for 10 min at 4 °C to remove cell debris. At the end of collection, cells were harvested in RIPA buffer with protease and phosphatase inhibitors (1 μg ml−1, Sigma). For immunoprecipitation of tau, magnetic anti-mouse protein A dynabeads (Life Technologies) were incubated with human tau-specific antibody (CP27, mouse) at a 2:1 ratio O/N, washed 3 times with 0.1% BSA in PBS and incubated with either conditioned media or cell lysates for 2 h at 4 °C. Beads were isolated using a magnetic stir bar and resuspended in loading buffer for western blot analysis.
Immunoblot
Immunoprecipitated samples were prepared and analyzed by immunoblot as previously described with the exception that membranes were probed with rabbit anti-tau (1:2000, Dako) and anti-actin (1:5000) antibodies. For human iPSC western blot, cells were harvested in buffer containing protease and phosphastase inhibitors (Roche). Equal amounts of protein were dephosphorylated using lambda protein phosphatase (NEB) prior to separation on 4–12% NuPage gels (Invitrogen). Recombinant tau ladder (Sigma) was used to identify specific tau isoforms. Proteins were transferred to nitrocellulose membranes and blocked with 3% milk in PBS before incubation with anti-tau antibody (total tau, DAKO) O/N at 4 °C. Membranes were incubated with IRDye 800 conjugated goat anti-rabbit (Rockland Inc) and proteins were visualized using the Odyssey Infrared imaging system (LiCor Biosciences). For western blot analysis of human iPSC neurons, cells were lysed in 10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% deoxycholate, plus protease and phosphastase inhibitors (Roche) for 1h at 4 °C. Proteins were separated on SDS-PAGE BisTris gels (NuPAGE Novex, 4–12%, Invitrogen) and subsequently transferred onto nitrocellulose membranes. Membranes were blocked in phosphate buffered saline containing 3% milk (PBS-M) for 1hr at RT. Membranes were incubated in primary antibody (DAKO polyclonal antibody to total tau, dilution 1:10,000) in PBS-M overnight at 4 °C. Blots were developed with IRDye 800 conjugated goat anti-rabbit (Rockland Inc) or IRDye 680 conjugated goat anti-mouse (Molecular Probes, Eugene, OR, USA) and visualized using an Odyssey Infrared Imaging System (Li-Cor Biosciences). For analysis of tau isoforms, samples were dephosphorylated prior to electrophoresis using lambda protein phosphatase and separated by SDS-PAGE alongside a recombinant tau ladder (Sigma).
Tau ELISA
Mature tau-KO-GFP neurons were exposed to tau-conditioned media for 6 h, 12 h, 1 and 6 days, washed 3× with warmed PBS, trypsinized for 2 min, collected and homogenized in buffer supplemented with protease and phosphatase inhibitors at 4 °C. Protein concentration was determined by BCA Assay (Pierce) and normalized with 1 × PBS. Sandwich ELISA was performed as previously described using tau monoclonal antibodies DA31 and DA9-HRP50.
Viral vectors
Tau RD-P301S YFP (aa 244–372 of the 441 full length tau, mutations P301S “PS”), YFP, mCherry viruses were prepared by transient co-transfection of HEK293T cells with indicated vectors using calcium phosphate method. Media containing virus was collected at 48 h post transfection and purified using Lenti-X-Concentrator according to manufacturer’s protocol (Clontech). Wild type and P301L 4R2N tau constructs were cloned into an eGFP-AAV1 vector (chicken beta-actin promoter), and all sequences were verified. AAV was subsequently produced and the genomic titer of each virus was determined by quantitative PCR.
RNA Extraction and Quantitative RT-PCR
RNA was extracted from 5–10mg hippocampal tissue from three, 7 month old mice (rTg4510 line, male and female) that were optogenetically stimulated for 20 days as describe before, plus three 2–3 month old animals, stimulated for five days, using a standard trizol procedure. SuperScript® III RT enzyme and SuperMix (Invitrogen™, Carlsbad, CA) containing 2.5 μM oligo(dT)20, 2.5 ng μl−1, random hexamers, 10 nM MgCl2 and dNTPs, was used for first strand cDNA synthesis in a 20 μL reaction volume. Gene expression was assessed using TaqMan® probes (ThermoFisher Scientific, Waltham, MA) for the human tau transgene (Hs00902194_m1), mouse NeuN (Rbfox3) (Mm01248771_m1) and the mouse hypoxanthine guanine phosphoribosyl transferase gene (Hprt) (Mm03024075). PCRs were performed by monitoring in real time the increase in fluorescence of the FAM and VIC fluorophores, using an iQ5 detector system (Bio-Rad, Hercules, CA). All samples were run in triplicate 20 μL duplexed PCR reactions using 1 μL each of FAM and VIC labeled TaqMan® probes, 10 μL of TaqMan® Gene Expression Master Mix (Applied Biosystems, Foster City, CA), and 8 μL of diluted cDNA (1:40), using amplification conditions as follows: 50 °C for 2 mins, followed by 95 °C for 10 mins and 40 cycles of denaturation at 95 °C (15 s), and priming, extension and data capture 60 °C (60 s). A standard curve was generated for each gene using dilutions of cDNA pooled from each sample. Relative expression changes were calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001), defined as the ΔCT of the sample normalized to the housekeeping gene Hprt, minus the ΔCT of the normalized reference sample from the standard curve.
In vitro stimulation and cell viability assessment
To induce activity, neurons at 12–14 DIV were treated with picrotoxin (100 μM) for 30 min without changing the medium. For optogenetic stimulation, neurons were transduced with tau (tau-P301L-GFP, 2 × 109 particles) and ChR2 (AAV9.CaMKIIa.hChR2(H134R)-mCherry.WPRE.hGH; Penn Vector Core, 3.98 × 1011 particles) at 3–5 DIV. After 9–12 DIV, control wild type or transduced neuronal cultures were placed on an upright microscope (Olympus) and stimulated with blue light (470 nm) at 0.1 mW mm−2 (40 mA) for 30 min at 2 sec intervals. The emission power was measured using a power meter (ThorLabs) that is placed directly below, and at the same distance from the objective lens as the cultures. The chamber temperature was maintained at 32 °C ± 2 °C. Media was collected before and after stimulation, centrifuged to remove cell debris, and analyzed by Sandwich ELISA using tau monoclonal antibodies DA31 and DA9-HRP50 in duplicates. Cell death was quantified by measuring LDH release in the same neuronal populations prior to, and after treatments, according to manufacturer’s protocol (Promega). Each experiment was performed three times in triplicates (n = 6).
Whole-cell patch clamp recordings
Mature neuronal cultures (DIV 12–14) were placed on the recording chamber (Warner Instruments) of an upright microscope equipped with a 10 × and a 40 × oil-immersion objectives (Olympus). Control and transduced neurons were identified by DIC optics and fluorescences, respectively. Cells were perfused with artificial cerebrospinal fluid (ACSF) containing (in mM): 119 NaCl, 26.2 NaHCO3, 10 glucose, 2.4 CaCl2, 3.6 KCl, 1.2 MgCl2-6H20, and 1.0 NaH2PO4-6H20 at < 0.5 ml min−1 flow rate. The chamber temperature was maintained at 32 °C ± 2 °C. Whole-cell patch clamp recordings were made with pulled borosilicate glass pipettes (G150F-4, Warner Instruments, tip resistance 2–4 MΩ) filled with intracellular solution containing (in mM): 115 K-gluconate, 20 KCl, 10 HEPES, 2 MgCl2, 2 ATP-Mg, 2 ATP-Na2, 0.3 GTP-Na, (pH=7.25, ~300 mOsm). Whole cell current clamp recording were performed with an Axopatch 200B amplifier (Molecular Devices) and digitized at 10 kHz with a Digidata 1332 (Molecular Devices). Data were acquired using Clampex 8.2 software (Molecular Devices) for subsequent analysis. Blue light (470 nm) or control red light (545 ± 30 nm) were delivered by an LED controller (Thorlabs, 40 mA) and triggered with a Master-8 pulse generator (A.M.P.I. Labs). Synaptic inputs were blocked with picrotoxin (50 μM) and NBQX (20 μM) (Sigma Aldrich). The input resistance and the baseline resting membrane potential were monitored throughout the recording. Current-voltage relationships were measured by injecting step current from −300 to + 150 pA with + 50 pA increments. Data were analyzed in Clampfit (Molecular Devices).
Surgery
Surgical procedures were performed following NIH guidelines in accordance with IACUC protocols. Mice were anesthetized with ketamine/xylazine (100 mg ml−1, 15 mg ml−1, respectively). AAV5 CamKII.hM3Dq-mCherry (1.7 × 1012 particles) and AAV9/CamKIIa.hChR2-mCherry (2 × 1012 particles) virus was injected into the MEC (0.2–0.3 mm in front of the transverse sinus (AP), 3.0–3.1 mm (ML), and 1.5 mm below the dura (DV), line rTg4510) of the left, or right hemisphere of the mice respectively. For in vivo optogenetic experiments, AAV9/CamKIIa.hChR2-mCherry (2 × 1012 particles) virus was injected into the hippocampus (1.5 mm (DV), 1.8 mm (ML), 1.8 mm (AP)) of both the left and right hemispheres of the mice (line rTg4510). A 20 mm cannula was implanted slightly above the injection site (~1.2 mm) in both brain hemispheres. For one mouse, a microdrive was implanted in the left hemisphere (~1.2 mm depth) to record neuronal activity. Dental cement was used to secure the cannulae and microdrive.
In vivo chemogenetic stimulation and electrophysiology
Ten days following viral DREADD transduction, clozapine-N-oxide (CNO) was given to mice systemically via intraperitoneal injections at 5 mg kg−1, two times per day (6 – 7 hrs in between injections), five days per week for either two or six weeks. Total stimulation was either 20 or 60 times. IP injections were alternated between left and right sides of the mouse to minimize scarring. CNO (C-929) was generously provided by the National Institutes of Mental Health Chemical Synthesis and Drug Supply Program. Both male and female mice were used.
In vivo optogenetic stimulation and electrophysiology
Ten days following ChR2 viral transduction, the left hemisphere of mice was optically stimulated with four pulses of LED light (470 nm) (Thorlabs) at 30 Hz frequency, three times with one -minute interval. Mice were stimulated three times per day, five times per week for four weeks (60 stimulations total). The right hemisphere was not stimulated to provide a within-animal control. To record neuronal activity, the mouse implanted with electrodes was plugged into the electrophysiology setup (Axona) during stimulation and neuronal activity was recorded. Individual neurons from the recording data were separated using the spike sorting software Tint (Axona). Neurons were identified based on spike- firing rate, amplitude, waveform and their refractory periods. sigTOOL (a Matlab-based signal analysis tool) was used to visualize changes in neuronal firing during optical stimulation. Both male and female mice were used.
Immunofluorescence, Nissl and DAB staining
Mouse brains were collected as described previously. Free-floating sections in either coronal or horizontal plane (35 μm) were used for immunofluorescence. Human iPSC neurons were grown in 8-well chamber slides (Ibidi) and primary neurons were grown as previously described. Neurons and free-floating tissues were immunolabeled and mounted as previously described with the following antibodies: mouse anti-tau (CP27, 1:1000, MC1, 1:2000, (gifts of P. Davies)), rabbit anti-GFP (1:500, abcam), total Tau (DAKO, 1:5000), 3R tau (RD3, Millipore, 1:1000), Pax6 (Covance, 1:300), Ki67 (BD Biosciences, 1:500). Fluorescent-conjugated secondary antisera mixtures containing Alexa 488 IgG, Alexa 594 IgG, or Alexa 657 IgG (Invitrogen) were used. Antigen retrieval was performed on tissues by heating in sodium citrate for 5 min at 95 °C. Tissues were treated with sudan black (0.1% in PBS) for 10 – 15 min to eliminate autofluorescence from lipofuscin. For Nissl staining, sections were stained with 0.1% cresyl violet in 70% EtOH for 10 min at RT, rinsed with distilled H20, washed three times with PBS and mounted with Prolong anti-fade medium (Invitrogen). DAB staining on tissues was performed as previously described2. Briefly, tissues were treated with 5% H2O2 in PBS for 10 minutes to quench endogenous peroxidase activity, blocked in PBS containing 5% horse serum and 0.3% Triton, and incubated at 4 °C overnight as previously described2 with the following antibodies: c-Fos (1:200, Santa Cruz, G3115), AT8 (S202/205, 1:1000, ThermoFisher, MN1020), MC1 (conformationally abnormal human tau, 1:1000). After three washes with PBS-T, tissues were developed using Super Picture HRP (Invitrogen) for 15 mins at room temperature on a rotator and detection of the chromagen with DAB (Sigma-Aldrich) was done according to the manufacturer’s manual. In the case of c-Fos staining, mice were sacrificed 75 minutes after their last CNO injection. ABC kit (Vectastain, Vector Labs) was used to develop the tissues according to the manufacturers manual. The sections were mounted, and visualized by light microscopy.
Confocal and light microscopy
Immunolabeled neurons and tissues were examined with a Zeiss LSM710 confocal microscope at 10× dry and 63× oil immersion objectives as previously described. Sequential tile scans were performed to capture wide field images of microfluidics (at 1024 × 1024 resolution) and tissues (512 × 512). Sequential scans were performed to capture images of cells grown on coverslips (1024 × 1024). All images from the same experiment were taken at the same laser intensity and detector gain. 3D co-localization analysis was performed using Volocity 4.0 Restoration software (Volocity, Improvision). All DAB sections were examined with a Zeiss AxioObserver.Z1 inverted microscope at 10×. Sequential tile scans were performed to capture wide field images of the whole section at 10×. Immunofluorescent images of co-culture experiments were captured at 20× magnification, 10 images per coverslip at either 512 × 512 resolution or 1024 × 1024 resolution, randomized and quantified by a technician who did not know the identity of the samples (n = 2 coverslips, 10 images per coverslip).
Statistical analyses and sample sizes
Statistical analyses were performed using GraphPad Prism 5.0 (Graph Pad Software, La Jolla, CA, USA) and Stata 12.1 (College Station, TX). No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those previously reported22. Normality of the data and homogeneity of group variances were assessed using the Shapiro-Wilk W test and Levine’s test respectively. When assumptions were not met or sample sizes were insufficient to test assumptions, non-parametric tests were employed. Adjustments were made for multiple testing under each hypothesis, via Bonferroni correction. Statistical significance was determined if adjusted p < 0.05. Data are represented in dot plot graphs with mean ± s.e.m. To investigate tau uptake into recipient neurons via the extracellular media we measured tau levels in neurons from MAPT −/− (knock out) mice at baseline (no incubation, n = 4 coverslips), 1 – 6 hours after the conditioned media was added (n = 6 independent coverslips), and 1 – 6 days after addition of conditioned media (n = 4 independent coverslips). A Wilcoxon rank-sum test was employed to test for differences in the levels of tau, comparing tau levels at baseline levels with levels at 6 hours or less, and tau at baseline with levels at 1 – 6 days. To examine whether tau release is enhanced following neural activity in vitro, primary neurons were stimulated with picrotoxin. Levels of mouse tau in wt neurons (mTau) (n = 9 separate coverslips), mouse tau plus human tau in rTg4510 neurons (mTau+ hTau) (n = 9 separate coverslips) and human tau in iPSC cultures (hTau) (n = 6 separate coverslips) was assessed. For each model, additional and independent cultures (mTau n = 9; mtau + hTau n = 9; hTau n = 6) were utilized as the non-stimulated control group. Experiments were performed over two days, with 50% of the independent samples assessed on each experimental day. Samples were analyzed as a single group (regardless of experimental day), using individual Student t-tests (with Satterthwaite’s approximation for unequal group variance) to determine differences between control and picrotoxin treatment in the three models. In vitro optogenetic stimulation was performed using four separate cultures for each of three experimental conditions: control, non-stimulated ChR and simulated ChR. Differences in tau release across conditions was tested using a Kruskal-Wallis omnibus test, followed by Dunn’s post hoc tests with a Bonferroni correction. To test the hypothesis that transfer of tau is enhanced by stimulating neuronal activity, the percent of recipient neurons with human tau from the donor was calculated (the number of cells expressing mCherry that were YFP positive/total mCherry positive cells × 100%). For each condition (stimulated and non-stimulated), neurons were plated on n = 5 independent coverslips. To obtain an accurate representation of neurons across each coverslip, multiple images were used for cell counting. The percent of total neurons with tau transferred from the donor were compared between stimulated and non-stimulated conditions using a Student’s t-test. Gene expression (Supplementary Figure 1) was compared between the optogenetically stimulated side of the hippocampus (S, n = 6 animals) and the non-stimulated sides (NS, n = 6 animals), using a paired sample t-test. To test the hypothesis that atrophy due to stimulation would be significantly different in the tau mice (n = 4), as compared to tau-KO mice (n = 4), left and right cell body layers in the hippocampus were measured in Nissl-stained sections using ImageJ. Nissl signal above threshold levels were compared between the regions of interest on each side of the brain slice. The threshold value was kept the same between slices, within an animal. The value for the left, stimulated hippocampus was divided by the right, non-stimulated hippocampus to obtain a ratio of left divided by right signal in each slice. Ratios of all slices from each animal were averaged. A Wilcoxon rank-sum test was employed to compare the ratio of left/right staining across mouse groups. The Wilcoxon-rank test statistic would be expected to approximate to the normal distribution with this sample size. In figures, calculated probabilities are symbolized by asterisks as follows: *P < 0.05; **P < 0.0; *** P < 0.001. Optogenetic stimulation and immunoflorescent image analysis was performed by a blinded observer. Other blinded authors analyzed DAB and immunofluorescent stains of brain tissues (non-stimulated hemisphere versus stimulated hemisphere). Statistical analysis was not performed blinded to the experimental conditions. For optogenetic and chemogenetic experiments, mice of the appropriate age were selected from several different litters and randomly assigned as control or experimental. No other randomization in experimental conditions or stimulus presentations was performed.
A Supplementary Methods Checklist is available.
Data Availability
The data that support the findings of this study are available from the corresponding author upon request. The authors declare that data supporting the findings of this study are available within the article. Source data for figures 4b, 5a, b, g, h, 6b, and 7h are provided within the article.
Supplementary Material
Supplementary Figure 1. Increased neuronal activity does not change human tau expression in the stimulated mice (line rTg4510, n = 6). To adjust for total neuron count, the expression ratio of hTau/NeuN was plotted for animals 1- 6. There was no significant difference between stimulated (mean ratio = 1.92 ± 1.28) versus non-stimulated (mean ratio = 2.43 ± 1.54) hemispheres; t(5) = −1.57, P = 0.18.
Supplementary Figure 2. Immunohistochemistry image of brain tissue of two-week stimulated EC-Tau mice stained with anti-human tau antibody, MC1 (green). DAPI (blue). Scale bar, 500 μm, n = 1 mouse.
Supplementary Figure 3. Immunohistochemistry image of brain tissue of six-week stimulated EC-Tau mice stained with (a) anti-human tau antibody, MC1 (green) acquired using lower green laser power, or (b) CP27. DAPI (blue). Scale bar, 500 μm, n = 3 mice.
Supplementary Figure 4. Full-length images of western blots presented in Fig. 4a. Lysates from tau-expressing neurons (rTg4510 primary neurons and P301L-GFP transduced neurons) and conditioned media from the same cells was immunoprecipitated with anti-hTau antibody (CP27) and analyzed by immunoblot with total tau antibody (TauC). Actin shows amounts of protein loaded and absence of cell contamination in conditioned media.
Acknowledgments
We thank C. Acker for help with Sandwich ELISA and P. Davies (Litwin Zucker Center for Alzheimer’s Research, Feinstein Institute Long Island) for providing tau antibodies. K. Jansen-West, E. Perkerson and L. Petrucelli (Mayo Clinic Jacksonville) are thanked for providing additional tau viruses. D. Sulzer is thanked for helpful discussions regarding cell electrophysiology. We also thank L. Liu for assistance with mouse tissue collection, C. Profaci for assistance with optogenetic experiments and L. Shi for administrative assistance. This work was supported by BrightFocus Foundation fellowship to J.W., NIH/NINDS grants NS081555 and NS074874 to K.D., Cure Alzheimer’s Fund to K.D. and NIH/NIA grant AG050425 to S.A.H. and K.D. A.M. is supported by funds from NIH/NIA AA19801 and the Parkinson’s Disease Foundation to D.S. S.W. is supported by the NIHR Queen Square Dementia Biomedical Research Unit.
Footnotes
Author contributions:
J.W.W. and K.E.D. designed experiments. J.W.W., S.A.H., I.M.B., A.M., S.W. conducted the experiments and data analyses. J.W.W, C.L.C and K.E.D. wrote the manuscript.
M.H, E.N, S.E and Y.H.F provided technical assistance.
S.A.H, I.M.B, G.A.R performed mouse surgery, in vivo recordings, in vivo stimulations, immunohistochemistry
A.M performed in vitro patch-clamp experiments and providing LED microscope, optimization of in vitro optogenetic stimulation
K.R and C.L.C performed the RT PCR experiment and performed AAV P301L-GFP, GFP, and WT-GFP virus cloning, packaging, and titration
C.L.C performed statistical analyses
D.W.S. and M.I.D. provided cell lysates containing tau seeds and RD PSY, YFP, and mCherry viruses.
I.M.B performed Nissl and immunofluorescence analysis.
S.W performed iPSC differentiation, data analysis and provided conditioned media.
R.A.C.M.B performed immunoprecipitation of tau from conditioned media and cell lysates
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 2.Liu L, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Calignon A, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris JA, et al. Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One. 2012;7:e45881. doi: 10.1371/journal.pone.0045881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clavaguera F, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–9540. doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iba M, et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33:1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanders DW, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu JW, et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem. 2013;288:1856–1870. doi: 10.1074/jbc.M112.394528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caillierez R, et al. Lentiviral delivery of the human wild-type tau protein mediates a slow and progressive neurodegenerative tau pathology in the rat brain. Mol Ther. 2013;21:1358–1368. doi: 10.1038/mt.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osinde M, Clavaguera F, May-Nass R, Tolnay M, Dev KK. Lentivirus Tau (P301S) expression in adult amyloid precursor protein (APP)-transgenic mice leads to tangle formation. Neuropathol Appl Neurobiol. 2008;34:523–531. doi: 10.1111/j.1365-2990.2008.00936.x. doi:NAN936[pii] 10.1111/j.1365-2990.2008.00936.x. [DOI] [PubMed] [Google Scholar]
- 12.Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012 doi: 10.1074/jbc.M112.346072. doi:M112.346072 [pii] 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gousset K, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol. 2009;11:328–336. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 14.Saman S, et al. Exosome-associated Tau Is Secreted in Tauopathy Models and Is Selectively Phosphorylated in Cerebrospinal Fluid in Early Alzheimer Disease. J Biol Chem. 2012;287:3842–3849. doi: 10.1074/jbc.M111.277061. doi:M111.277061 [pii] 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asai H, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon D, et al. Tau Overexpression Results in Its Secretion via Membrane Vesicles. Neurodegener Dis. 2012 doi: 10.1159/000334915. doi:000334915 [pii] 10.1159/000334915. [DOI] [PubMed] [Google Scholar]
- 17.Dujardin S, et al. Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One. 2014;9:e100760. doi: 10.1371/journal.pone.0100760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamada K, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31:13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barten DM, et al. Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J Alzheimers Dis. 2011;24(Suppl 2):127–141. doi: 10.3233/JAD-2011-110161. doi:W3V1180601X3532U [pii] 10.3233/JAD-2011-110161. [DOI] [PubMed] [Google Scholar]
- 20.Takeda S, et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat Commun. 2015;6:8490. doi: 10.1038/ncomms9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurz A, et al. Tau protein in cerebrospinal fluid is significantly increased at the earliest clinical stage of Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12:372–377. doi: 10.1097/00002093-199812000-00020. [DOI] [PubMed] [Google Scholar]
- 22.Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389–394. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamada K, et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014;211:387–393. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busche MA, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. doi:321/5896/1686 [pii] 10.1126/science.1162844 [doi] [DOI] [PubMed] [Google Scholar]
- 25.Busche MA, et al. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2012;109:8740–8745. doi: 10.1073/pnas.1206171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siskova Z, et al. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron. 2014;84:1023–1033. doi: 10.1016/j.neuron.2014.10.024. [DOI] [PubMed] [Google Scholar]
- 27.Minkeviciene R, et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hall AM, et al. Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s disease. J Neurosci. 2015;35:6221–6230. doi: 10.1523/JNEUROSCI.2552-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santacruz K, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. doi:309/5733/476 [pii] 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- 31.Taylor AM, et al. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. doi:nmeth777 [pii] 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Styren SD, Hamilton RL, Styren GC, Klunk WE. X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer’s disease pathology. J Histochem Cytochem. 2000;48:1223–1232. doi: 10.1177/002215540004800906. [DOI] [PubMed] [Google Scholar]
- 33.Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc. 2012;7:1836–1846. doi: 10.1038/nprot.2012.116. [DOI] [PubMed] [Google Scholar]
- 34.Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci. 2012;15:477–486, S471. doi: 10.1038/nn.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sposito T, et al. Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10 + 16 splice-site mutation in MAPT. Hum Mol Genet. 2015;24:5260–5269. doi: 10.1093/hmg/ddv246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoon KW, Covey DF, Rothman SM. Multiple mechanisms of picrotoxin block of GABA-induced currents in rat hippocampal neurons. J Physiol. 1993;464:423–439. doi: 10.1113/jphysiol.1993.sp019643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 38.Zhu H, Roth BL. DREADD: a chemogenetic GPCR signaling platform. Int J Neuropsychopharmacol. 2015;18 doi: 10.1093/ijnp/pyu007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. doi:M808759200 [pii] 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. doi:M110.209296 [pii] 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michel CH, et al. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J Biol Chem. 2014;289:956–967. doi: 10.1074/jbc.M113.515445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamada K, et al. Analysis of in vivo turnover of tau in a mouse model of tauopathy. Mol Neurodegener. 2015;10:55. doi: 10.1186/s13024-015-0052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holmes BB, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–3147. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mirbaha H, Holmes BB, Sanders DW, Bieschke J, Diamond MI. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J Biol Chem. 2015;290:14893–14903. doi: 10.1074/jbc.M115.652693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santa-Maria I, et al. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J Biol Chem. 2012;287:20522–20533. doi: 10.1074/jbc.M111.323279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bomben V, et al. Bexarotene reduces network excitability in models of Alzheimer’s disease and epilepsy. Neurobiol Aging. 2014;35:2091–2095. doi: 10.1016/j.neurobiolaging.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanchez PE, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Busche MA, et al. Rescue of long-range circuit dysfunction in Alzheimer’s disease models. Nat Neurosci. 2015;18:1623–1630. doi: 10.1038/nn.4137. [DOI] [PubMed] [Google Scholar]
- 49.Thom M, et al. Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post-mortem study. Brain. 2011;134:2969–2981. doi: 10.1093/brain/awr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Acker CM, Forest SK, Zinkowski R, Davies P, d’Abramo C. Sensitive quantitative assays for tau and phospho-tau in transgenic mouse models. Neurobiol Aging. 2013;34:338–350. doi: 10.1016/j.neurobiolaging.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Increased neuronal activity does not change human tau expression in the stimulated mice (line rTg4510, n = 6). To adjust for total neuron count, the expression ratio of hTau/NeuN was plotted for animals 1- 6. There was no significant difference between stimulated (mean ratio = 1.92 ± 1.28) versus non-stimulated (mean ratio = 2.43 ± 1.54) hemispheres; t(5) = −1.57, P = 0.18.
Supplementary Figure 2. Immunohistochemistry image of brain tissue of two-week stimulated EC-Tau mice stained with anti-human tau antibody, MC1 (green). DAPI (blue). Scale bar, 500 μm, n = 1 mouse.
Supplementary Figure 3. Immunohistochemistry image of brain tissue of six-week stimulated EC-Tau mice stained with (a) anti-human tau antibody, MC1 (green) acquired using lower green laser power, or (b) CP27. DAPI (blue). Scale bar, 500 μm, n = 3 mice.
Supplementary Figure 4. Full-length images of western blots presented in Fig. 4a. Lysates from tau-expressing neurons (rTg4510 primary neurons and P301L-GFP transduced neurons) and conditioned media from the same cells was immunoprecipitated with anti-hTau antibody (CP27) and analyzed by immunoblot with total tau antibody (TauC). Actin shows amounts of protein loaded and absence of cell contamination in conditioned media.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request. The authors declare that data supporting the findings of this study are available within the article. Source data for figures 4b, 5a, b, g, h, 6b, and 7h are provided within the article.