

Abstract
Amplified neuroinflammatory responses following an immune challenge occur with normal aging and can elicit or exacerbate neuropathology. The mechanisms mediating this sensitized or “primed” immune response in the aged brain are not fully understood. The alarmin high mobility group box 1 (HMGB1) can be released under chronic pathological conditions and initiate inflammatory cascades. This led us to investigate whether HMGB1 regulates age-related priming of the neuroinflammatory response. Here, we show that HMGB1 protein and mRNA were elevated in the hippocampus of unmanipulated aged rats (24-month-old F344XBN rats). Furthermore, aged rats had increased HMGB1 in the CSF, suggesting increased HMGB1 release. We demonstrate that blocking HMGB1 signaling with an intracisterna magna (ICM) injection of the competitive antagonist to HMGB1, Box-A, downregulates basal expression of several inflammatory pathway genes in the hippocampus of aged rats. This indicates that blocking the actions of HMGB1 might reduce age-associated inflammatory priming. To test this hypothesis, we evaluated whether HMGB1 antagonism blocks the protracted neuroinflammatory and sickness response to peripheral Escherichia coli (E. coli) infection in aged rats. ICM pretreatment of aged rats with Box-A 24 h before E. coli infection prevented the extended hippocampal cytokine response and associated cognitive and affective behavioral changes. ICM pretreatment with Box-A also inhibited aging-induced potentiation of the microglial proinflammatory response to lipopolysaccharide ex vivo. Together, these results suggest that HMGB1 mediates neuroinflammatory priming in the aged brain. Blocking the actions of HMGB1 appears to “desensitize” aged microglia to an immune challenge, thereby preventing exaggerated behavioral and neuroinflammatory responses following infection.
SIGNIFICANCE STATEMENT The world's population is aging, highlighting a need to develop treatments that promote quality of life in aged individuals. Normal aging is associated with precipitous drops in cognition, typically following events that induce peripheral inflammation (e.g., infection, surgery, heart attack). Peripheral immune stimuli cause exaggerated immune responses in the aged brain, which likely underlie these behavioral deficits. Here, we investigated whether the alarmin high mobility group box 1 (HMGB1) mediates age-associated “priming” of the neuroinflammatory response. HMGB1 is elevated in aged rodent brain and CSF. Blocking HMGB1 signaling downregulated expression of inflammatory pathway genes in aged rat brain. Further, HMGB1 antagonism prevented prolonged infection-induced neuroinflammatory and sickness responses in aged rats. Overall, blocking HMGB1 “desensitized” microglia in the aged brain, thereby preventing pathological infection-elicited neuroinflammatory responses.
Keywords: danger-associated molecular patterns, healthy aging, microglia, neuroimmunology, sickness behavior
Introduction
Cognitive function gradually declines as people age. With normal healthy aging, this decline is typically minor; however, aged individuals are susceptible to delirium and precipitous drops in cognitive function. In aged individuals, delirium is associated with innate immune system activation (Simone and Tan, 2011) and frequently follows events that induce peripheral inflammation, such as infection (Ehlenbach et al., 2010), surgery (Bekker and Weeks, 2003), or injury (Cape et al., 2014). Peripheral immune stimuli cause exaggerated immune responses in the aged brain and these exaggerated neuroinflammatory responses appear fundamental to age-associated cognitive declines (Godbout et al., 2005; Barrientos et al., 2006).
Altered inflammatory potential in the aged brain is due, in part, to changes in microglia, the predominant innate immune cells of the brain and the major CNS source of inflammatory molecules (Barrientos et al., 2015b). Microglia exhibit a quiescent surveillant phenotype under nonpathological conditions. Upon detecting a pathogen or endogenous danger signal, microglia undergo rapid morphological and functional changes that initiate an inflammatory response (Ransohoff and Perry, 2009). These functional and morphological changes, however, are not “all or nothing.” Microglia challenged with an initial minor physiological or inflammatory stimulus can enter a primed (sensitized) state. Primed microglia display a functional inflammatory phenotype similar to quiescent microglia, but produce an exaggerated inflammatory response to secondary immune stimulation (Perry and Teeling, 2013). Microglia from aged animals display this primed immunophenotype, which has several features: (1) upregulation of key proinflammatory immune receptors [e.g., toll-like receptors (TLRs)], (2) increased major histocompatibility complex II (MHCII), and (3) downregulation of factors involved in terminating inflammatory responses, including CD200 and CX3CR1 (Henry et al., 2009; Frank et al., 2010b; Wynne et al., 2010). Microglia from aged animals do not release more inflammatory molecules under basal conditions (but see Griffin et al., 2006) but demonstrate a potentiated proinflammatory response to immune activation (Frank et al., 2010b).
Interestingly, there is a higher density of microglia in the hippocampus (Maier and Watkins, 1998) and prolonged neuroinflammatory responses in the aged hippocampus following peripheral immune stimuli are largely mediated by microglia (Chen et al., 2008; Frank et al., 2010b). These heightened neuroinflammatory responses in aged animals likely underlie cognitive impairments induced by peripheral immune challenges (Barrientos et al., 2006, 2009, 2012; Frank et al., 2006b, 2010a). For example, blocking the actions of interleukin (IL)-1β with an IL-1 receptor antagonist prevents postoperative cognitive decline in aged rats (Barrientos et al., 2012). However, the neuroimmune processes that initiate age-related microglial priming remain unclear.
Innate immune cells can be activated in the absence of pathogens by endogenous molecules, such as danger-associated molecular patterns (DAMPs), acting on pattern-recognition receptors (Schaefer, 2014). Microglial priming in the aged brain could be caused by atypical accumulation of DAMPs, including high mobility group box 1 (HMGB1) protein (Kapetanovic et al., 2015). HMGB1, a chromatin-binding factor localized to the nucleus, is released under pathological conditions (e.g., sepsis, stroke, Alzheimer's disease; Lotze and Tracey, 2005). Extracellular HMGB1 interacts with TLR2, TLR4, and the receptor for advanced glycation end products (RAGE), thereby driving proinflammatory responses (Yang and Tracey, 2010). HMGB1 is released in the brain both actively by microglia and passively by necrotic or damaged cells (Scaffidi et al., 2002).
Importantly, extranuclear HMGB1 does not necessarily induce a proinflammatory cytokine response in the brain; rather, HMGB1 can potentiate the effects of a subsequent neuroinflammatory challenge (Weber et al., 2015). This priming effect implicates HMGB1 in the sensitized inflammatory response in the aged brain. In support of this prediction, here we show that HMGB1 is elevated in the CSF and hippocampus of aged rats. Furthermore, we demonstrate that blocking HMGB1 actions with the competitive antagonist Box-A (described below) in the aged CNS can prevent the heightened neuroinflammatory and sickness responses to a peripheral immune challenge.
Materials and Methods
Animals.
Young (3-month-old) and old (24-month-old) male F344XBN F1 rats were used in these experiments. Rats of this age and strain were selected to study normal, non-neurodegenerative aging because they are relatively healthy and not near senescence (these rats live for well over 30 months). Rats were received from the National Institute on Aging and pair-housed (cages measuring 52 (length) × 30 (width) × 21 (height) cm) with an age-matched conspecific. Food and water were available ad libitum and rats were maintained at an ambient temperature of 22 ± 2°C. Rats were given ≥2 weeks to acclimate to colony conditions before experimentation began. All rats were maintained on a 12:12 light/dark cycle with lights on at 7:00 A.M. All experimental procedures were conducted in accordance with the University of Colorado Institutional Animal Care and Use Committee and the Guide of the Care and Use of Laboratory Animals (eighth edition). Efforts were made to minimize animal use and discomfort.
Box-A intracisterna magna injections and CSF collection.
Intracisterna magna (ICM) injections of the HMGB1 antagonist Box-A (HMGBiotech) were conducted as previously described (Weber et al., 2015). HMGB1 is composed of three distinct protein domains: two DNA-binding sequences termed Box-A and Box-B, and a negatively charged C terminus. The proinflammatory effects of this molecule reside in Box-B, whereas the isolated Box-A portion of HMGB1 is not immunoreactive and acts as a competitive antagonist to HMGB1. Indeed, Box-A protein significantly displaces saturable 125I-labeled HMGB1 binding in cultured macrophages and blocks HMGB1 from binding to and activating its cognate immune receptors (RAGE, TLR4, and TLR2; Andersson et al., 2002; Yang et al., 2004). For the ICM injections, rats were anesthetized with 5% isoflurane in oxygen and then maintained on 1.5% isoflurane during the brief procedure (∼3 min). The dorsal aspect of the skull was shaved and swabbed with 70% EtOH. A sterile 27 gauge needle attached via PE50 tubing to a 25 μl Hamilton syringe was inserted into the cisterna magna (verified by withdrawing 2 μl of clear CSF). Ten micrograms of Box-A [HMGBiotech; certified lipopolysaccharide (LPS) free] in 5 μl of pyrogen-free sterile water or vehicle was delivered.
The CSF collection followed the same procedure except instead of an injection, 0.2 ml of clear CSF was collected via PE50 tubing attached to a sterile 1 ml syringe. CSF was then spun down at 1000 × g for 10 min and the supernatant was collected and stored at −80°C.
Escherichia coli administration.
Rats received a single intraperitoneal injection of either Escherichia coli [2.5 × 109 colony-forming units (CFUs) suspended in 0.25 ml of sterile saline; ATCC, #15746[ or vehicle 2 h after lights on. E. coli was used rather than the more typical immune stimulant LPS for the following reasons: (1) E. coli affects a broad range of immune targets (LPS mostly acts via TLR4); (2) E. coli is more ecologically relevant; and (3) this maintains consistency with our previous studies (Barrientos et al., 2006, 2009). All rats received the same dose of E. coli as this is the more conservative procedure since aged rats are much heavier and would receive more E. coli if the dose was adjusted for body mass.
ELISA and Western blot.
Hippocampal samples were sonicated on ice using a tissue-extraction reagent (Invitrogen) supplemented with protease inhibitor mixture (Sigma-Aldrich). Homogenates were centrifuged (14,000 × g for 10 min at 4°C) and supernatants collected and stored at −20°C. Total protein was quantified using a Bradford assay. ELISAs for rat IL-1β (R&D Systems) were run in duplicate according to the manufacturer's instructions. An ELISA for rat HMGB1 (LifeSpan BioSciences) was used to measure HMGB1 in CSF according to the manufacturer's instructions.
For Western blot, samples were heated to 75°C for 10 min and loaded into a standard polyacrylamide Bis-Tris gel (Invitrogen). SDS-PAGE was performed in 3-(N-morpholino)-propanesulfonic acid running buffer (Invitrogen) at 175 V for ∼75 min. Protein was transferred onto a nitrocellulose membrane using an iBlot dry transfer system (Invitrogen). The membrane was then blocked with Odyssey blocking buffer (LI-COR Biosciences) for 1 h and incubated overnight at 4°C in block buffer with the following primary antibodies: rabbit anti-rat HMGB-1 monoclonal antibody (1:4000; Abcam) and mouse anti-rat β-actin (1:200,000; Sigma-Aldrich). The membrane was washed in 1× PBS + 0.1% Tween and then incubated in blocking buffer containing either goat anti-rabbit or goat anti-mouse (LI-COR) IRDye 800CW secondary antibody at a concentration of 1:10,000 (LI-COR) for 1 h at room temperature. Protein expression was quantified using an Odyssey Infrared Imager (LI-COR) and expressed relative to the housekeeping protein β-actin.
Quantitative PCR.
Rat primers were previously designed using GenBank at the National Center for Biotechnology Information (NCBI), the Operon Oligo Analysis Tool, and the Basic Local Alignment Search Tool at NCBI and obtained from Invitrogen. Primers were designed to span exon/exon boundaries and thus exclude amplification of genomic DNA. Primer specificity was verified by melt curve analysis. Primers included β-actin (forward, TTCCTTCCTGGGTATGGAAT; reverse, GAGGAGCAATGATCTTGATC), HMGB1 (forward, GAGGTGGAAGACCATGTCTG; reverse, AAGAAGAAGGCCGAAGGAGG), IL-1β (forward, CCTTGTGCAAGTGTCTGAAG; reverse, GGGCTTGGAAGCAATCCTTA), IL-6 (forward, AGAAAAGAGTTGTGCAATGGCA; reverse, GGCAAATTTCCTGGTTATATCC), IL-10 (forward, GGACTTTAAGGGTTACTTGGG; reverse, AGAAATCGATGACAGCGTCG), IL-18 (forward, GACCGAACAGCCAACGAATC; reverse, GGGTAGACATCCTTCCATCC), MHCII (forward, AGCACTGGGAGTTTGAAGAG; reverse, AAGCCATCACCTCCTGGTAT), NLRP3 (forward, AGAAGCTGGGGTTGGTGAATTR; reverse, GTTGTCTAACTCCAGCATCTG), P2X7 (forward, TTTCGGTTTGGCCACCGTGT; reverse, ACTTTAACGTCGGCTTGGGC), TLR2 (forward, TGGAGGTCTCCAGGTCAAATC; reverse, ACAGAGATGCCTGGGCAGAAT), TLR4 (forward, TCCCTGCATAGAGGTACTTC; reverse, CACACCTGGATAAATCCAGC), and TNFα (forward, CAAGGAGGAGAAGTTCCCA; reverse, TTGGTGGTTTGCTACGACG). RNA was extracted from hippocampal homogenates using TRIzol reagent and 2 μg of RNA was reversed transcribed to cDNA using Superscript II (Life Technologies) according to the manufacturer's instructions. RNA was isolated from microglia and reversed transcribed to cDNA using the SuperScript III CellsDirect cDNA Synthesis System (Life Technologies). PCR amplification of cDNA was performed using the Quantitect SYBR Green PCR Kit (Qiagen) with a MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad). Gene expression was determined in duplicate and expressed relative to β-actin. There were no group differences in β-actin expression.
Immunohistochemistry.
Rats received a lethal injection of sodium pentobarbital. After rats were completely unresponsive, they were perfused transcardially with ice-cold saline followed by 4% paraformaldehyde. Brains were removed, postfixed overnight in 4% paraformaldehyde, cyroprotected in 30% sucrose, and frozen with dry ice and isopentane. Brains were sectioned onto Superfrost Plus slides at 18 μm using a cryostat and stored at 20°C. Slides were warmed for 10 min and then washed in 1× PBS for 5 min. Slides were blocked with 10% NGS in 1× PBS with 0.2% Triton X (PBS + Tx) for 1 h at room temperature. Slides were incubated at room temperature overnight in 1× PBS + Tx with the following primary antibodies: HMGB1 (rabbit, ab18256, 1:1000), CD11b (mouse, bd554859, 1:1000), NeuN (mouse, ab104224, 1:1000), GFAP (mouse, MP691102, 1:100). Slides were washed three times in 1× PBS and then incubated in secondaries (A11037: 594 goat anti-rabbit; A11029: 488 goat anti-mouse) diluted to 1:500 in 1× PBS + Tx for 2 h. Slides were then washed once in 1× PBS and coverslipped with Vectashield with DAPI. Images were captured using a Nikon A1R Laser Scanning Confocal and Total Internal Reflection Fluorescence (TIRF) Inverted Microscope and analyzed by a condition-blind observer using Fiji. Counts were established bilaterally in two sections per area and an average was generated for each animal.
Microglia isolations and ex vivo treatments.
Hippocampal microglia were isolated using a Percoll density gradient as described by Frank et al. (2006a). Rats were saline-perfused for 3 min, brains were removed, and the hippocampus was dissected out on ice. The hippocampus was then homogenized in 3 ml of 0.2% glucose in 1× Dulbecco's PBS (DPBS). The homogenate was passed through a 40 μm filter into a 50 ml conical, which was rinsed with an additional 2 ml of DPBS, and then the homogenate was transferred to a 5 ml falcon tube. Cells were pelleted at 1000 × g for 10 min at 22°C and then the supernatant was removed. A Percoll (GE Healthcare) gradient was created by resuspending the pellet in 2 ml of 70% isotonic Percoll (isotonic Percoll consists of a 9:1 ratio of Percoll and 10× DPBS; 100% isotonic Percoll is then diluted with 1× DPBS), followed by a layer of 2 ml of 50% isotonic Percoll and topped with 1 ml of DPBS. The gradient was spun at 1200 × g for 45 min at 22°C with no acceleration or brake. Myelin debris was removed and then microglia were extracted from the 50/70% interface. Microglia were washed in DPBS and pelleted at 1000 × g for 10 min at 22°C. Microglia were resuspended in media [filtered sterile high-glucose DMEM (Invitrogen, 11960-044) + 10% FBS (Atlanta Biological, S11050)] and microglia concentration was determined by trypan blue exclusion. Microglia were plated at a density of 6000 cells/100 μl in a 96-well v-bottom plate. To assess microglia cytokine responsiveness, cells were challenged ex vivo with 10 μl of LPS (E. coli serotype 0111:B4; Sigma-Aldrich) at a concentration of 10 or 100 ng/ml or media alone at 37°C, 5% CO2. The LPS concentrations, incubation time, and cell density were based on time course and LPS concentration curves previously published by our laboratory (Frank et al., 2006a, 2010b). After 3 h, plates were centrifuged at 1000 × g for 10 min at 4°C to pellet cells and the supernatant was removed. Cells were washed with 1× DPBS, centrifuged at 1000 × g for 10 min at 4°C, and RNA was isolated (described above). This procedure yields highly pure microglia (Iba-1+/MHCII+/CD163−/GFAP−), which was confirmed with quantitative PCR in this experiment.
Behavioral testing.
A pre-exposure fear-conditioning paradigm was used to evaluate hippocampal-dependent memory (Barrientos et al., 2006). The pre-exposure paradigm separates the construction of the conjunctive representation of the conditioning context and the association of that representation with the shock, allowing for detection of impairments more selective to the hippocampus than a standard fear-conditioning paradigm (Barrientos et al., 2006). Rats underwent pre-exposure training as described by Barrientos et al. (2015a; for experimental design, see Fig. 6A). In brief, rats were placed in a novel clear plastic conditioning chamber (26 (length) × 21 (width) × 24 (height) cm) with a removable floor of stainless steel rods 1.5 mm in diameter that was placed inside a white ice chest. Rats were exposed to the context six times (first exposure was 5 min and subsequent exposures were 40 s). Rats were carried to and from the context in a black bucket with a lid. Three days later, rats were transported to the conditioning context in the black bucket and immediately received a 2 s 1.5 mA shock (in context for <5 s) before being removed and transported back to their home cage. Freezing behavior was assessed, as an index of memory, over a 6 min period in the conditioned context and a novel control context by a condition-blind observer.
Figure 6.

Blocking HMGB1 prevents prolonged delirium-like behaviors in aged rats following infection. A, Experimental schematic: aged and young rats received an ICM injection of Box-A or vehicle followed by an intraperitoneal injection of saline or E. coli 24 h later (n = 7–8/group). Four days following E. coli injection, rats were trained in a pre-exposure fear-conditioning paradigm. B, Freezing behavior in the experimental context (context A) and control context (context B). C, Juvenile social exploration was evaluated in a separate cohort of young and aged rats (n = 6–8/group) that received an ICM injection of Box-A or vehicle followed 24 later by an intraperitoneal injection of saline or E. coli. Data are presented as mean ± SEM. *p < 0.05.
Juvenile social exploration, a measure of anxiety, and sucrose preference, a measure of anhedonia, were assessed as previously described (Fonken et al., 2015). To determine sucrose preference, rats were provided with two bottles (randomized placement), one containing water and one containing water supplemented with 2% sucrose. Rats were acclimated to the two-choice test for 8 h on 2 nights before any experimental manipulations and all rats showed a strong preference for the sucrose solution. Sucrose intake was then measured for 8 h each night following experimental manipulations and a percentage of relative sucrose intake was calculated: [sucrose intake/(sucrose intake + water intake)] × 100.
To assess the motivation to engage in social exploratory behavior, a juvenile rat was introduced to the test subject in a novel cage for 3 min. Experimental rats were acclimated to the cage for 30 min before testing. A condition-blind observer recorded the total time the experimental rat engaged in social investigation of the juvenile (sniffing, grooming, trailing, etc.). Baseline social behavior was established 24 h before experimental manipulations and then behavior was repeatedly assessed following immune challenge. Results are expressed as a percentage compared with respective baseline measures.
Statistics.
All data are presented as mean ± SEM. Statistical analysis consisted of Student's t test or ANOVA using Prism 6 (GraphPad Software). Omnibus F values are reported for ANOVAs and served as the criterion for post hoc comparisons (Tukey's multiple-comparisons test). Threshold for statistical significance was set at p < 0.05.
Results
HMGB1 gene and HMGB1 protein expression are basally elevated in the aged brain
To determine whether HMGB1 is a potential mediator of age-related neuroinflammatory priming, we first evaluated HMGB1 gene and HMGB1 protein expression in the hippocampus of old (n = 6) and young (n = 5) rats. We specifically focused on the hippocampus in this experiment because it is a brain region with a high concentration of inflammatory receptors (Maier and Watkins, 1998). Further, the hippocampus is considered critical for the behaviors investigated in subsequent experiments (Kim and Fanselow, 1992). Both HMGB1 gene (t(9) = 3.0, p < 0.05; Fig. 1A) and HMGB1 protein (t(9) = 3.3, p < 0.01; Fig. 1B) expression were significantly elevated in the hippocampus of old rats.
Figure 1.

HMGB1 is elevated in aged rodent brain and CSF. A, B, Relative HMGB1 gene (A) and HMGB1 protein (B) expression were measured in the hippocampus of young (3-month-old, n = 5) and aged (24-month-old, n = 6) F344xBN rats. C, In a separate cohort of young and aged rats (n = 8/group) HMGB1 was measured in the CSF. Gene-expression data are presented relative to β-actin. All data are presented as mean ± SEM. *p < 0.05.
Aged rats increased HMGB1 release
The inflammatory effects of HMGB1 are due to its movement from the nucleus to either the cytosolic or extracellular space (Yang and Tracey, 2010). Elevated HMGB1 in tissue lysates does not clarify whether more HMGB1 is being released. To determine whether there are differences in HMGB1 release, we first evaluated HMGB1 in the CSF of young and old rats (n = 8/group). HMGB1 was elevated in the CSF of old rats (t(14) = 2.4, p < 0.05; Fig. 1C). Next, to determine the cellular source of HMGB1, we evaluated nuclear colocalization of HMGB1 in neurons, microglia, and astrocytes in the brain of old and young rats (n = 8 rats/group, as described previously; Faraco et al., 2007; Qiu et al., 2008). There were no differences between young and old rats in HMGB1 colocalization with DAPI in GFAP+ and NeuN+ cells (p > 0.05; Fig. 2C,D,F,G) as colocalization was uniformly high in these cells (95% in NeuN+ cells; 88% in GFAP+ cells). In contrast, old rats exhibited reduced nuclear HMGB1 colocalization in hippocampal CD11b+ cells (F(1,27) = 23.8, p < 0.05; Fig. 2A,B,E). While 95.6% of CD11b+ cells from young animals expressed HMGB1 in the nucleus, only 83.9% of CD11b+ cells in the aged brain expressed nuclear HMGB1. Unfortunately, measuring HMGB1 release in microglia isolated from young and old rats was not possible because there is a higher percentage of cell death in aged microglia maintained ex vivo for prolonged periods of time (data not shown). Overall, these data suggest that aged rats release more HMGB1 and that the major cellular source is likely CD11b+ microglia.
Figure 2.

HMGB1 may be released from CD11b+ cells in the aged hippocampus. A–G, Nuclear colocalization of HMGB1 was quantified in the hippocampus of young and aged rats by triple-labeling DAPI and HMGB1 with either CD11b (microglia; A, B, E), GFAP (astrocytes; C, F), or NeuN (neurons; D, G; n = 8/group). Images were captured using a Nikon A1R Laser Scanning Confocal and TIRF Inverted Microscope and analyzed using Fiji (ImageJ) by a condition-blind observer. Arrows are highlighting triple-labeled cells and arrowheads represent DAPI+ cells lacking nuclear HMGB1. Data are presented as mean ± SEM. *p < 0.05.
Neuroinflammatory priming in aged rats causes prolonged hippocampal inflammation following peripheral infection
Next, the neuroinflammatory effects of peripheral infection were studied in the hippocampus of young and old rats. Previous work in our laboratory has primarily focused on infection-induced IL-1β (Barrientos et al., 2009). The present experiments sought to establish whether neuroinflammatory changes apply to other inflammatory factors. Four days following a single injection of vehicle or E. coli (2.5 × 109 CFUs, i.p.), hippocampal tissue was collected to evaluate expression of inflammatory cytokines and receptors (n = 6/group; one old animal died following E. coli injection). By 4 d postinfection, the hippocampus of E. coli-treated and saline-treated young rats had comparable expression of inflammatory cytokines, suggesting that infection-induced hippocampal inflammation had resolved by this time (Fig. 3). In contrast, in old rats, infection-induced hippocampal inflammation was sustained at 4 d after E. coli injection. In agreement with previous findings, IL-1β protein and IL-1β gene expression were elevated in the hippocampus of aged animals that received E. coli (interaction of age × E. coli: F(1,19) = 5.2 and 7.4 respectively and post hoc, p < 0.05; Fig. 3A,B). IL-1β was increased 55% in old rats injected with E. coli 4 d prior compared with aged saline-injected rats. IL-18 and TNFα mRNA expression were similarly elevated in the aged hippocampus following E. coli injection (interaction of age × E. coli: F(1,18) = 5.2 and 4.9, p < 0.05; Fig. 3B). IL-6 and IL-10 mRNA were affected by age but not by E. coli 4 d following the injection. IL-6 mRNA expression was increased in old animals (main effect of age: F(1,19) = 4.4, p < 0.05; Fig. 3B) and the anti-inflammatory cytokine IL-10 was reduced in the aged hippocampus (main effect of age: F(1,19) = 4.7, p < 0.05; Fig. 3B).
Figure 3.

Aged animals exhibit prolonged inflammation following infection. Hippocampal tissue was collected from young and aged rats 4 d following an E. coli or saline injection (n = 6/group). A–C, IL-1β protein and IL-1β gene-expression levels of inflammatory cytokines (B) and inflammatory receptors (C) were all evaluated. D–F, HMGB1 (D) and NLRP3 (E) mRNA expression were also evaluated and NLRP3 (F) was significantly associated with IL-1β protein. G, H, A separate cohort of rats (n = 8/group) underwent behavioral testing in a sucrose anhedonia (G) and juvenile social exploration task (H). Gene-expression data are presented relative to β actin. All data are presented as mean ± SEM. *, Main effect; †, simple effect of E. coli; in all cases p < 0.05.
The expression of several inflammatory pathway genes and receptors were also evaluated to determine potential mechanisms leading to heightened neuroinflammatory responses in aged rats. TLR2 (F(1,18) = 28.0), TLR4 (F(1,18) = 27.3), and the purinergic receptor P2X7R (F(1,18) = 25.9) were elevated in the aged hippocampus (Fig. 3C). Consistent with Figure 1, HMGB1 mRNA expression was increased in the aged hippocampus regardless of E. coli treatment (F(1,18) = 8.2, p < 0.05; Fig. 3D). Furthermore, NLRP3 mRNA expression was elevated in old rats 4 d following E. coli treatment (interaction of age × E. coli: F(1,18) = 6.3, p < 0.05; Fig. 3E). The NLRP3 inflammasome is a major pathway through which pro-IL-1β and pro-IL-18 are cleaved into their active forms. In agreement with this, NLRP3 mRNA expression significantly correlated with IL-1β protein (r = 0.54, p < 0.05; Fig. 3F). Thus, aged rats infected intraperitoneally with E. coli show prolonged hippocampal inflammatory responses compared with young rats. Importantly, genes for proteins that could prime microglia in aged rats—TLR2, TLR4, HMGB1, and P2X7R—were substantially elevated in aged hippocampus even without a peripheral challenge.
Infection causes prolonged affective changes in aged rats
Previous work in our laboratory suggests that prolonged neuroinflammatory responses are associated with impairments in hippocampal-dependent memory in aged rats (Barrientos et al., 2006). Importantly, in addition to cognitive impairments, delirium is characterized by a host of affective and behavioral changes, including anxiety, irritability, depression, and apathy. Thus, we next sought to determine whether aged rats demonstrate additional delirium-like behavioral responses following infection. To test whether neuroinflammatory changes are more broadly associated with other sickness/affective behavioral changes, young and old rats received either E. coli or saline and then underwent testing in juvenile social exploration and sucrose anhedonia tests (n = 8 per group). There were no differences in baseline sucrose intake in the sucrose anhedonia test (p > 0.05) and all groups showed a strong preference for the sucrose solution (Fig. 3G). On the day of E. coli administration, rats injected with E. coli compared with vehicle reduced their preference for the sucrose solution regardless of age (main effect of E. coli: F(1,28) = 20.5, p < 0.05). However, only old rats demonstrated a sustained reduction in sucrose preference 4 d after E. coli administration (interaction of age × E. coli: F(1,28) = 7.4, p < 0.05).
In the juvenile social exploration task, both young and old rats showed an initial drop in social investigation 8 h following the E. coli challenge (main effect of E. coli: F(1,27) = 18.4). By 24 h, social exploration was comparable to baseline levels in young but not old animals that received E. coli (main effect of age and E. coli: F(1,27) = 12.5 and 4.8 and post hoc, p < 0.05). Moreover, old rats demonstrated sustained reductions in social exploration 2 d following E. coli treatment (main effect of age and E. coli: F(1,27) = 9.7 and 4.2 and post hoc, p < 0.05). Overall, the results from the sucrose preference and juvenile social explorations tests are consistent with a prolonged sickness response in aged rats that received E. coli.
Blockade of HMGB1 with the antagonist Box-A prevents neuroinflammatory priming
To determine whether HMGB1 has a critical role in neuroinflammatory priming, we next gave a single ICM injection of the HMGB1 competitive antagonist Box-A and collected hippocampal tissue 24 h later. The ICM injection of Box-A downregulated expression of inflammatory pathway genes including MHCII (interaction of age × Box-A: F(1,18) = 5.1, p < 0.05; Fig. 4A) and TLR4 (interaction of age × Box-A: F(1,19) = 8.5, p < 0.05; Fig. 4A).
Figure 4.
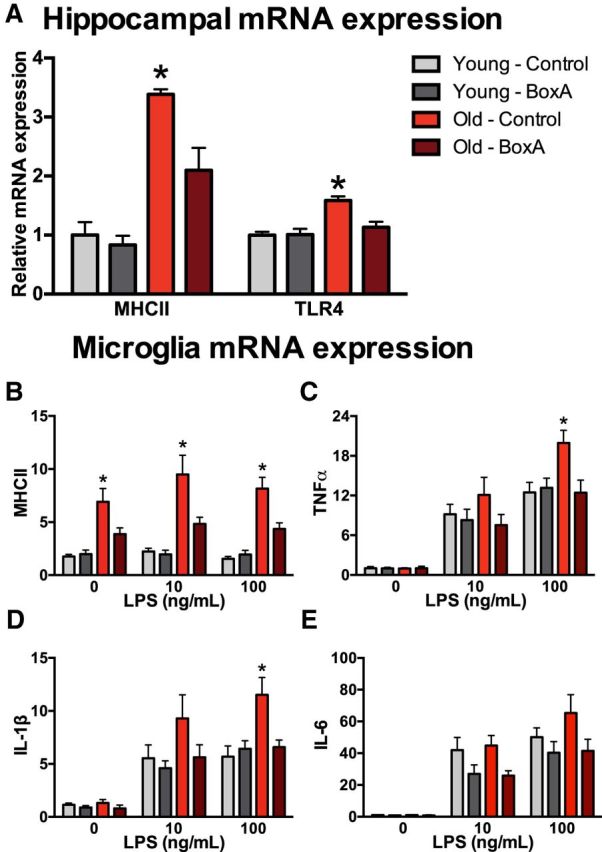
Blocking HMGB1 with Box-A prevents neuroinflammatory priming. A, Hippocampal tissue was collected 24 h after aged and young rats were treated with ICM Box-A and expression of inflammatory pathway genes was evaluated (n = 5–6/group). Microglia were isolated 24 h after aged and young rats were treated with ICM Box-A (n = 6/group). B–E, Microglia were stimulated with LPS ex vivo and relative gene expression for MHCII (B), TNFα (C), IL-1β (D), and IL-6 (E) was determined. Gene-expression data are presented relative to β actin. All data are presented as mean ± SEM. *p < 0.05.
Previous research demonstrated that aging primes the microglia proinflammatory response to an immune challenge ex vivo (Frank et al., 2010b). Thus, we next evaluated whether blocking HMGB1 would prevent aging-induced microglia priming. To test this, hippocampal microglia were isolated from young and old rats 24 h following an ICM injection of Box-A or saline (n = 6/group). After isolation, microglia were plated and stimulated ex vivo with LPS. In agreement with previous findings, microglia from old rats increased MHCII expression even in the absence of LPS stimulation, and Box-A ameliorated this increase (interaction of age × Box-A at 0, 10, and 100 μg of LPS: F(1,20) = 5.2, 4.9, and 10.7 and post hoc; p < 0.05; Fig. 4B). As previously reported, aging itself did not result in heightened proinflammatory cytokines (0 ng/ml LPS). However, microglia from old rats had elevated cytokine production in response to immune challenge (100 ng/ml LPS stimulation). More specifically, these results indicated that aging potentiated the LPS-induced increases in all three cytokines. Importantly, our results also indicate that the potentiation of the inflammatory response in microglia isolated from aged rats was blocked by prior Box-A for TNFα (interaction of age × Box-A at 100 μg of LPS: F(1,19) = 5.8 and post hoc; p < 0.05; Fig. 4C) and for IL-1β (interaction of age × Box-A at 100 μg of LPS: F(1,19) = 7.6 and post hoc; p < 0.05; Fig. 4D). While IL-6 mRNA results showed a similar trend, they were not statistically significant (p > 0.05). This suggests that microglia may upregulate inflammatory mechanisms upon detecting excess extracellular HMGB1 in old rats and that these effects can be blocked with the competitive antagonist Box-A.
Blockade of HMGB1 before an E. coli challenge prevents prolonged neuroinflammatory responses in aged rats
Next we tested whether blocking HMGB1 can also prevent prolonged in vivo neuroinflammatory responses in aged rats. Rats received a single ICM injection of Box-A or vehicle followed 24 h later by an intraperitoneal injection of E. coli (Fig. 5A). Hippocampal tissue was collected at baseline (time of intraperitoneal injection) or 24 h or 4 d after E. coli injection (n = 5–6 per group/time). There were no baseline differences in hippocampal IL-1β protein. Twenty-four hours following E. coli injection, IL-1β protein was elevated in aged but not young rats. Importantly, Box-A pretreatment blocked the age-related increase in hippocampal IL-1β that followed E. coli injection (main effect of drug: F(1,18) = 15.6). Furthermore, hippocampal IL-1β protein levels were still elevated in old rats 4 d after E. coli injection, and pretreatment with Box-A eliminated this age-related potentiation (interaction of age × drug: F(1,23) = 9.5, p < 0.05; Fig. 5B). While Box-A pretreatment also reduced IL-1β mRNA expression in aged and young rats 24 h following E. coli injection (F(1,18) = 5.9, p < 0.05), the results were highly variable (relative expression levels of the majority of the samples fell between 1 and 10; however, five samples ranged between 11 and 140). The pattern of hippocampal TNFα and IL-6 mRNA expression were similar to the IL-1β protein results. Box-A pretreatment prevented increases in TNFα mRNA expression in the hippocampus of aged rats both 24 h and 4 d after E. coli injection (interaction of age × drug: F(1,18) = 5.0; F(1,20) = 8.1, p < 0.05; Fig. 5D). Box-A pretreatment similarly prevented increases in IL-6 at 24 h and 4 d after E. coli injection (interaction of age × drug: F(1,18) = 5.5; F(1,19) = 6.9, p < 0.05; Fig. 5E). Box-A pretreatment also caused basal reductions in hippocampal IL-6 (main effect of drug: F(1,18) = 6.4, p < 0.05). Thus, Box-A pretreatment dampened the exaggerated infection-induced inflammatory response in the aged rat hippocampus.
Figure 5.

Box-A pretreatment prevented prolonged inflammatory responses in aged E. coli-treated rats. A, Experimental schematic: aged and young rats received an ICM injection of Box-A or vehicle and an intraperitoneal injection of E. coli 24 h later. Tissue was collected at 0 h (no E. coli control), 24 h, or 4 d after E. coli injection. Relative expression of IL-1β protein (B), IL-1β mRNA (C), TNFα mRNA (D), and IL-6 mRNA (E) were quantified in hippocampal tissue (n = 5–6/group/time). Gene-expression data are presented relative to β actin. All data are presented as mean ± SEM. *, Main effect; †, simple effect; in all cases p < 0.05.
HMGB1 blockade prevents prolonged impairments in hippocampal-dependent memory and sickness behavior following infection in aged rats
To test whether reduced neuroinflammatory responses are associated with reversal of behavioral changes in aged rats following E. coli injection, aged and young rats were trained and tested in a pre-exposure fear-conditioning paradigm (n = 7–8 per group; Fig. 6A). In agreement with previous findings, E. coli interfered with fear-context memory but only in old rats (interaction of age × drug: F(3,50) = 4.0, post hoc, p < 0.05; Fig. 6B). Interestingly, pretreatment with Box-A prevented the memory impairments in old rats (post hoc, p < 0.05; Fig. 6B). Post hoc analysis demonstrates that old rats pretreated with Box-A before E. coli injection did not significantly differ from young rats. Importantly, there were no group differences in a novel context (p > 0.05; Fig. 6B), suggesting that differences in freezing behavior were not due to altered activity levels. These data show that a single prior Box-A injection protects aged rats from prolonged infection-induced cognitive impairment.
Furthermore, a separate cohort of rats was tested in a juvenile social exploration task following ICM Box-A or vehicle and subsequent E. coli or saline treatment. All rats that received E. coli showed reduced juvenile social exploration 8 h following the injection (main effect of E. coli in aged and young: F(1,20) = 14.9 and 13.6, p < 0.05; Fig. 6C). By 24 h following E. coli injection, young animals no longer exhibited significant reductions in social explorations (p > 0.05). In contrast, aged rats that received E. coli demonstrated prolonged reductions in juvenile social exploration at 24 h and 4 d (main effect of E. coli in aged rats at 24 h: F(1,20) = 26.2, p < 0.05). Importantly, Box-A pretreatment ameliorated reductions in social exploration 4 d following E. coli treatment (interaction of Box-A × E. coli in aged rats: F(1,20) = 6.3, p < 0.05; Fig. 6C).
Discussion
Aged rats exhibit a “primed” neuroinflammatory response: aged animals do not release more inflammatory molecules under basal conditions, but demonstrate an enhanced neuroinflammatory response to an immune challenge. These experiments indicate that HMGB1 mediates this neuroinflammatory “priming” in aged animals. HMGB1 gene and HMGB1 protein expression were elevated under basal conditions in the hippocampus of aged rats. Moreover, aged animals had increased HMGB1 in the CSF. HMGB1 was likely released from microglia in the aged brain and may interact with upregulated innate immune receptors to prime neuroinflammatory responses. Importantly, increases in HMGB1 and upregulation of innate immune receptors occurred in the absence of immune stimulation in aged rats. Blocking HMGB1 signaling with an ICM injection of the competitive antagonist Box-A downregulated expression of several inflammatory pathway genes. Furthermore, aged rats exhibited prolonged elevations in proinflammatory cytokines following a peripheral immune challenge and pretreatment of aged rats with Box-A before E. coli injection prevented protracted hippocampal inflammatory responses, cognitive impairments, and sickness responses. HMGB1 likely mediates inflammatory priming through autoregulatory effects on microglia: ICM pretreatment with Box-A prevented priming in microglia stimulated ex vivo with LPS.
HMGB1 appears to have a key role in microglial priming that occurs with age. Microglia phenotype and function change with age, with microglia in aged animals exhibiting a reduced threshold for activation and enhanced proinflammatory responses despite similar basal levels of inflammatory cytokines. Exaggerated inflammatory responses in aged rodents and humans contribute to a host of maladaptive behavioral changes, including delirium and affective disorders (Ehlenbach et al., 2010; Simone and Tan, 2011; Cape et al., 2014). Microglia function is tightly regulated by the CNS microenvironment. In the aged CNS, microglia may become primed via the accumulation of endogenous DAMPs (Kapetanovic et al., 2015). In support of this hypothesis, we show that the DAMP HMGB1 is elevated in the aged brain. Both HMGB1 gene and HMGB1 protein expression were elevated in the hippocampus of aged rats in the absence of any experimental treatment.
HMGB1 is a ubiquitously expressed DNA binding protein that regulates inflammatory processes when released from the cell's nucleus (Yang and Tracey, 2010). While HMGB1 is passively released in the brain by necrotic or damaged cells, it appears to only be actively released by innate immune cells (Wang et al., 1999). Thus, we next established whether there are differences in HMGB1 release in the aged and young rat brain. HMGB1 protein was elevated in the CSF of aged compared with young rats, indicating increased HMGB1 release in aged rats. Increases in HMGB1 in the CSF have previously been associated with a decrease in nuclear colocalization of HMGB1, suggesting HMGB1 is released from cells in which HMGB1 is not detectable (Faraco et al., 2007; Qiu et al., 2008). The colocalization of HMGB1 in the nucleus (DAPI) in microglia (CD11b+), astrocytes (GFAP+), and neurons (NeuN+) was evaluated as an additional measure of release and as an indicator of the cellular source of HMGB1 (Faraco et al., 2007; Qiu et al., 2008). Nuclear HMGB1 colocalization was uniformly high in hippocampal neurons and astrocytes and there were no age-associated differences. In contrast, nuclear HMGB1 colocalization was reduced in CD11b+ microglia in old compared with young rats. Together, the overall increases in hippocampal and CSF HMGB1 protein expression, combined with the reductions in nuclear HMGB1 in microglia, suggest increases in HMGB1 release in the aged brain. Moreover, the inhibitory effects of Box-A (which presumably acts extracellularly) strongly suggest cellular release of HMGB1. Of note, there are two alternative sources of HMGB1 in the aged brain: (1) paravascular clearance pathways are impaired in the aged brain, indicating that pathological accumulation of inflammatory molecules, including HMGB1, may also occur (Kress et al., 2014); (2) aged animals may have increases in cell death, which would also cause HMGB1 release (Jackson et al., 2009).
Once released from the nucleus, HMGB1 activates inflammatory cells by interacting with several receptors for DAMPs, including RAGE, TLR2, and TLR4 (Park et al., 2004; Yu et al., 2006; van Beijnum et al., 2008). Importantly, TLR2 and TLR4 are expressed by microglia/macrophages and astrocytes and are upregulated in the aged hippocampus (Fig. 3). Increases in these innate immune receptors may leave aged rats particularly susceptible to the damaging effects of excess HMGB1. Indeed, here we show that basal increases in HMGB1 in aged rats are associated with an exaggerated and prolonged inflammatory response. Young rats resolve neuroinflammatory responses within ∼24 h following an E. coli injection (Barrientos et al., 2009), whereas several proinflammatory cytokines remain elevated in the hippocampus of aged rats 4 d after E. coli injection (Fig. 3). These results build on previous findings from our group, and demonstrate that prolonged neuroinflammatory responses following infection in aged rats are not specific to IL-1β. Several other inflammatory cytokines, including TNFα and IL-18, were elevated in the aged hippocampus 4 d following E. coli injection. Furthermore, NLRP3 mRNA expression was increased in the hippocampus of aged rats 4 d following E. coli injection and significantly correlated with IL-1β protein expression. The NLRP3 inflammasome is a major pathway through which proIL-1β and pro-IL-18 are cleaved into their active forms. Exogenous ICM administration of recombinant HMGB1 increases NLRP3 protein and mRNA concomitant with priming the neuroinflammatory response to a peripheral immune challenge (Frank et al., 2016). This indicates that infection-induced elevations in NLRP3 may be the proximal mediator for the extended IL-1β and IL-18 response in aged rats. However, the present results do not exclude the possibility that inflammasomes other than NLRP3, as well as inflammasome-independent pathways, which also drive IL-1β maturation, may be involved in aging-induced priming phenomena.
To determine whether HMGB1 is critical for priming immune responses in the aged brain, we blocked HMGB1 with an ICM injection of the selective competitive antagonist to HMGB1, Box-A. Box-A downregulated expression of several inflammatory pathway genes in the hippocampus of aged rats and prevented the protracted hippocampal inflammatory response. Box-A pretreatment also blocked E. coli-induced memory impairments in aged rats in a fear-conditioning paradigm. In agreement with previous research, context fear memory was significantly reduced in aged rats trained in a pre-exposure fear-conditioning paradigm 4 d following an E. coli challenge (Barrientos et al., 2006, 2009, 2012; Frank et al., 2010a). However, aged rats that received Box-A before the peripheral immune challenge performed comparably to aged saline-treated rats. Furthermore, Box-A pretreatment similarly prevented prolonged changes in other sickness behaviors in aged rats 4 d following E. coli injection (Fig. 6).
Finally, we investigated whether microglia are the effector cells targeted by HMGB1, as microglia likely regulate neuroinflammatory priming in aged animals (Frank et al., 2010b). ICM pretreatment with Box-A prevented priming in microglia stimulated ex vivo with LPS, suggesting that HMGB1 likely mediates inflammatory priming through autoregulatory effects on microglia (Fig. 7). Of note, extranuclear HMGB1 that remains in the cell can also activate immune responses through intracellular pattern-recognition receptors (Yanai et al., 2009). For example, cytosolic HMGB1 regulates autophagy (Tang et al., 2010; Yanai et al., 2013). This indicates that even in the absence of active secretion from glial cells, HMGB1 can act in an autoregulatory feedback manner to enhance innate immune responses. One limitation to these results is that age-associated inflammatory priming was only evaluated in the context of infection. However, it is likely that neuroinflammatory priming by HMGB1 would generalize to other types of immune challenges. For example, aged rodents also demonstrate enhanced neuroinflammatory responses following stroke, spinal cord injury, and laparotomy (Manwani et al., 2011; Barrientos et al., 2012; Fenn et al., 2014).
Figure 7.

Proposed mechanism of microglia inflammatory priming in aged brain.
The present work shows that excess HMGB1 accumulation is critical for priming neuroinflammatory responses in the aged brain. Furthermore, our results indicate that microglia are likely the cellular source and cognate target of increases in HMGB1 with aging. The exact mechanism(s) that lead to HMGB1 release is unclear. However, there are several possible sources. Importantly, aging increases hippocampal corticosterone concentrations and blocking corticosterone with an ICM injection of the glucocorticoid receptor antagonist mifepristone can prevent neuroinflammatory priming in aged rats (Barrientos et al., 2015a). Stress causes HMGB1 release (Weber et al., 2015), suggesting that glucocorticoids may play a role in HMGB1-induced neuroinflammatory priming in the aged brain. Additionally, increases in misfolded protein aggregates due to impaired glymphatic clearance may trigger DAMP signaling (Kress et al., 2014).
Conclusions
Aged microglia are primed cells that have the potential to amplify neuroinflammatory responses in a maladaptive manner. Excess HMGB1 accumulation occurs during normal aging and may represent the top of an age-related neuroinflammatory hierarchy (Fig. 7). These results indicate that blocking the actions of HMGB1 may “desensitize” microglia in the aged brain and prevent exaggerated pathology-related neuroinflammation, cognitive decline, and affective disturbances following infection.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant R01AG028271 to M.G.F., R.M.B., L.R.W., and S.F.M.; NIH Grant F32AG048672 to L.K.F.; and a National Alliance for Research on Schizophrenia and Depression Young Investigator Grant sponsored by the New York Women's Committee to L.K.F. We thank Andrew D. Gaudet for helpful comments and the University of Colorado for animal care.
The authors declare no competing financial interests.
References
- Andersson U, Erlandsson-Harris H, Yang H, Tracey KJ. HMGB1 as a DNA-binding cytokine. J Leukoc Biol. 2002;72:1084–1091. [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Biedenkapp JC, Sprunger DB, Wright-Hardesty KJ, Watkins LR, Rudy JW, Maier SF. Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol Aging. 2006;27:723–732. doi: 10.1016/j.neurobiolaging.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Frank MG, Hein AM, Higgins EA, Watkins LR, Rudy JW, Maier SF. Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav Immun. 2009;23:46–54. doi: 10.1016/j.bbi.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Hein AM, Frank MG, Watkins LR, Maier SF. Intracisternal interleukin-1 receptor antagonist prevents postoperative cognitive decline and neuroinflammatory response in aged rats. J Neurosci. 2012;32:14641–14648. doi: 10.1523/JNEUROSCI.2173-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Thompson VM, Kitt MM, Amat J, Hale MW, Frank MG, Crysdale NY, Stamper CE, Hennessey PA, Watkins LR, Spencer RL, Lowry CA, Maier SF. Greater glucocorticoid receptor activation in hippocampus of aged rats sensitizes microglia. Neurobiol Aging. 2015a;36:1483–1495. doi: 10.1016/j.neurobiolaging.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Kitt MM, Watkins LR, Maier SF. Neuroinflammation in the normal aging hippocampus. Neuroscience. 2015b;309:84–99. doi: 10.1016/j.neuroscience.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker AY, Weeks EJ. Cognitive function after anaesthesia in the elderly. Best Pract Res Clin Anaesthesiol. 2003;17:259–272. doi: 10.1016/S1521-6896(03)00005-3. [DOI] [PubMed] [Google Scholar]
- Cape E, Hall RJ, van Munster BC, de Vries A, Howie SE, Pearson A, Middleton SD, Gillies F, Armstrong IR, White TO, Cunningham C, de Rooij SE, MacLullich AM. Cerebrospinal fluid markers of neuroinflammation in delirium: a role for interleukin-1beta in delirium after hip fracture. J Psychosom Res. 2014;77:219–225. doi: 10.1016/j.jpsychores.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2008;22:301–311. doi: 10.1016/j.bbi.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlenbach WJ, Hough CL, Crane PK, Haneuse SJ, Carson SS, Curtis JR, Larson EB. Association between acute care and critical illness hospitalization and cognitive function in older adults. JAMA. 2010;303:763–770. doi: 10.1001/jama.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraco G, Fossati S, Bianchi ME, Patrone M, Pedrazzi M, Sparatore B, Moroni F, Chiarugi A. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem. 2007;103:590–603. doi: 10.1111/j.1471-4159.2007.04788.x. [DOI] [PubMed] [Google Scholar]
- Fenn AM, Hall JC, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique arginase+/IL-1β+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Rα after traumatic spinal cord injury. J Neurosci. 2014;34:8904–8917. doi: 10.1523/JNEUROSCI.1146-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK, Frank MG, Kitt MM, Barrientos RM, Watkins LR, Maier SF. Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain Behav Immun. 2015;45:171–179. doi: 10.1016/j.bbi.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Wieseler-Frank JL, Watkins LR, Maier SF. Rapid isolation of highly enriched and quiescent microglia from adult rat hippocampus: immunophenotypic and functional characteristics. J Neurosci Methods. 2006a;151:121–130. doi: 10.1016/j.jneumeth.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW, Watkins LR, Maier SF. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging. 2006b;27:717–722. doi: 10.1016/j.neurobiolaging.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Hein AM, Biedenkapp JC, Watkins LR, Maier SF. IL-1RA blocks E. coli-induced suppression of Arc and long-term memory in aged F344xBN F1 rats. Brain Behav Immun. 2010a;24:254–262. doi: 10.1016/j.bbi.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Watkins LR, Maier SF. Aging sensitizes rapidly isolated hippocampal microglia to LPS ex vivo. J Neuroimmunol. 2010b;226:181–184. doi: 10.1016/j.jneuroim.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Weber MD, Fonken LK, Hershman SA, Watkins LR, Maier SF. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: a role for the NLRP3 inflammasome. Brain Behav Immun. 2016;55:215–224. doi: 10.1016/j.bbi.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- Griffin R, Nally R, Nolan Y, McCartney Y, Linden J, Lynch MA. The age-related attenuation in long-term potentiation is associated with microglial activation. J Neurochem. 2006;99:1263–1272. doi: 10.1111/j.1471-4159.2006.04165.x. [DOI] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TC, Rani A, Kumar A, Foster TC. Regional hippocampal differences in AKT survival signaling across the lifespan: implications for CA1 vulnerability with aging. Cell Death Differ. 2009;16:439–448. doi: 10.1038/cdd.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapetanovic R, Bokil NJ, Sweet MJ. Innate immune perturbations, accumulating DAMPs and inflammasome dysregulation: a ticking time bomb in ageing. Ageing Res Rev. 2015;24:40–53. doi: 10.1016/j.arr.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R, Nedergaard M. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76:845–861. doi: 10.1002/ana.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Maier SF, Watkins LR. Cytokines for psychologists: implications of bidirectional immune-to-brain communication for understanding behavior, mood, and cognition. Psychol Rev. 1998;105:83–107. doi: 10.1037/0033-295X.105.1.83. [DOI] [PubMed] [Google Scholar]
- Manwani B, Liu F, Xu Y, Persky R, Li J, McCullough LD. Functional recovery in aging mice after experimental stroke. Brain Behav Immun. 2011;25:1689–1700. doi: 10.1016/j.bbi.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- Perry VH, Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. 2013;35:601–612. doi: 10.1007/s00281-013-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Nishimura M, Wang Y, Sims JR, Qiu S, Savitz SI, Salomone S, Moskowitz MA. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab. 2008;28:927–938. doi: 10.1038/sj.jcbfm.9600582. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem. 2014;289:35237–35245. doi: 10.1074/jbc.R114.619304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone MJ, Tan ZS. The role of inflammation in the pathogenesis of delirium and dementia in older adults: a review. CNS Neurosci Ther. 2011;17:506–513. doi: 10.1111/j.1755-5949.2010.00173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ, 3rd, Lotze MT. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1) Angiogenesis. 2008;11:91–99. doi: 10.1007/s10456-008-9093-5. [DOI] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF. Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci. 2015;35:316–324. doi: 10.1523/JNEUROSCI.3561-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne AM, Henry CJ, Huang Y, Cleland A, Godbout JP. Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav Immun. 2010;24:1190–1201. doi: 10.1016/j.bbi.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, Savitsky D, Ronfani L, Akira S, Bianchi ME, Honda K, Tamura T, Kodama T, Taniguchi T. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462:99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]
- Yanai H, Matsuda A, An J, Koshiba R, Nishio J, Negishi H, Ikushima H, Onoe T, Ohdan H, Yoshida N, Taniguchi T. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci U S A. 2013;110:20699–20704. doi: 10.1073/pnas.1320808110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Tracey KJ. Targeting HMGB1 in inflammation. Biochim Biophys Acta. 2010;1799:149–156. doi: 10.1016/j.bbagrm.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Wang H, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]