

Abstract
Acute intermittent hypoxia (AIH) induces phrenic long-term facilitation (pLTF), a form of spinal motor plasticity. Competing mechanisms give rise to phrenic motor facilitation (pMF; a general term including pLTF) depending on the severity of hypoxia within episodes. In contrast, moderate acute sustained hypoxia (mASH) does not elicit pMF. By varying the severity of ASH and targeting competing mechanisms of pMF, we sought to illustrate why moderate AIH (mAIH) elicits pMF but mASH does not. Although mAIH elicits serotonin-dependent pLTF, mASH does not; thus, mAIH-induced pLTF is pattern sensitive. In contrast, severe AIH (sAIH) elicits pLTF through adenosine-dependent mechanisms, likely from greater extracellular adenosine accumulation. Because serotonin- and adenosine-dependent pMF interact via cross talk inhibition, we hypothesized that pMF is obscured because the competing mechanisms of pMF are balanced and offsetting during mASH. Here, we demonstrate the following: (1) blocking spinal A2A receptors with MSX-3 reveals mASH-induced pMF; and (2) sASH elicits A2A-dependent pMF. In anesthetized rats pretreated with intrathecal A2A receptor antagonist injections before mASH (PaO2 = 40–54 mmHg) or sASH (PaO2 = 25–36 mmHg), (1) mASH induced a serotonin-dependent pMF and (2) sASH induced an adenosine-dependent pMF, which was enhanced by spinal serotonin receptor inhibition. Thus, competing adenosine- and serotonin-dependent mechanisms contribute differentially to pMF depending on the pattern/severity of hypoxia. Understanding interactions between these mechanisms has clinical relevance as we develop therapies to treat severe neuromuscular disorders that compromise somatic motor behaviors, including breathing. Moreover, these results demonstrate how competing mechanisms of plasticity can give rise to pattern sensitivity in pLTF.
SIGNIFICANCE STATEMENT Intermittent hypoxia elicits pattern-sensitive spinal plasticity and improves motor function after spinal injury or during neuromuscular disease. Specific mechanisms of pattern sensitivity in this form of plasticity are unknown. We provide evidence that competing mechanisms of phrenic motor facilitation mediated by adenosine 2A and serotonin 2 receptors are differentially expressed, depending on the pattern/severity of hypoxia. Understanding how these distinct mechanisms interact during hypoxic exposures differing in severity and duration will help explain interesting properties of plasticity, such as pattern sensitivity, and may help optimize therapies to restore motor function in patients with neuromuscular disorders that compromise movement.
Keywords: hypoxia, long-term facilitation, motor control, pattern sensitivity, phrenic, spinal cord
Introduction
The neural system controlling breathing (including spinal respiratory motor neurons) undergoes considerable plasticity when animals experience intermittent periods of low oxygen or intermittent hypoxia (Mitchell and Johnson, 2003; Devinney et al., 2013). Intermittent hypoxia-induced respiratory plasticity may be a key respiratory control strategy beyond simple chemo-feedback, conferring more robust and effective homeostatic regulation of breathing (Mitchell and Johnson, 2003).
Although hypoxia triggers plasticity in both respiratory and nonrespiratory motor systems (EA Dale et al., 2014; Gonzalez-Rothi et al., 2015), which arise from multiple, discrete cellular mechanisms (Dale-Nagle et al., 2010; Devinney et al., 2013), we know little concerning the functional significance of mechanistic heterogeneity, or the nature of interactions between these mechanisms. By greater understanding of interactions between mechanisms, we hope to gain insight concerning the biological significance and clinical relevance of intermittent hypoxia-induced respiratory motor plasticity. Indeed, intermittent hypoxia has been used as a therapeutic tool to restore respiratory and nonrespiratory motor function with spinal injury or neuromuscular disease (N Dale and Frenguelli, 2014; Navarrete-Opazo and Mitchell, 2014b; Gonzalez-Rothi et al., 2015).
A prominent example of intermittent hypoxia-induced respiratory motor plasticity is phrenic long-term facilitation (pLTF), a sustained increase in phrenic activity lasting hours after exposure to acute intermittent hypoxia (AIH). pLTF is one of multiple forms of phrenic motor facilitation (pMF), a more general term that refers to any long-lasting increase in phrenic motor output caused by spinal plasticity in or near the phrenic motor nucleus, regardless of the initiating stimulus. pLTF is pattern-sensitive because it is observed following moderate acute intermittent (mAIH), but not moderate acute sustained hypoxia (mASH) (Baker and Mitchell, 2000).
We have learned a great deal about cellular mechanisms giving rise to pMF in recent years (Dale-Nagle et al., 2010; Devinney et al., 2013; Fields and Mitchell, 2015). When pLTF is initiated by mAIH, it requires: intermittent activation of spinal Gq protein coupled serotonin 2 (5-HT2) receptors (Fuller et al., 2001b; Baker-Herman and Mitchell, 2002; Macfarlane and Mitchell, 2009, 2011), ERK MAP kinase activity (Hoffman et al., 2012), new brain derived neurotrophic factor synthesis (Baker-Herman et al., 2004), TrkB activation (Dale et al., 2016), and PKCθ activity (Devinney et al., 2015). In contrast, when pLTF is initiated by AIH consisting of severe hypoxic episodes (sAIH), it requires spinal adenosine 2A (A2A) receptor activation and is serotonin-independent (Nichols et al., 2012). Our working model is that sAIH-induced pLTF arises from Gs protein-coupled receptor activation, triggering a distinct mechanism that requires exchange protein activated by cAMP (EPAC) (Fields et al., 2015), Akt and mammalian target of rapamycin signaling (Dougherty et al., 2015) and new TrkB synthesis (vs BDNF) (Golder et al., 2008). These diverse mechanisms interact via cross talk inhibition (Dale-Nagle et al., 2010), with the serotonin-dependent mechanism predominant following mAIH (Hoffman et al., 2010; Navarrete-Opazo and Mitchell, 2014a), and the adenosine-dependent mechanism predominant following sAIH (Nichols et al., 2012). The shift from serotonin-dependent to adenosine-dependent pLTF with the progression from mAIH to sAIH may result from greater extracellular adenine nucleotide accumulation during severe hypoxia, shifting the balance between the competing serotonin- and adenosine-dependent mechanisms (Conde and Monteiro, 2004; Gourine et al., 2005; Dale and Frenguelli, 2009). When these mechanisms are activated equally, they may cancel one another, thereby obscuring pMF.
Here we reasoned that extracellular adenosine accumulation is greater with more severe (i.e., sAIH and sASH vs mAIH and mASH), or longer cumulative durations of hypoxia with the same severity (i.e., mASH vs mAIH; sASA vs sAIH). We hypothesized that greater A2A receptor activation masks pMF following mASH and that A2A receptor inhibition would reveal serotonin-dependent pMF. Further, we hypothesized that both sAIH and sASH elicit A2A receptor-dependent pMF and that this pMF is enhanced by 5-HT2 receptor inhibition. Overall, we demonstrate that competing adenosine- and serotonin-dependent mechanisms contribute differentially to pMF depending on the severity and duration of hypoxia.
Materials and Methods
Animals.
Adult (12–16 weeks; 280–500 g) male Sprague Dawley rats (Harlan, Colony 211, Colony 218a) were studied. Rats from each colony were evenly distributed among the experimental groups to mitigate any impact from substrain differences on experimental results (Fuller et al., 2001a). The University of Wisconsin Animal Care and Use committee approved all experimental procedures.
Intrathecal drug injections.
Treatment groups were given intrathecal injections of 10% DMSO in ACSF (vehicle, 12 μl ACSF; in mm as follows: 120 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 23 NaHCO3, 10 glucose, equilibrated with 95% O2, 5% CO2 for 15 min), the A2A receptor antagonist MSX-3 (Sigma-Aldrich, 200 μm in 12 μl of 10% DMSO in ACSF), or the broad-spectrum serotonin receptor antagonist methysergide maleate (Sigma-Aldrich, 20 mm in 12 μl ACSF). Injections were made 12 min before exposure to mASH, sASH, or sham (i.e., no hypoxia).
Experimental preparation.
Rats were induced with isoflurane in a closed chamber and placed on a heated surgical table to maintain body temperature; anesthesia was maintained via nose cone (3.5% isoflurane, 50% O2). Tracheotomy was performed and rats were pump-ventilated through a tracheal tube (Rodent Ventilator 683, Harvard Apparatus; tidal volume 2.2–2.7 ml; frequency 70–75 breaths/min) with a gas mixture of ∼50% O2, 50% N2, and 3.5% isoflurane. End-tidal CO2 was maintained at 40–44 mmHg by adjusting the ventilator and/or adding CO2 to the inspired gas mixture. Tracheal pressure was continuously monitored. A catheter was placed in the tail or femoral vein to administer intravenous fluids (1.5–3.0 ml/h of 64%–75% lactated Ringer's solution, 4%–10% HCO3, and 15%–30% of 6% hetastarch in 0.9% NaCl) to maintain arterial fluid and acid base balance (base excess change <±3 mEq/L during protocol). A catheter was placed in the femoral artery to monitor blood pressure and draw arterial blood samples for analysis (PaO2, PaCO2, pH, base excess; ABL800 Radiometer).
Using a dorsal approach, the left phrenic nerve was isolated, cut, and the central end was desheathed and protected with saline-soaked cotton until nerve recordings were initiated. For intrathecal drug delivery, a C2 laminectomy was performed and a silicone catheter (2 Fr; Access Technologies) attached to a 50 μl Hamilton syringe that was inserted through the dura and advanced until the tip was over the C4 spinal segment. The rats were converted to urethane anesthesia by slowly withdrawing inhaled isoflurane while administering intravenous urethane (1.7–1.9 mg/kg). After ∼1 h, the rat was paralyzed with pancuronium bromide (2.5 mg/kg, i.v.). Body temperature was maintained at 37.5 ± 1°C (rectal thermometer; Fisher Scientific). Blood pressure was monitored to ensure physiological stability (80–150 mmHg baseline, <30 mmHg change at 60 min). Adequate anesthesia was tested by toe pinch induced responses in blood pressure or phrenic nerve activity. Maximal CO2 responses were elicited at the end of each experiment to verify adequate nerve responses and preparation stability. Rats with ≤30% hypoxic response during hypoxia or ≤50% maximal hypercapnic response were eliminated from analysis.
Neurophysiological measurements.
At ∼1 h following conversion to urethane anesthesia, the desheathed phrenic nerve was covered with mineral oil and placed on bipolar silver electrodes. Nerve activity was amplified (10,000×), bandpass filtered (300–10,000 Hz model 1800, A-M Systems), rectified and integrated with a continuous moving averager (time constant: 50 ms; CWE, MA-821 filter). The integrated signal was digitized and analyzed with a data acquisition system (WINDAQ, DATAQ Instruments). To determine the CO2 apneic and recruitment thresholds, inspired CO2 was lowered and/or ventilator rate increased until phrenic nerve bursting ceased; CO2 was then slowly raised by increasing inspired CO2 and/or decreasing ventilator rate until phrenic nerve bursting resumed. Baseline end-tidal CO2 was set 2–3 mmHg above the recruitment threshold. Baseline nerve activity was recorded for at least 20 min.
Rats then received 12 μl intrathecal injections of vehicle (10% DMSO in ACSF), methysergide (20 mm), or MSX-3 (200 μm) at C4 delivered as 2 μl boluses every 30 s. At 12 min following MSX-3 or vehicle injections, a blood sample was drawn to assess baseline conditions. For methysergide, baseline was recorded for 15–20 min after injection, consistent with previous studies (Baker-Herman and Mitchell, 2002). In rats where both methysergide and MSX-3 were delivered, MSX-3 was delivered 5–8 min after methysergide. Rats were then exposed to mASH (12%–14% O2; PaO2 = 40–54 mmHg), sASH (6%–8% O2; PaO2 = 25–36 mmHg), or no hypoxia (i.e., time controls, ∼50% O2). Arterial PCO2 was maintained within ±1.5 mmHg of baseline. PaO2 was >150 mmHg at baseline and after hypoxia in all studies. Blood samples were drawn at 5 and 20 min of ASH. After ASH, baseline conditions were restored and confirmed with blood gas analysis at 15, 30, and 60 min after ASH (PaO2 > 180 mmHg, PaCO2 ± 1.5 mmHg baseline, and base excess ±3 mEq/L baseline).
Data analyses.
Integrated phrenic burst amplitude and frequency were averaged in 60 s bins at baseline, during ASH, and at 15, 30, and 60 min after ASH (or similar times in time control experiments). Burst amplitudes were normalized as a percentage change from baseline in each rat. Frequency was expressed as change from baseline value (burst/min). Two-way ANOVA with a repeated-measures design was used to analyze phrenic burst amplitude, burst frequency, PaCO2, PaO2, mean arterial pressure, and base excess at baseline, during ASH, and 15, 30, and 60 min after ASH. Post hoc comparisons were made with Fisher's LSD (SigmaPlot version 12.0; Systat Software). In some cases, MSX-3 and methysergide time controls were grouped together as drug time control experiments because no significant differences were found between groups. For PaO2 during hypoxia, all rats exposed to mASH or sASH were grouped together because no significant differences were found between drug treatment groups. Differences between groups were considered significant if p < 0.05. Listing all p values for post hoc comparisons was done sparingly because of the number of significant differences in individual comparisons; specific p values are given when appropriate. Values are mean ± SEM.
Experimental design.
We hypothesize that, while spinal adenosine and serotonin are each capable of inducing pMF alone, they exhibit mutual inhibition during sustained hypoxia, and this balance varies with the severity of hypoxia. In Figure 1A, we illustrate the hypothetical adenosine 2A and serotonin receptor activation during increasingly severe and/or prolonged hypoxic episodes; implicit in this schema is the assumption that adenosine progressively rises with increasing severity and/or duration of hypoxia, whereas serotonin effects are already maximal with moderate hypoxia.
Figure 1.

Hypotheses providing the rationale for experimental design. A, The cumulative stimulus driving pMF mechanisms during hypoxic exposures increases, depending on pattern (sustained > intermittent) and severity (severe > moderate) of hypoxia. Hypoxia causes the following: (1) adenosine accumulation and activation of spinal A2A receptors, which we illustrate here as increasing with hypoxia “intensity” (solid line); and (2) serotonin (5-HT) release from descending raphe neurons, which reaches a maximum during mASH, with fixed serotonin receptor activation during sAIH and sASH (dashed line). pMF is not observed with mASH because both mechanisms are equal and offsetting due to balanced cross talk inhibition. Overall pMF expression may depend on the relative activation of A2A versus 5-HT receptors during hypoxia. B, Representative phrenic nerve trace demonstrating experimental protocol. During phrenic nerve recordings, intrathecal drug injections are delivered 12–20 min before exposure to 25 min of mASH or sASH. pMF is observed 60 min after mASH or sASH. C, Experimental conditions are listed for clarification.
The experimental design is illustrated in Figure 1B, showing timing of intrathecal drug injections before mASH or sASH exposures. Experimental groups included 6–10 rats pretreated with spinal injections of vehicle, MSX-3, or methysergide, or sequential injections of methysergide followed by MSX-3 before exposure to sham (time control; no hypoxia), mASH, or sASH while recording phrenic nerves.
The experimental groups studied (with n values) were as follows: (1) vehicle before mASH (veh + mASH, n = 10); (2) MSX-3 before mASH (MSX-3 + mod ASH, n = 8); (3) methysergide (methy) before mASH (methy + mASH, n = 8); (4) methysergide before MSX-3 followed by mASH (methy + MSX-3 + mASH, n = 6); (5) vehicle before sASH (veh + sASH, n = 8), (6) MSX-3 before sASH (MSX-3 + sASH, n = 8); (7) methysergide before sASH (methy + sASH, n = 6); (8) vehicle without hypoxia (veh time control, n = 8); (9) MSX-3 without hypoxia (MSX-3 time control, n = 6); and (10) methysergide without hypoxia (methy time control, n = 7).
Results
Blood gases, mean arterial pressure, and base excess
Blood gases were similar between all groups throughout the protocol, except when inspired oxygen was purposefully adjusted during ASH. Average PaO2 was >200 mmHg during baseline and after hypoxia, demonstrating that changes in PaO2 are not influencing phrenic activity at these times. Average PaCO2 was not significantly different among groups at baseline, except for vehicle-treated rats that were to be exposed to mASH or sASH, which had slightly lower PaCO2 versus rats later given MSX-3 and exposed to mASH (p = 0.04, p = 0.05, respectively; Table 1); there is no clear rationale for these differences because the rats had not yet received differential treatments. As expected, PaO2 during ASH was significantly decreased from baseline and time controls (p < 0.001, Table 1). During mASH, PaO2 was between 40 and 54 mmHg and was significantly higher than PaO2 during sASH (26–36 mmHg; p < 0.001, Table 1). Rats given vehicle injections had significantly lower PaCO2 values during sASH (p = 0.002, Table 1). However, PaCO2 was not significantly different from baseline in any group at 60 min after hypoxia, demonstrating that blood gas differences cannot explain observed changes in phrenic nerve burst amplitude.
Table 1.
Arterial partial pressure of carbon dioxide (PaCO2) and oxygen (PaO2), mean arterial pressure (MAP), and base excess during baseline, hypoxia, and 60 min after hypoxiaa
| Treatment groups | PaCO2 (mmHg) |
PaO2 (mmHg) |
MAP (mmHg) |
Base excess (mEq/L) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Hypoxia | 60 min | Baseline | Hypoxia | 60 min | Baseline | Hypoxia | 60 min | Baseline | Hypoxia | 60 min | |
| mASH | ||||||||||||
| Vehicle | 46.0 ± 1.1* | 46.2 ± 1.1 | 45.9 ± 1.2 | 313 ± 9 | 46.2 ± 1**,†,†† | 294 ± 8** | 108 ± 4 | 81 ± 6** | 100 ± 4** | −0.2 ± 0.6 | −0.6 ± 0.6 | 1.2 ± 0.5 |
| MSX-3 | 48.6 ± 0.9 | 48.8 ± 1.0 | 49.2 ± 0.9 | 313 ± 5 | 44.5 ± 1**,†,†† | 295 ± 5** | 118 ± 9 | 77 ± 8** | 106 ± 7** | −0.4 ± 0.5 | −1.3 ± 0.5 | 0.7 ± 0.5 |
| Methy | 46.0 ± 1.3 | 45.7 ± 1.2 | 46.4 ± 1.3 | 295 ± 6 | 44.7 ± 1**,†,†† | 263 ± 11** | 106 ± 4 | 66 ± 4** | 91 ± 3** | −1.0 ± 0.5 | −1.5 ± 0.5 | 0.0 ± 0.5 |
| Methy + MSX-3 | 46.3 ± 0.5 | 47.0 ± 0.6 | 46.4 ± 0.7 | 290 ± 12*,††† | 45.4 ± 2**,†,†† | 278 ± 10 | 97 ± 8* | 68 ± 7** | 94 ± 5 | 0.3 ± 0.7 | −0.3 ± 1.0 | 1.6 ± 1.0 |
| sASH | ||||||||||||
| Vehicle | 45.7 ± 0.9* | 44.2 ± 0.8** | 46.4 ± 0.8 | 310 ± 5 | 30.4 ± 1**,***,†† | 291 ± 9** | 110 ± 8 | 52 ± 9** | 94 ± 9** | −0.9 ± 0.5 | −8.9 ± 1.6** | 0.9 ± 1.0 |
| MSX-3 | 46.9 ± 1.0 | 46.7 ± 0.9 | 47.0 ± 0.9 | 309 ± 4 | 31.8 ± 1**,***,†† | 291 ± 6** | 106 ± 6 | 49 ± 5** | 83 ± 5** | −0.2 ± 0.3 | −7.1 ± 1.1** | −0.1 ± 0.4 |
| Methy | 48.1 ± 1.3 | 48.2 ± 1.0 | 48.5 ± 1.3 | 262 ± 13‡ | 30.3 ± 1**,***,†† | 238 ± 9** | 99 ± 6 | 40 ± 5** | 91 ± 5 | 1.0 ± 0.5 | −8.4 ± 0.9** | −0.5 ± 1.2 |
| Time control | ||||||||||||
| Vehicle | 46.0 ± 0.9 | 46.1 ± 0.8 | 45.3 ± 0.8 | 311 ± 10 | 310 ± 8 | 301 ± 7 | 101 ± 7* | 99 ± 7 | 93 ± 7 | −0.1 ± 0.7 | 0.0 ± 0.6 | 1.1 ± 0.6 |
| MSX-3 | 48.3 ± 0.4 | 48.5 ± 0.5 | 48.9 ± 0.5 | 310 ± 7 | 306 ± 6 | 304 ± 8 | 112 ± 4 | 109 ± 4 | 100 ± 5** | 0.5 ± 0.6 | 1.1 ± 0.3 | 2.5 ± 0.4 |
| Methy | 47.0 ± 0.9 | 47.1 ± 1.0 | 46.9 ± 0.8 | 299 ± 9 | 299 ± 11 | 276 ± 9** | 103 ± 6 | 101 ± 6 | 104 ± 8 | −0.2 ± 0.4 | 0.1 ± 0.4 | 1.1 ± 0.2 |
aValues are mean ± SE.
Significant difference (p < 0.05) from MSX3 + mASH (*), baseline (**), all mASH groups (***), all sASH groups (†), all time control groups (††), vehicle + mASH (†††), or all groups (‡).
Mean arterial pressure (MAP) at baseline was the same in most groups, although, for unknown reasons, it was slightly lower in vehicle time control rats (p = 0.045; Table 1), and rats later injected with methysergide plus MSX-3 and exposed to mASH (p = 0.028; Table 1). As expected, MAP decreased significantly from baseline values during ASH (p < 0.001; Table 1). All rats exposed to ASH, except for rats given methysergide and MSX-3 and exposed to mASH, or methysergide and exposed to sASH, exhibited slight but significant decreases in MAP at 60 min (8–23 mmHg, p < 0.05; Table 1). MSX-3-injected time control rats also exhibited a slight decrease in MAP at 60 min after sham ASH (12 mmHg, p = 0.005; Table 1). Such time-dependent MAP changes are characteristic of this experimental preparation (Fuller et al., 2001b; Baker-Herman and Mitchell, 2008). Previously, we demonstrated that MAP changes of ∼20 mmHg had minimal effect on respiratory activity in this experimental preparation (Bach and Mitchell, 1996). Overall, there is no clear evidence that changes in MAP from the beginning to the end of experiments, or among experimental groups, were sufficient to influence the basic conclusions of this study.
Base excess was held within 1.5 mEq/L of baseline throughout protocols, except during sASH, when base excess values significantly decreased (p < 0.001; Table 1). This decrease most likely results from transient lactic acidosis due to severe hypoxemia. However, no groups had significantly different base excess values 60 min after hypoxia versus baseline; thus, acidosis is not directly responsible for enhanced phrenic nerve activity following sASH. Each value is the average from multiple rats (vehicle, n = 10; MSX-3, n = 8; MSX-3 time control, n = 6).
Short-term hypoxic responses
ASH increased phrenic nerve burst amplitude versus baseline or time controls (p ≤ 0.002; Fig. 2). Vehicle, MSX-3, or methysergide injections had no significant effects on phrenic nerve burst amplitude during ASH (p > 0.05; Fig. 2). However, rats injected with methysergide plus MSX-3 exposed to mASH showed a significantly decreased phrenic burst amplitude response versus rats injected with vehicle (p = 0.047) or methysergide (p = 0.044) and exposed to mASH (Fig. 2). Phrenic responses to severe ASH were greater than responses to mASH; rats injected with vehicle, MSX-3, or methysergide and exposed to sASH exhibited significantly greater amplitude response versus the same drug groups during moderate ASH (p ≤ 0.036; Fig. 2), similar to the short-term hypoxic phrenic response in a prior study comparing mAIH versus sAIH (Nichols et al., 2012). No significant differences in the short-term hypoxic phrenic response were found between rats injected with vehicle, MSX-3, or methysergide during sASH (p > 0.05; Fig. 2).
Figure 2.

Short-term hypoxic phrenic responses during mASH or sASH after intrathecal injections of vehicle (open square), MSX-3 (gray square), methysergide (filled square), and methysergide + MSX-3 (square filled with diagonal crossing lines). All groups are significantly increased from time controls (no ASH, 2.5 ± 1.6%; p ≤ 0.002). &Significantly decreased versus vehicle + mASH. #Significantly decreased versus methysergide + mASH. *Significantly increased versus vehicle + mASH. †Significantly increased versus MSX-3 + mASH. ‡Significantly increased versus methysergide + mASH. Values are mean ± SE. Significance is p < 0.05.
Spinal A2A receptor blockade reveals pMF following moderate ASH
Compressed traces of phrenic neurograms illustrate phrenic burst amplitude before, during, and after mASH in rats pretreated with intrathecal injections of vehicle or MSX-3 (A2A receptor antagonist; injections indicated in Fig. 3A, black arrows). In rats given vehicle injections and exposed to mASH, there was no significant pMF versus vehicle time controls (15 ± 4% vs 10 ± 3%, respectively; p = 0.40; Fig. 3B), in agreement with previous reports from our laboratory (Baker and Mitchell, 2000; Wilkerson et al., 2008). In contrast, rats given intrathecal MSX-3 exhibited significant pMF following mASH (39 ± 5%), an effect significantly greater than rats injected with vehicle and exposed to mASH (15 ± 4%, p < 0.001; Fig. 3B), or MSX-3 time controls (0 ± 3%, p < 0.001; Fig. 3B). There was a slight, but significant, increase in phrenic burst frequency (frequency LTF) at 60 min after ASH in rats injected with MSX-3 and exposed to mASH (3.2 ± 1.5 bursts/min) versus time controls (−1.4 ± 2.8 bursts/min at 60 min, p = 0.007; Fig. 3C), but not versus rats given vehicle injections and exposed to mASH (0.4 ± 1.8 bursts/min, p = 0.06; Fig. 3C). Small, significant changes in frequency following hypoxic exposures are consistent with findings of other studies on pMF/pLTF (Baker-Herman and Mitchell, 2002, 2008; Nichols et al., 2012).
Figure 3.

Pretreatment with spinal A2A receptor antagonist (200 mm MSX-3) reveals pMF following mASH. A, Representative phrenic neurograms after mASH (PaO2 = 40–50 mmHg, 25 min) from rats given intrathecal injections of vehicle or MSX-3 (↑) compared with rats treated as time controls and not exposed to ASH after MSX-3 delivery. Dotted line indicates baseline. Gray represents pMF. B, Summary data for δ phrenic amplitude following mASH exposure in rats given vehicle (inverted gray triangle) or MSX-3 (gray circle) and exposed to mASH. MSX-3 time controls are shown for comparison (no mASH, filled circle). C, Summary data for Δ phrenic burst frequency following mASH exposure. n = 10, vehicle; n = 8, MSX-3; n = 6, MSX-3 time control (no ASH). *Significantly increased from MSX-3 time control. #Significantly increased from MSX-3 + mASH. All significant differences are p < 0.05.
Spinal serotonin receptor blockade does not reveal pMF following moderate ASH
Intrathecal methysergide injections before mASH revealed a slight but significant increase in phrenic burst amplitude (17 ± 7%) versus methysergide time controls (1 ± 3%, p = 0.007; Fig. 4B). However, rats injected with methysergide and then exposed to mASH were not significantly different from rats injected with vehicle and exposed to moderate ASH (15 ± 4%, p = 0.688), suggesting that spinal serotonin receptor blockade with methysergide does not reveal pMF following mASH. However, in addition to its effects on 5-HT2 receptors, it blocks multiple other serotonin receptor subtypes that may contribute to the adenosine-dependent S pathway to pMF, such as 5-HT7 receptors (Hoffman et al., 2011). There was no significant phrenic long-term facilitation in these groups (p > 0.05, Fig. 4C).
Figure 4.

Pretreatment with spinal 5-HT receptor antagonist (20 mm methysergide, 20 min prior ASH; methy) prevents pMF in rats injected with spinal A2A receptor antagonist (200 mm MSX-3, 12 min prior mASH) and exposed to mASH. A, Representative phrenic neurograms after mASH from rats given intrathecal injections of methysergide (↑) or both methysergide and MSX-3 (↑) compared with rats treated as time controls and not exposed to mASH after methysergide delivery. The dotted line indicates baseline. Gray represents pMF. B, Summary data for Δ phrenic amplitude in rats given methysergide (inverted gray triangle) or methysergide + MSX-3 (gray circle) and exposed to mASH or methysergide time controls (no ASH, filled circle). Rats given MSX-3 and exposed to mASH (from Fig. 1, open circle) are shown for comparison. C, Summary data for change in phrenic burst frequency (Δ frequency) following mASH exposure in the same groups. n = 8, methysergide; n = 7, methy + MSX-3; n = 7, methysergide time control (no ASH). *Significant difference versus methysergide + MSX-3 + mASH. #Significant difference versus methysergide time controls (no ASH). †Significant difference versus methysergide + mASH. All significant differences are p < 0.05.
pMF following A2A blockade and moderate ASH is serotonin-dependent
Compressed phrenic neurograms illustrate phrenic burst amplitude before, during, and after mASH in rats pretreated with intrathecal methysergide, MSX-3 or both (methysergide injections denoted by black arrows; MSX-3 injections denoted by gray arrows; Fig. 4A). Methysergide prevented pMF in rats given MSX-3 and mASH (0 ± 6%, p < 0.001 vs MSX-3 + mASH and p > 0.05 vs time controls; Fig. 4B), demonstrating that mASH-induced pMF revealed by A2A receptor inhibition is serotonin-dependent. Thus, MSX-3 revealed pMF is similar to pLTF following mild or moderate AIH (Bach and Mitchell, 1996; Fuller et al., 2001b; Baker-Herman and Mitchell, 2002; Nichols et al., 2012) (PaO2 35–55 mmHg). Methysergide also prevented frequency LTF in rats given MSX-3 plus mASH (−1.4 ± 1.6 bursts/min; p = 0.005 vs MSX-3 + mASH, p > 0.05 vs all other groups; Fig. 4C); this effect is consistent with previous reports concerning the impact of spinal methysergide on frequency LTF in previous studies of mild and moderate AIH (Baker-Herman and Mitchell, 2002; Nichols et al., 2012).
Severe ASH causes pMF via an A2A-dependent mechanism
Compressed phrenic neurograms illustrate phrenic burst amplitude before, during, and after sASH in rats pretreated with vehicle, MSX-3, or methysergide (injections denoted in Fig. 5A, black arrows). Vehicle-injected rats exhibited significant pMF following sASH (53 ± 12%) versus vehicle (10 ± 3%, p < 0.001) or drug time controls (MSX-3 or methysergide, 1 ± 2%; p < 0.001; Fig. 5B). In contrast to mASH, rats given MSX-3 and sASH no longer exhibit significant pMF versus vehicle (10 ± 3%, p = 0.966) or drug time controls (MSX-3 or methysergide, 1 ± 2%; p = 0.394; Fig. 5B). Thus, sASH-induced pMF requires spinal A2A receptor activation, similar to sAIH-induced pLTF (Nichols et al., 2012). Significant frequency LTF was not observed in rats pretreated with vehicle or MSX-3 and sASH (p > 0.05; Fig. 5C), unlike one earlier study on sAIH (Nichols et al., 2012). This discrepancy might be due to pattern-sensitive effects of severe hypoxia on frequency LTF, rat strain differences, or variability between studies because frequency LTF is typically small and inconsistent (Baker-Herman and Mitchell, 2008).
Figure 5.

sASH elicits A2A receptor-dependent pMF, which is enhanced by pretreatment with spinal 5-HT receptor blockade (20 mm methysergide, 20 min prior ASH; methy). A, Representative phrenic neurograms after sASH (PaO2 = 25–35 mmHg, 25 min) from rats given intrathecal injections of vehicle, MSX-3, or methysergide (↑). Dotted line indicates baseline. Gray represents pMF. B, Summary data for Δ phrenic amplitude in rats given vehicle (gray circle), MSX-3 (inverted gray triangle), or methysergide (gray triangle) and exposed to sASH or no hypoxia (time controls; methysergide or MSX-3 injections, filled circle). C, Summary data for change in phrenic burst frequency (Δ frequency) for the same groups. n = 8, vehicle; n = 8, MSX-3; n = 6, methysergide; n = 13, time controls. *Significant difference versus time controls. #Significant difference versus MSX-3 + sASH. ‡Significant difference versus vehicle + sASH. All significant differences are p < 0.05.
Spinal serotonin receptor blockade enhances sASH-induced pMF
Rats pretreated with spinal methysergide (Fig. 4A, black arrow) exhibited significantly higher pMF (87 ± 26%) versus vehicle-injected rats after sASH (53 ± 12%, p = 0.011; Fig. 5B). Enhanced pMF was present at each time after sASH (p ≤ 0.011 vs vehicle + sASH), suggesting that the enhancement occurs during sASH per se. In contrast, pLTF in rats exposed to sAIH is unaffected by spinal methysergide (Nichols et al., 2012). Spinal methysergide revealed a slight, but significant, frequency LTF (6.2 ± 2.2 bursts/min) versus drug time controls (MSX-3 or methysergide, −0.4 ± 1.3 bursts/min; p < 0.001; Fig. 5C), or vehicle-injected rats exposed to sASH (2.6 ± 1.1 bursts/min, p = 0.049; Fig. 5C).
sASH elicits pMF, but moderate ASH does not
Although mASH did not elicit significant pMF (15 ± 4%) in vehicle-injected rats (vs time controls), sASH did (severe ASH: 53 ± 12%; time controls: 10 ± 3%, p < 0.001). Responses to mAIH and sAIH were significantly different from one another (p < 0.001; Fig. 6A), suggesting that the severity of hypoxia in addition to pattern (intermittent vs sustained) is a key factor regulating phrenic motor plasticity.
Figure 6.
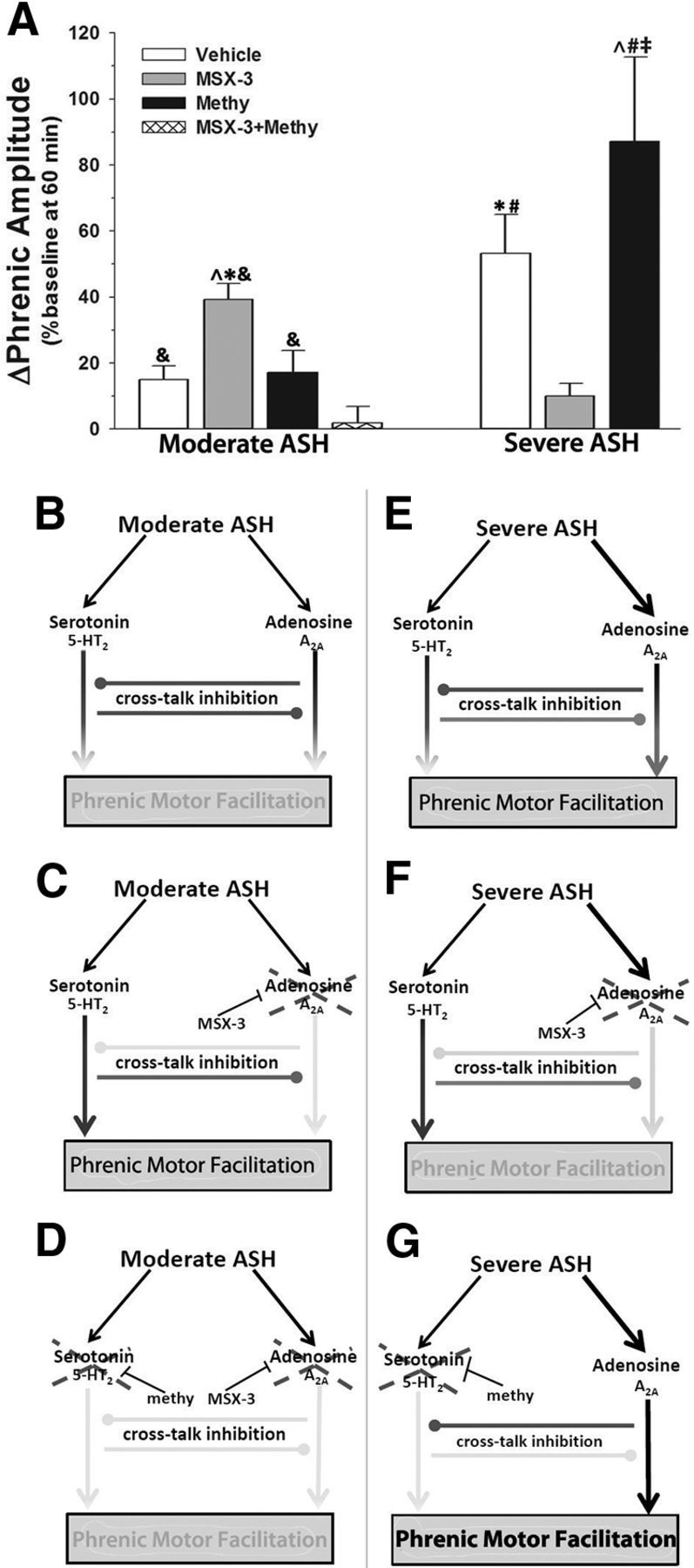
Competing 5-HT2/A2A-dependent mechanisms could explain pattern sensitivity of pMF induced by moderate hypoxia. A, Summary of pMF following mASH or sASH in rats treated with vehicle, 200 mm MSX-3, 20 mm methysergide, or 20 mm methysergide + 200 mm MSX-3. B, Proposed 5-HT2/A2A interactions predict magnitude of pMF. ↓ indicates activation. Gray line with gray circle represents inhibition. Gray line with intersecting bar represents blockade by intrathecal drug. During mASH, balanced activation of 5-HT2- and A2A-dependent pathways allows for cross talk inhibition to constrain expression of pMF. C, Spinal blockade of A2A pathway with MSX-3 during mASH prevents cross talk inhibition of the 5-HT2 pathway, revealing pMF. D, Blockade of serotonin receptors with methysergide (methy) before A2A receptor blockade and mASH exposure prevents pMF, demonstrating that cross talk inhibition from A2A-dependent pathway restrains 5-HT2-dependent pMF during mASH. E, sASH causes pMF because of greater relative A2A receptor activation, overcoming cross talk inhibition. F, Blockade of spinal A2A receptors prevents expression of pMF following severe ASH, demonstrating that pMF following severe ASH is A2A-dependent. G, Blockade of spinal serotonin receptors prevents cross talk inhibition of the A2A pathway during severe ASH, causing enhanced pMF. *Significant versus vehicle + mASH. ∧Significant versus methysergide + mASH. &Significant versus methysergide + MSX-3 + mASH. #Significant versus MX3 + sASH. ‡Significant versus vehicle + sASH.
Discussion
The fundamental conclusion of this study is that both moderate and severe hypoxia evoke pMF via competing serotonin- and adenosine-dependent mechanisms. Interactions between these pathways ultimately determine pMF expression. The magnitude of adenosine and serotonin-dependent pathway activation, and their relative contributions to pMF depend on the pattern and severity of hypoxia. Our findings are summarized in a simplified model (Fig. 6). During mASH, serotonin and adenosine receptor activation are balanced, preventing pMF due to offsetting cross talk inhibition (Fig. 6B). Thus, by blocking spinal A2A receptors during mASH, a serotonin-dependent pMF is revealed (Fig. 6C). This pMF revealed by MSX-3 is serotonin dependent because it was blocked by methysergide, similar to serotonin-dependent, mAIH-induced pLTF (Fig. 6D).
In contrast to mASH, sASH elicits A2A-dependent pMF (Fig. 6E,F); because sASH induced pMF is enhanced by spinal methysergide, serotonin receptor activation inhibits A2A receptor-dependent pMF (Fig. 6G). Together, these results suggest mutual inhibitory interactions between serotonin- and adenosine-dependent pathways to pMF, and that these inhibitory interactions modulate pMF with both mAIH and sASH. We conclude that pattern sensitivity in hypoxia-induced pMF/pLTF occurs when the cross talk inhibition is balanced and offsetting, negating the expression of either mechanism (i.e., no pMF). The offsetting effects (i.e., pattern sensitivity) can be explained by inhibitory interactions between serotonin- and adenosine-dependent pathways to pMF. In contrast, severe ASH induces pMF whether delivered as AIH or ASH, likely because adenosine receptor activation exceeds serotonin receptor activation in both cases (for clarification of this concept, see Fig. 1A).
Serotonin receptor activation during sASH constrains A2A-dependent pMF
Although several studies demonstrated that A2A receptors inhibit 5-HT2 receptor-dependent pMF following mAIH (Hoffman et al., 2010; Nichols et al., 2012; Navarrete-Opazo and Mitchell, 2014), less information is available concerning the impact of serotonin receptors on adenosine-dependent pMF with sAIH or sASH. Here we demonstrate that spinal serotonin receptor activation with sASH constrains A2A-dependent pMF (Fig. 4). However, both A2A (Golder et al., 2008; Hoffman et al., 2010) and 5-HT7 receptors (Hoffman and Mitchell, 2011, 2013) activate Gs-coupled receptors (S pathway) and constrain Gq-protein coupled receptor-dependent pLTF (Q pathway) following mAIH (Dale-Nagle et al., 2010). Because methysergide is a potent inhibitor of both 5-HT2 (Gq) and 5-HT7 (Gs) receptors (Peroutka, 1988; Villalón et al., 1997), we may have underestimated the magnitude of serotonin (5-HT2) receptor inhibition of severe hypoxia-induced pMF because methysergide is also expected to reduce 5-HT7 contributions to pMF with severe hypoxia. Nevertheless, inhibitory interactions between serotonin- and adenosine-dependent pathways to pMF appear to be bidirectional, assuring that either serotonergic or adenosinergic contributions to pMF predominate, except when these cross talk interactions are balanced and offsetting, cancelling pMF.
Because methysergide did not reveal significant pMF following mASH (Fig. 5A), A2A receptor activation alone is not sufficient to elicit pMF with mASH. However, the failure of methysergide to reveal pMF after mASH could be due to known methysergide effects on 5-HT7 receptors (see above). These receptors signal differently from 5-HT2 receptors that underlie mAIH-induced pLTF (Fuller et al., 2001b; Macfarlane et al., 2011) but signal more like the Gs-coupled A2A receptors. Indeed, 5-HT7 receptor activation alone elicits pMF (Hoffman and Mitchell, 2011) and constrains mAIH-induced pMF (Hoffman and Mitchell, 2013). Because A2A and 5-HT7 receptors activate similar (cAMP-dependent) signaling cascades, coactivation may be necessary for pMF following mASH. Because 5-HT7 receptors may contribute to the A2A-dependent pMF with mASH, selective 5-HT2 receptor inhibition (e.g., ketanserin) during mASH may reveal pMF.
Cross talk interactions via downstream signaling molecules
Inhibitory coupling between A2A and 5-HT2 receptor-induced pMF likely occurs via downstream signaling cascades, such as protein kinase C and adenylate cyclase activation. Because spinal protein kinase A (PKA) attenuates, and PKA inhibition enhances mAIH-induced pLTF (Hoffman and Mitchell, 2013), PKA is essential for S (A2A, 5-HT7) to Q pathway inhibition (5-HT2). Alternative cAMP signaling underlies the S pathway to pMF, specifically EPAC (Fields et al., 2015). In contrast, 5-HT2-dependent pMF following mAIH requires protein kinase Cθ activity (Devinney et al., 2015). Cross talk interactions between PKC and PKA pathways are known in other model systems (e.g., Aplysia) where PKC constrains PKA activity and prevents sensorimotor long-term facilitation (Farah et al., 2009).
PKA also negatively regulates PKC activity via modulation of reactive oxygen species formation. In cell cultures, PKA decreases ROS production by phosphorylating the NADPH oxidase A1 subunit (Kim et al., 2007), or via downregulation of the p47 subunit (Bengis-Garber and Gruener, 1996). NADPH oxidase activity is required for pLTF induced by mAIH, most likely because reactive oxygen species inhibit constraining protein phosphatases (Wilkerson et al., 2007; Macfarlane et al., 2009). Multiple inhibitory cross talk interactions may occur between PKC-dependent (e.g., 5-HT2) and PKA-dependent (e.g., A2A and 5-HT7 receptor) signaling, allowing for multilevel control of different cellular pathways to pMF.
Pattern sensitivity
Patterned stimuli have a profound influence on the expression of plasticity. The benefits of “spaced” training on long-term memory formation have long been appreciated and are being actively explored to refine therapeutic approaches to learning disorders (Ebbinghaus, 1913; Philips et al., 2013a). However, the cellular and molecular correlates of pattern sensitivity have seldom been explored and have been mostly studied in invertebrate model systems, such as sensorimotor long-term facilitation in Aplysia (Mauelshagen et al., 1998). Studies of pattern sensitivity in pLTF demonstrated that okadaic acid-sensitive serine/threonine phosphatases constrain (and prevent) serotonin-dependent pMF with mASH, suggesting that serotonin-dependent mechanisms may be activated but undermined by concurrent phosphatase activity (Wilkerson et al., 2007, 2008).
Here, we present evidence for a novel mechanism of pattern sensitivity in spinal neuroplasticity: mutual, inhibitory coupling between competing 5-HT2 and A2A-induced signaling cascades. Pattern sensitivity in Aplysia sensorimotor long-term facilitations occurs at multiple levels, including G-proteins (Ye et al., 2008), temporal integration of PKA and MAPK signaling (Philips et al., 2013b), and PKA inhibition from increased PKC activity (Farah et al., 2009). An emerging principle from all of these studies is that pattern sensitivity arises from inhibitory interactions between signaling molecules involved in different forms of plasticity. The present study is consistent with this principle and indicates that pattern sensitivity in hypoxia-induced pMF emerges from inhibitory coupling between Gq and Gs protein-coupled signaling cascades.
Some forms of memory and neuroplasticity lack apparent pattern sensitivity. For example, short-term memory, which lasts on the order of minutes, is not subject to spacing effects (Xia et al., 1998). Similarly, traumatic/emotional memories (e.g., flashbulb memory) are induced by a single event and form long-term memories via mechanisms distinct from long-term memories produced by spaced trials (Irvine et al., 2005, 2006; Diamond et al., 2007). Plasticity without apparent pattern sensitivity may allow storage of critical memories following a single trial of intense stimuli, such as remembering never to touch a hot stove again.
Here, sASH elicited pMF by a mechanism insensitive to the pattern of hypoxia. We postulate that sASH causes greater extracellular adenosine accumulation and, thus, greater A2A receptor activation (vs 5-HT2 receptors; Fig. 1A). This pattern insensitivity might elevate respiratory motor output after even a single, intense hypoxic episode, analogous to touching a hot stove. Similar to “flashbulb memories” after traumatic events, sASH-induced plasticity may permit significant adaptation in response to life-threatening events, such as transient respiratory failure, severely hypoxic environments, or end stages of respiratory disease. Pattern-insensitive plasticity may allow more rapid and complete adaptations in response to such life-threatening events.
Clinical relevance
In recent years, we have elucidated multiple mechanisms, each capable of giving rise to spinal, respiratory plasticity. We are just now beginning to appreciate how these mechanisms interact to influence the expression of plasticity after a given stimulus (including its pattern, severity, and duration). We demonstrate that pharmacological inhibition of spinal A2A receptors disrupts cross talk inhibition, revealing pMF following mASH, a stimulus that does not ordinarily trigger pMF (Baker and Mitchell, 2000; Wilkerson et al., 2008). Furthermore, sASH elicits A2A-dependent pMF, an effect enhanced by spinal serotonin receptor inhibition. A detailed understanding of mechanisms giving rise to pMF may guide future clinical applications based on the ability to elicit motor plasticity with hypoxia and/or small molecules that manipulate this intricate system (Mahamed and Mitchell, 2007). Such therapeutic approaches are currently in development for diverse clinical disorders that challenge respiratory (Mahamed and Mitchell, 2007; Lovett-Barr et al., 2012; Nichols et al., 2013) and nonrespiratory motor function (Trumbower et al., 2012; Hayes et al., 2014), including spinal cord injury, amyotrophic lateral sclerosis, and sleep apnea.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants HL080209, HL111598, and HL69064. M.J.D. was supported by NIH Grants T32 007654 and T32 GM007507. N.L.N. was supported by the Francis Families Foundation. We thank Bradley Wathen for expert technical assistance.
The authors declare no competing financial interests.
References
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol. 2000;529:215–219. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Determinants of frequency long-term facilitation following acute intermittent hypoxia in vagotomized rats. Respir Physiol Neurobiol. 2008;162:8–17. doi: 10.1016/j.resp.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Bengis-Garber C, Gruener N. Protein kinase A downregulates the phosphorylation of p47 phox in human neutrophils: a possible pathway for inhibition of the respiratory burst. Cell Signal. 1996;8:291–296. doi: 10.1016/0898-6568(96)00052-6. [DOI] [PubMed] [Google Scholar]
- Conde SV, Monteiro EC. Hypoxia induces adenosine release from the rat carotid body. J Neurochem. 2004;89:1148–1156. doi: 10.1111/j.1471-4159.2004.02380.x. [DOI] [PubMed] [Google Scholar]
- Dale EA, Ben Mabrouk F, Mitchell GS. Unexpected benefits of intermittent hypoxia: enhanced respiratory and nonrespiratory motor function. Physiology (Bethesda) 2014;29:39–48. doi: 10.1152/physiol.00012.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale EA, Fields DP, Devinney MJ, Mitchell GS. Phrenic motor neuron TrkB expression is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. Exp Neurol. 2016;S0014–4886(16):30123–30126. doi: 10.1016/j.expneurol.2016.05.012. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Frenguelli BG. Release of adenosine and ATP during ischemia and epilepsy. Curr Neuropharmacol. 2009;7:160–179. doi: 10.2174/157015909789152146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale-Nagle EA, Hoffman MS, MacFarlane PM, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv Exp Med Biol. 2010;669:225–230. doi: 10.1007/978-1-4419-5692-7_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinney MJ, Huxtable AG, Nichols NL, Mitchell GS. Hypoxia-induced phrenic long-term facilitation: emergent properties. Ann N Y Acad Sci. 2013;1279:143–153. doi: 10.1111/nyas.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinney MJ, Fields DP, Huxtable AG, Peterson TJ, Dale EA, Mitchell GS. Phrenic long-term facilitation requires PKCθ activity within phrenic motor neurons. J Neurosci. 2015;35:8107–8117. doi: 10.1523/JNEUROSCI.5086-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond DM, Campbell AM, Park CR, Halonen J, Zoladz PR. The temporal dynamics model of emotional memory processing: a synthesis on the neurobiological basis of stress-induced amnesia, flashbulb and traumatic memories, and the Yerkes-Dodson law. Neural Plast. 2007;2007:60803. doi: 10.1155/2007/60803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty BJ, Fields DP, Mitchell GS. Mammalian target of rapamycin is required for phrenic long-term facilitation following severe but not moderate acute intermittent hypoxia. J Neurophysiol. 2015;114:1784–1791. doi: 10.1152/jn.00539.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbinghaus H. Memory: a contribution to experimental psychology. New York: Columbia University; 1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah CA, Weatherill D, Dunn TW, Sossin WS. PKC differentially translocates during spaced and massed training in Aplysia. J Neurosci. 2009;29:10281–10286. doi: 10.1523/JNEUROSCI.1533-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields DP, Mitchell GS. Spinal metaplasticity in respiratory motor control. Front Neural Circuits. 2015;9:2. doi: 10.3389/fncir.2015.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields DP, Springborn SR, Mitchell GS. Spinal 5-HT7 receptors induce phrenic motor facilitation via EPAC-mTORC1 signaling. J Neurophysiol. 2015;114:2015–2022. doi: 10.1152/jn.00374.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Baker TL, Behan M, Mitchell GS. Expression of hypoglossal long-term facilitation differs between substrains of Sprague-Dawley rat. Physiol Genomics. 2001a;4:175–181. doi: 10.1152/physiolgenomics.2001.4.3.175. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol. 2001b;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. discussion 2000. [DOI] [PubMed] [Google Scholar]
- Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, Baker-Herman TL, Mitchell GS. Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J Neurosci. 2008;28:2033–2042. doi: 10.1523/JNEUROSCI.3570-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rothi EJ, Lee KZ, Dale EA, Reier PJ, Mitchell GS, Fuller DD. Intermittent hypoxia and neurorehabilitation. J Appl Physiol. 2015;119:1455–1465. doi: 10.1152/japplphysiol.00235.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Llaudet E, Dale N, Spyer KM. Release of ATP in the ventral medulla during hypoxia in rats: role in hypoxic ventilatory response. J Neurosci. 2005;25:1211–1218. doi: 10.1523/JNEUROSCI.3763-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes HB, Jayaraman A, Herrmann M, Mitchell GS, Rymer WZ, Trumbower RD. Daily intermittent hypoxia enhances walking after chronic spinal cord injury: a randomized trial. Neurology. 2014;82:104–113. doi: 10.1212/01.WNL.0000437416.34298.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Mitchell GS. Spinal 5-HT7 receptor activation induces long-lasting phrenic motor facilitation. J Physiol. 2011;589:1397–1407. doi: 10.1113/jphysiol.2010.201657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Mitchell GS. Spinal 5-HT7 receptors and protein kinase A constrain intermittent hypoxia-induced phrenic long-term facilitation. Neuroscience. 2013;250:632–643. doi: 10.1016/j.neuroscience.2013.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Golder FJ, Mahamed S, Mitchell GS. Spinal adenosine A2(A) receptor inhibition enhances phrenic long term facilitation following acute intermittent hypoxia. J Physiol. 2010;588:255–266. doi: 10.1113/jphysiol.2009.180075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Nichols NL, Macfarlane PM, Mitchell GS. Phrenic long-term facilitation after acute intermittent hypoxia requires spinal ERK activation but not TrkB synthesis. J Appl Physiol. 2012;113:1184–1193. doi: 10.1152/japplphysiol.00098.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine EE, Vernon J, Giese KP. AlphaCaMKII autophosphorylation contributes to rapid learning but is not necessary for memory. Nat Neurosci. 2005;8:411–412. doi: 10.1038/nn1431. [DOI] [PubMed] [Google Scholar]
- Irvine EE, von Hertzen LS, Plattner F, Giese KP. alphaCaMKII autophosphorylation: a fast track to memory. Trends Neurosci. 2006;29:459–465. doi: 10.1016/j.tins.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Kim JS, Diebold BA, Babior BM, Knaus UG, Bokoch GM. Regulation of Nox1 activity via protein kinase A-mediated phosphorylation of NoxA1 and 14-3-3 binding. J Biol Chem. 2007;282:34787–34800. doi: 10.1074/jbc.M704754200. [DOI] [PubMed] [Google Scholar]
- Lovett-Barr MR, Satriotomo I, Muir GD, Wilkerson JE, Hoffman MS, Vinit S, Mitchell GS. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. J Neurosci. 2012;32:3591–3600. doi: 10.1523/JNEUROSCI.2908-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane PM, Mitchell GS. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J Physiol. 2009;587:5469–5481. doi: 10.1113/jphysiol.2009.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane PM, Satriotomo I, Windelborn JA, Mitchell GS. NADPH oxidase activity is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. J Physiol. 2009;587:1931–1942. doi: 10.1113/jphysiol.2008.165597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane PM, Vinit S, Mitchell GS. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neuroscience. 2011;178:45–55. doi: 10.1016/j.neuroscience.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol. 2007;92:27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- Mauelshagen J, Sherff CM, Carew TJ. Differential induction of long-term synaptic facilitation by spaced and massed applications of serotonin at sensory neuron synapses of Aplysia californica. Learn Mem. 1998;5:246–256. [PMC free article] [PubMed] [Google Scholar]
- Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol. 2003;94:358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Navarrete-Opazo A, Mitchell GS. Recruitment and plasticity in diaphragm, intercostal and abdominal muscles in unanesthezised rats. J Appl Physiol. 2014a;117:180–188. doi: 10.1152/japplphysiol.00130.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete-Opazo A, Mitchell GS. Therapeutic potential of intermittent hypoxia: a matter of dose. Am J Physiol Regul Integr Comp Physiol. 2014b;307:R1181–R1197. doi: 10.1152/ajpregu.00208.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Dale EA, Mitchell GS. Severe acute intermittent hypoxia elicits phrenic long-term facilitation by a novel adenosine-dependent mechanism. J Appl Physiol. 2012;112:1678–1688. doi: 10.1152/japplphysiol.00060.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN, Mitchell GS. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am J Respir Crit Care Med. 2013;187:535–542. doi: 10.1164/rccm.201206-1072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroutka SJ. Antimigraine drug interactions with serotonin receptor subtypes in human brain. Ann Neurol. 1988;23:500–504. doi: 10.1002/ana.410230512. [DOI] [PubMed] [Google Scholar]
- Philips GT, Kopec AM, Carew TJ. Pattern and predictability in memory formation: from molecular mechanisms to clinical relevance. Neurobiol Learn Mem. 2013a;105:117–124. doi: 10.1016/j.nlm.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips GT, Ye X, Kopec AM, Carew TJ. MAPK establishes a molecular context that defines effective training patterns for long-term memory formation. J Neurosci. 2013b;33:7565–7573. doi: 10.1523/JNEUROSCI.5561-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumbower RD, Jayaraman A, Mitchell GS, Rymer WZ. Exposure to acute intermittent hypoxia augments somatic motor function in humans with incomplete spinal cord injury. Neurorehabil Neural Repair. 2012;26:163–172. doi: 10.1177/1545968311412055. [DOI] [PubMed] [Google Scholar]
- Villalón CM, Heiligers JP, Centurión D, De Vries P, Saxena PR. Characterization of putative 5-HT7 receptors mediating tachycardia in the cat. Br J Pharmacol. 1997;121:1187–1195. doi: 10.1038/sj.bjp.0701260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JE, Macfarlane PM, Hoffman MS, Mitchell GS. Respiratory plasticity following intermittent hypoxia: roles of protein phosphatases and reactive oxygen species. Biochem Soc Trans. 2007;35:1269–1272. doi: 10.1042/BST0351269. [DOI] [PubMed] [Google Scholar]
- Wilkerson JE, Satriotomo I, Baker-Herman TL, Watters JJ, Mitchell GS. Okadaic acid-sensitive protein phosphatases constrain phrenic long-term facilitation after sustained hypoxia. J Neurosci. 2008;28:2949–2958. doi: 10.1523/JNEUROSCI.5539-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia SZ, Feng CH, Guo AK. Multiple-phase model of memory consolidation confirmed by behavioral and pharmacological analyses of operant conditioning in Drosophila. Pharmacol Biochem Behav. 1998;60:809–816. doi: 10.1016/S0091-3057(98)00060-4. [DOI] [PubMed] [Google Scholar]
- Ye X, Shobe JL, Sharma SK, Marina A, Carew TJ. Small G proteins exhibit pattern sensitivity in MAPK activation during the induction of memory and synaptic facilitation in Aplysia. Proc Natl Acad Sci U S A. 2008;105:20511–20516. doi: 10.1073/pnas.0808110105. [DOI] [PMC free article] [PubMed] [Google Scholar]