

Abstract
Purpose: Recombinant human endostatin (rhEs) is an angiogenesis inhibitor which is used as a specific drug in the treatment of non-small-cell lung cancer. In the current research, we developed an efficient method for expressing soluble form of the rhEs protein in the periplasmic space of Escherichia coli via fusing with pelB signal peptide.
Methods: The human endostatin (hEs) gene was amplified using synthetic (hEs) gene as a template; then, cloned and expressed under T7 lac promoter. IPTG was used as an inducer for rhEs expression. Next, the osmotic shock was used to extraction of protein from the periplasmic space. The presence of rhEs in the periplasmic space was approved by SDS-PAGE and Western blotting.
Results: The results show the applicability of pelB fusion protein system usage for secreting rhEs in the periplasm of E. coli in the laboratory scale. The rhEs represents approximately 35 % (0.83mg/l) of the total cell protein.
Conclusion: The present study apparently is the first report of codon-optimized rhEs expression as a fusion with pelB signal peptide. The results presented the successful secretion of soluble rhEs to the periplasmic space.
Keywords: Angiogenesis, Endostatin, Signal peptide, E. coli, Gene expression, Periplasm
Introduction
Endostatin (Es), an angiogenesis inhibitor, is firstly identified by O′Reilly et al. It is a 20 kDa protein generated from the non-collagenous carboxyl-terminal end of collagen XVIII by proteolysis from large precursor proteins.1 Es has a compact structure composed of two heparin- binding sites, two disulfide bonds, and a zinc binding site at the N-terminus. The disulfide bonds are essential for the compactness, stability, and activity of Es.2
Recombinant human Endostatin (rhEs) is a specific drug for treatment of non-small-cell lung cancer that was approved by the State FDA in China. The main advantage of rhEs administration as an angiogenesis inhibitor is the lack of toxicity and drug resistance that makes it a broad-spectrum anti-angiogenesis and tumor suppressing agent in a variety of cancers. Meanwhile, as a protein, its clinical application has some problems such as insolubility, instability, high price and need for high dosage.3
The rhEs has been expressed by eukaryotic and prokaryotic expression systems such as yeast, mammalian cell, insect cell, baculovirus, and Escherichia coli (E. coli).4-6 E. coli expression system has many advantages including, rapid growth, high expression level, and well-characterized genetic background, a large number of mutant host strains and expression vectors and cheap cultivation. Thus, it is widely used for recombinant protein production.4,7,8
However, a significant disadvantage of this system is that heterologous proteins are often folded incorrectly (especially in proteins with disulfide bonds) and deposited as insoluble inclusion bodies in the cytoplasm.9 Also, as hydrophobic protein, expression of rhEs in E. coli produces insoluble inclusion bodies in the cytoplasm.10,11 The expression of proteins in periplasm via fusing with signal peptide in N-terminal residue, is one of the common methods for avoidance of inclusion bodies in the cytoplasm.12 In the most cases, the periplasmic expressions of recombinant proteins facilitate downstream process, including, folding and in-vivo stability as well as production of soluble and bioactive proteins at a reduced process cost. A variety of methods have been reported to solve these obstacles in rhEs production.13-16 Regarding the advantages of periplasmic expression of recombinant proteins and clinical importance of rhEs, the aim of this research was to express recombinant rhEs in the periplasmic space of E. coli.
Material and Methods
Reagents
Enzymes including, NcoI, XhoI, T4DNA ligase, Pfu DNA polymerase, Gene Ruler DNA Ladder Mix (Cat. No. SM0334) and PageRuler Unstained Protein (Cat. No. SM0661) were obtained from Thermo Scientific (USA). Mini PlusTM -Plasmid DNA Extraction and Viogene® Gel/PCR DNA Isolation System kits were purchased from Viogene (New Taipei City, Taiwan). PCR reagents were purchased from Cinnagen (Tehran, Iran). Agar, tryptone, yeast extract, kanamycin, ampicillin and IPTG were obtained from Sigma-Aldrich (St. Louis, MO, USA). Prestained Protein Ladder was from Sinaclon (Tehran, Iran). All chemicals used in SDS-PAGE were purchased from Merck (Darmstadt, Germany). Endostatin Human ELISA kit was obtained from Abcam (Cambridge, UK).
Bacterial strains, plasmids, Cell lines, media
The E. coli strains DH5α and BL21 (DE3) (Pasteur Institute, Iran) were used as cloning and expression hosts respectively, pET26b (+) as expression vector was obtained from Pasteur Institute of Iran. Clone JET™ PCR Cloning Kit was from Thermo Scientific Company. Primers were from Shine Gene Company (Shanghai, China). In addition, the interest synthetic construct prepared from Shine Gene Company (Shanghai, China) as cloned sequence in pUC57 plasmid.
Construction of synthetic gene and amplification
The human Endostatin coding sequence from human collagen XVIIIa gene (GenBank accession No. AF184060.1) was taken for codon optimization. Gene optimization and synthesis was done by Shine Gene Bio-Technologies Company (Shanghai, China). This construct was sub cloned in pUC57 including these sequences; NdeI restriction site, alkaline phosphate signal peptide coding sequence (GenBank accession No. M 13763.1), hEs coding sequence and XhoI restriction site.
Further, the hEs gene was amplified using pUC57-phoA sp-hEs as a template. PCR was performed using the Eppendorf thermal cycler with a set of primers in a total volume of 40 μL. The reaction mixture contained 4 μL of 10 × PCR buffer, 4 μL MgCl2 (20 mM), 0.7 μL for each dNTP (2.5mM) ,1.5μL plasmid DNA, 0.6 μL of each primer and 1 μL of Pfu DNA polymerase (5 units/ μL). The program for PCR was one cycle at 95°C for 5 minutes, 30 cycles with denaturation at 95°C for 35 seconds, annealing at 60°C for 40 s, then 1 minute at 72°C and final extension at 72°C for 7 minutes. The amplification products were electrophoresed on the 1% agarose gel and visualized after SYBR Green I staining. The sequences of primers were hEs F, TCATCACCACCATGGCGCACTCTCACCG and hEs R: GATCCGATAATTTGGCTCGAGTCA. The Unique NcoI and XhoI sites (underlined bases) were added to amplified hEs synthetic gene, using these primers. In addition, for avoiding of frame shifting during the translation of hEs transcript, two nucleotides (CG, Bolded bases) were added after the NcoI site.
Constructing of the hEs expression vector
The NcoI-hEs-XhoI amplified fragments were purified with Viogene® Gel/PCR DNA Isolation kit and then ligated into pJET1.2/blunt cloning vector, using Clone JET™ PCR Cloning Kit (Thermo Scientific Company) following manufacturer's instructions. The E. coli DH5α competent cells were transformed by calcium chloride method.17 With the recombinant plasmid (pJET-hEs) for plasmid amplification and selected on LB agar with 100 µg /ml ampicillin. For confirmation of successful transformation, a single colony of transformants was grown in 3 of ml LB medium containing ampicillin, followed by colony PCR. For further confirmation, clones containing recombinant vectors were analyzed by sequencing.
Further, the pJET-hEs and the pET26b (+) vector were double-digested with the NcoI and the XhoI restriction enzymes. Following, the hEs gene was inserted just after a pelB leader sequence in the expression vector pET26b (+). The recombinant vectors were transformed into the competent E. coli BL21 (DE3) by calcium chloride method.17 Transformants were selected on LB agar containing kanamycin (50 µg /ml). Then, the clones containing hEs gene were confirmed by colony PCR. Subsequently, the accuracy of the inserted fragment in positive clone was confirmed by DNA sequencing. The bacterial stocks were kept at −70 °C in 20 % (v/v) glycerol for long-term usage. The detail of construction steps is demonstrated in Figure 1.
Figure 1.
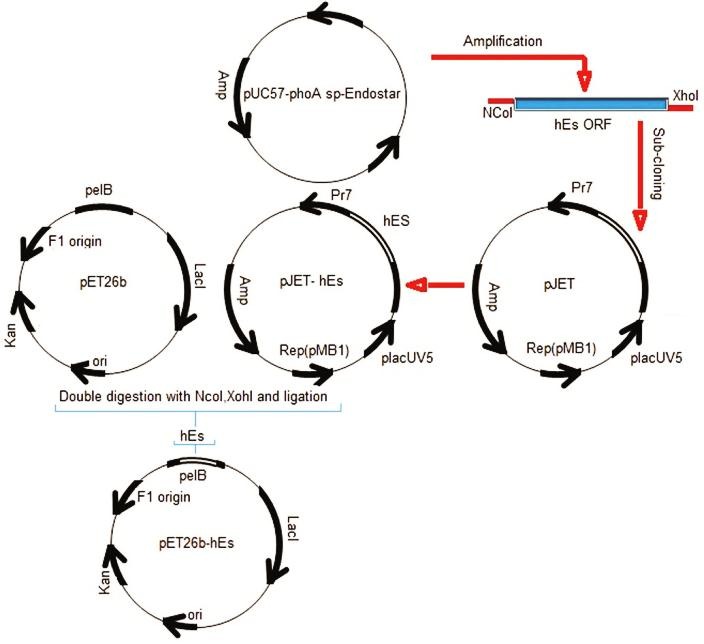
Construction of the expression vector (pET26b-hEs).
Expression of rhEs
A single colony of E. coli BL21 (pET-hEs) was inoculated in 5 ml of LB medium that supplemented with kanamycin (50µg/ml) and incubated overnight at 37 °C in a shaking (180 rpm) incubator. Then, 2 ml of starter culture was transferred into 500 ml Erlenmeyer flask with 50 ml of the same broth containing kanamycin (50µg/ml) and was incubated in a shaker incubator (37°C, 180rpm). The cell growth rate was checked during cultivation using optical density measurements at 600 nm every one hour. In following, the cells were induced with 0.5 mM IPTG at OD600 of 0.6. A part of the cultivation was used as negative control without adding IPTG. After 7 hours of additional growth at 37 °C, cells were harvested through centrifugation at 8000 g for 15 min at 4 °C.
Isolation of the periplasmic rhEs
The isolation of the rhEs, which was expressed in the periplasm, was done by preparing cell fractions according to the literature.18,19 Briefly, the culture was centrifuged at 8000g for 15 minutes at 4 °C. Then, the pellet was suspended in 10ml of buffer (50 mM Tris–HCl, 5 mM MgSO4,18% sucrose, 0.1 mM EDTA, pH 8.0) and stirred slowly with magnetic stirrer at 4 °C for 10 minutes to mix thoroughly. The mixture was centrifuged at 10000 g for 15 minutes and the supernatant was collected as the periplasmic fraction. The total soluble protein was analyzed by the Bradford method (Quick Start™ Bradford Protein Assay, BioRad) using serum bovine albumin as a standard.20
Purification
The supernatant contains rhEs was applied for purification. The crude supernatant containing soluble rhEs was concentrated using an Amicon Ultra centrifugal filter (Ultra-15, MWCO 10 kDa, Z706345 Sigma). Concentrated sample was loaded onto a 1mL SP Sepharose (GE Healthcare Life Science) column that had been equilibrated with 50 mM Tris–HCl, 5 mM MgSO4, 0.1 mM EDTA, pH 6.0 at a flow rate of 0.5 mL/min. The column was washed with 10 mL of the equilibration buffer. The bound rhEs eluted sequentially with different concentrations of NaCl (100 to 1000mM) in Tris buffer. The peak fraction containing the rhEs was collected, pooled and desalted by dialyzing (overnight) at 4°C against 10mmol/L Tris buffer (pH 6.0). The dialyzed sample was then loaded onto a HiPrep 16/60 Sephacryl S-100 HR (GE Healthcare Life Science) equilibrated with Phosphate buffer for further purification. All chromatography was performed at 4°C. Also, Endostatin Human ELISA kit was used to obtain rhEs concentrations, following manufacturer's instructions.
SDS-PAGE and Western blot analysis
The expressed and purified rhEs was checked using sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) under reduced conditions as described by Laemmli.21 Briefly, about 10-20 µl of samples were homogenized in SDS sample loading buffer. The total polyacrylamide concentration was 12% for separating gel and 4% for stacking gel. Protein bands were imagined using Coomassie Brilliant Blue R250. In addition, western blot analysis was applied for further identification of produced rhEs. Briefly, Proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane. The membranes were blocked with blocking buffer for 2 h at 37°C and incubated with rabbit anti-endostatin polyclonal antibody (ab3453) overnight at 37°C. After four times washing with phosphate-buffered saline (PBS), the membranes were incubated with Goat Anti-Rabbit IgG H&L (HRP) (ab6721) for 1 h at room temperature and followed by three times washing with phosphate-buffered saline (PBS). Proteins were visualized by incubation with ECL Western Blotting Substrate (ab65623).
Results
Amplification of the hEs gene
The wild type hEs gene has 31 rare codons including: 3CGG, 3AGG for Arginine, 8CCC for proline, 4 TCC, 2 TCA, 1 AGT, 4TCG for serine and 6 GGG for glycine. These codons were replaced with preferred E. coli codons in synthetic hEs. A summary of codon usage in wild type and codon optimized synthetic hEs gene is shown in Table 1. CAI applied to estimate the adaptation of codon optimized hEs to E. coli codons, was 0.878. The optimized gene had 75.8% of identity with respect to the wild type hEs gene. The construct analyzing showed that there is no cryptic splice donor, cryptic splice acceptor, prokaryotic ribosome binding site, RNA destabilizes sequence and shin-dalgarn sequence. Additionally, restriction analyses showed no restriction sites that may interfere with cloning procedure.
Table 1. Comparison of codon usage for E. coli in wild and optimized hEs gene based on Ikemura classification.
|
Codons
(Number) |
a E . coli Kazusa (%) | b E . coli high exp (%) |
Wt.hEs
(%) |
Opt.hEs
(%) |
Codons
(Number) |
a E . coli Kazusa (%) | b E . coli high exp. (%) | Wt.hEs (%) |
Opt.hEs
(%) |
| Ala (19) | - | - | - | - | Pro (10) | - | - | - | - |
| GCC | 23.87 | 16.14 | 57.89 | 0.00 | *CCC | 5.63 | 1.63 | 80 | 0 |
| GCG | 27.99 | 32.30 | 21.05 | 5.26 | CCG | 19.35 | 71.89 | 20 | 100 |
| GCT | 17.36 | 27.54 | 10.52 | 84.21 | Ser (20) | - | - | - | - |
| GCA | 21.60 | 24.01 | 10.52 | 10.52 | AGC | 15 | 24.33 | 40 | 0 |
| Arg (15) | - | - | - | - | *TCG | 8.52 | 7.39 | 20 | 0 |
| CGC | 18.38 | 32.97 | 37.5 | 0 | *TCC | 9.29 | 26.56 | 20 | 0 |
| *CGG | 6.49 | 0.80 | 20 | 0.00 | *TCA | 9.94 | 4.79 | 10 | 0 |
| *AGG | 2.56 | 0.29 | 20 | 0 | *TCT | 10.94 | 32.41 | 5 | 100 |
| CGT | 18.92 | 64.25 | 6.66 | 100 | *AGT | 10.73 | 4.52 | 5 | 0 |
| Asp (8) | - | - | - | - | Trp (4) | - | - | - | - |
| GAC | 18.83 | 46.05 | 100 | 12.5 | TGG | 13.78 | 100.00 | 100 | 100 |
| GAT | 32.38 | 53.95 | 0 | 87.5 | Val (9) | - | - | - | - |
| Asn (4) | - | - | - | - | GTG | 23.47 | 26.81 | 66.66 | 0 |
| AAC | 21.20 | 82.75 | 100 | 100 | GTC | 14.04 | 13.45 | 22.22 | 0 |
| Cys (4) | - | - | - | - | GTT | 20.04 | 39.77 | 11.11 | 100 |
| TGC | 5.99 | 61.15 | 75 | 100 | Lys (5) | - | - | - | - |
| TGT | 5.35 | 38.85 | 25 | 0 | AAG | 13.05 | 21.45 | 100 | 0 |
| Gly (16) | - | - | - | - | AAA | 35.60 | 78.55 | 0 | 100 |
| GGC | 25.66 | 42.83 | 56.25 | 0 | Thr (7) | - | - | - | - |
| *GGG | 11.58 | 4.36 | 37.5 | 0 | ACC | 21.39 | 53.60 | 42.86 | 100 |
| GGT | 24.93 | 50.84 | .6.25 | 93.75 | ACG | 13.76 | 12.65 | 42.86 | 0 |
| *GGA | 1.61 | 1.97 | 0 | 6.25 | ACT | 11.02 | 29.08 | 14.28 | 0 |
| Glu (7) | - | - | - | - | Phe (10) | - | - | - | - |
| GAG | 18.80 | 24.65 | 100 | 85.72 | TTC | 15.62 | 70.92 | 80 | 90 |
| GAA | 18.83 | 75.35 | 0 | 14.28 | TTT | 22.46 | 29.08 | 20 | 10 |
| Gln (8) | - | - | - | - | Ile (6) | - | - | - | - |
| CAG | 28.12 | 81.35 | 100 | 100 | ATC | 22.69 | 65.94 | 83.33 | 100 |
| Tyr (3) | - | - | - | - | ATT | 29.67 | 33.49 | 16.67 | 0 |
| TAC | 12.01 | 64.77 | 100 | 100 | Met (3) | - | - | - | - |
| Leu (20) | - | - | - | - | ATG | 25.95 | 100.00 | 100 | 100 |
| CTG | 46.4 | 76.67 | 75 | 100 | Stop | - | - | - | - |
| CTC | 10.08 | 8.03 | 25 | 0 | TAA | 1.99 | - | 0 | 50 |
| His (7) | - | - | - | - | TGA | 1.04 | - | 0 | 50 |
| CAC | 8.82 | 70.23 | 71.42 | 100 | TAG | 0.29 | - | 100 | 0 |
| CAT | 12.47 | 29.77 | 28.58 | 0 | GC% | - | - | 68% | 55% |
Codons with low frequency (<10%) are highlighted in yellow (light) and the most preferred codon for each amino acid is highlighted in gray (dark). The optimized hEs gene has lower preferred codons with low frequency than native gene. For gene optimization, rare codons frequency can cause translation error (3.94%) were set to 0.18 and the GC content reduced from 68 % to 55 %, closer to the average GC content of other highly expressed genes in E. coli.
aTaken and adapted from http://www.kazusa.or.jp/codon, bthegenes correspond to genes highly expressed during exponential growth and *Rare codon.
Subsequently, the hEs was amplified using specific primers and pUC57- phoA sp- hEs as a template that showed a 592bp band on 1% agarose gel (Figure 2). This fragment contains hEs coding sequence (555bp), NcoI and XhoI restriction sites.
Figure 2.
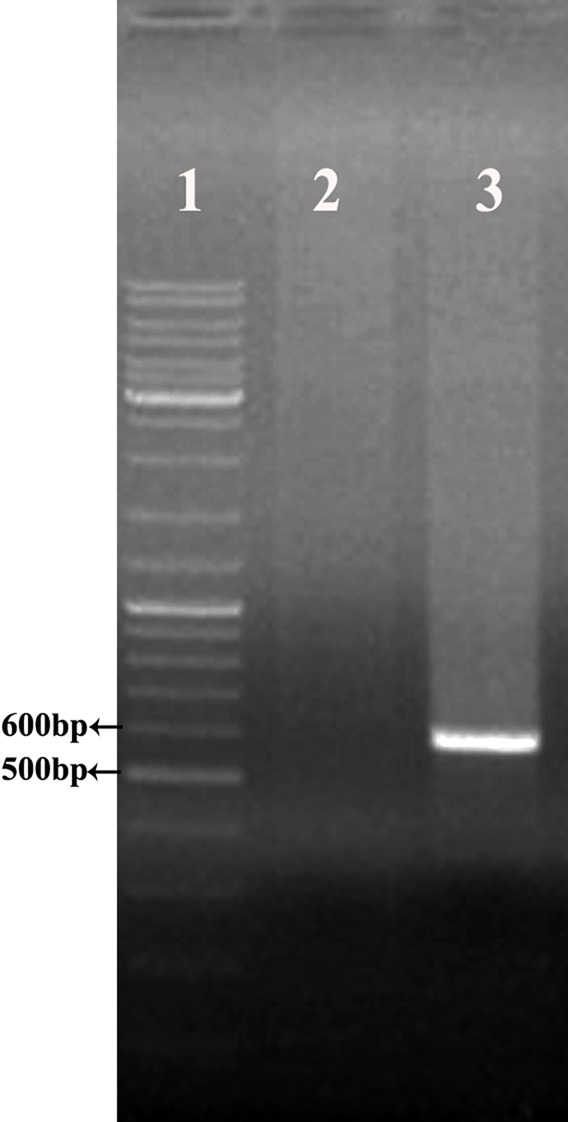
Amplification of hEs coding sequence using Pfu DNA polymerase. (lane1) Gene Ruler DNA Ladder Mix (Thermo scintific), (lane 2) negative control, (lane 3) amplified ORF of hEs (592 bp).
The PCR product was inserted into pJET1.2/blunt cloning vector (between the nucleotides 371 and 372) and transformed into competent cells of E. coli DH5α for propagation (as mentioned in Material and Methods). White- red screening method confirmed the authenticity of transformation. Following, colonies containing recombinant plasmids were confirmed by colony PCR (data are not shown). Also, the positive clones were further confirmed by DNA sequencing (ShineGene Bio-Technologies, Inc., China).
Construction of the pET26b-rhEs expression vector
The plasmid pET26b (+) was used to create expression vectors that targets rhEs into the E. coli periplasm. The protein-coding sequence of the hEs gene was placed between the NcoI and XhoI sites of the pET26 (+). The recombinant pET26b (+) - hEs contained a T7 promoter, Lac operon, N-terminal pelB signal sequence, multiple cloning sites, rhEs encoding gene and T7 terminator. Following the transformation of E. coli BL21 (DE3), the positive clones were selected on LB agar containing kanamycin (50µg/ml). The transformed colonies that contain recombinant pET26b (+) - hEs plasmids were PCR positive against the hEs gene specific primers (Figure 3). The correct orientation of the hEs gene in the pET26b (+) - hEs plasmid was confirmed by double digestion with NcoI and XhoI restriction enzymes. Digestion of the pET26b (+) - hEs plasmid using restriction enzymes (Xho I, Nco I) produced two expected fragments (data are not shown). Digestion results showed the hEs gene had been inserted into the pET26b+ plasmid. Sequencing analysis of the recombinant pET26b (+) - hEs plasmid with the mentioned primers established that there were no amplification mistakes and frame shift in sequence of the cloned hEs gene. Also, the coding sequence of hEs was placed correctly in downstream of pelB signal sequence at the5′ region and the two stop codons placed at the 3′end of gene before the XhoI site.
Figure 3.
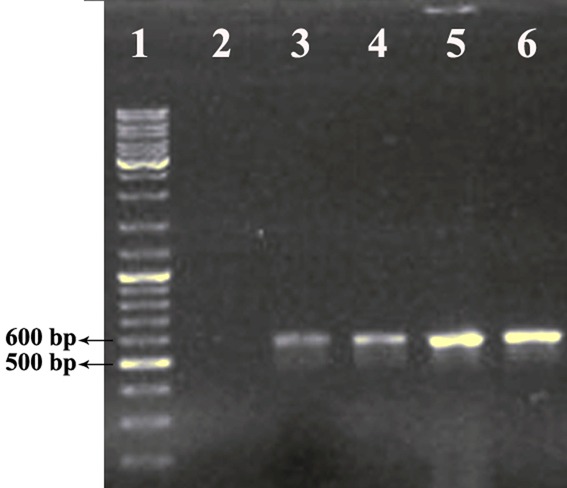
Colony-PCR reaction for randomly selected transformants (four colonies). (lane1) Gene Ruler DNA Ladder Mix (Thermo scientific), (lane 2) negative control, (lane3-6) PCR products at 592 bp representing positive colonies that had pET26b-hEs plasmid.
Expression and analysis of recombinant human Endostatin
In order to analyze the expression of the rhEs protein, a single colony of E. coli BL21 (DE3) carrying the pET26b (+)-hEs vector was grown and induced with IPTG. The final concentration of IPTG was 0.5 mM with a 7 h incubation time at 37 °C. Following the induction, total protein was extracted and determined by the Bradford method showing a 2.37 mg/l concentration. Also, the total periplasmic protein of induced and non-induced cells were run and compared on a 12% SDS-PAGE (Figure 4). The presence of corresponding protein bands approved the expression of the rhEs protein. The result from SDS-PAGE showed that there is a single band of 20 kDa corresponding to hEs (pET26b (+) - hEs). The identity and quantity of the expressed protein was further confirmed by an endostatin specific ELISA test. The rhEs represents about 35 % (0.83mg/l) of the total protein according to ELISA and Bradford results.
Figure 4.
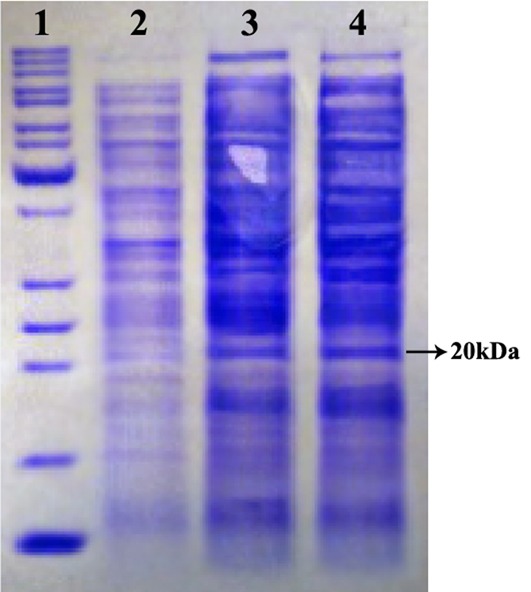
Expression analysis of rhEs in periplasmic space using 12% SDS-PAGE under reduced condition. The band about 20 kDa represents the produced rhEs. Lane 1, protein marker is 10 to 200 kDa from down to up; lane 2, total protein before induction with IPTG as a control; lane 3 and 4, total protein after induction with IPTG in recombinant pET26b.
Purification and identification of rhEs
After isolating the periplsmic proteins, rhEs was purified using cation exchange chromatography and size exclusion chromatography sequentially. The SP- Sepharose column was used for rhEs purification as the first step of chromatography.
The recombinant protein bound strongly, and washing with the low-salt Phosphate buffer (0.1-0.3 M) removed other E. coli-derived proteins. Protein eluted with the 0.4 M NaCl fraction had a maximum amount of rhEs, but was contaminated with another low molecular weight protein (Figure 5). The protein fractions eluted at 0.4 M NaCl were pooled, concentrated and dialyzed against Tris Puffer; pH 6.5. The purified protein was further separated by size exclusion chromatography using a Sephacryl S-100 column. The purified rhEs presented approximately 20 kDa on SDS–PAGE (Figure 5) that was compatible with the theoretical molecular mass, and was found to be immune-reactive when evaluated by Western blot through using rabbit anti-endostatin polyclonal antibody (Figure 6). Finally, the results of SDS-PAGE, Western blot and ELISA assay confirmed the successful secretion of the expressed hEs protein into the periplasm.
Figure 5.
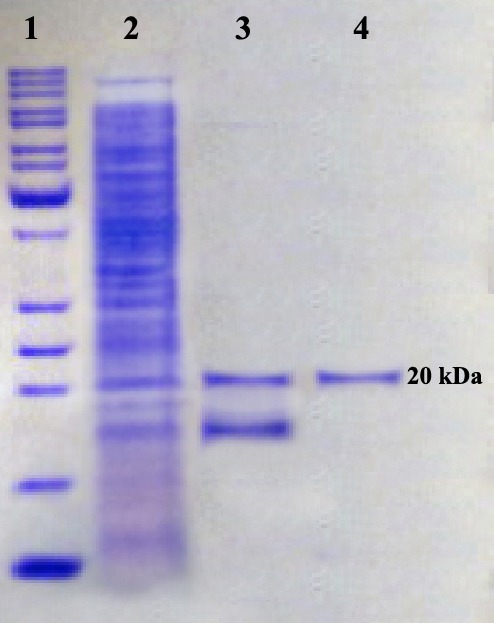
SDS–PAGE analysis of the purified rhEs. Lane 1, protein marker; lane 2, crude protein; lane 3, rhEs protein purified by SP Sepharose column; lane 4, rhEs protein purified by Sephacryl S-100.
Figure 6.
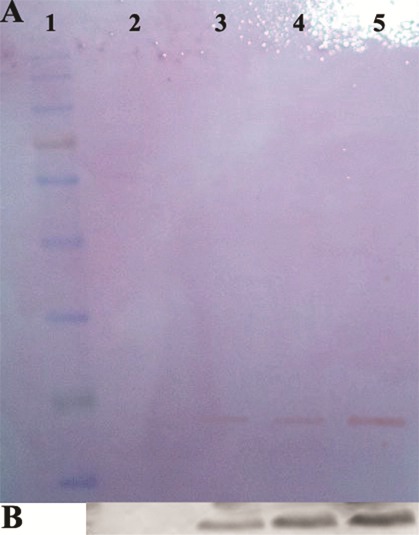
Western-blot analysis with specific rabbit anti-endostatin polyclonal antibody. Panel (A) is the ponceaus staining of pvdf membrane. Lane 2 is the control sample; lane 3-5 represent the sample related to periplasmic proteins after induction, cation exchange purified rhEs and size exclusion purified rhEs, respectively. Panel (B) is the immunoblotting image of samples shown in panel A. Lanes 2, 3, 4 and 5 correspond to the lanes 2, 3, 4 and 5 in panel A. Lane 1 is protein marker.
Discussion
Recently, numerous studies have revealed the important role of angiogenesis in tumor growth. Therefore, inhibition of angiogenesis with anti-angiogenic agents is an attractive field of medical research. Among the anti-angiogenic agents, Es is an attractive candidate as anti- cancer agent with high efficacy, fewer side effects and no reported drug resistance.3,5
Several hosts, including E. coli, yeast, mammalian cell and baculovirus were used to produce rhEs.4,6 Expressing heterologous proteins in E. coli system has some advantages to other systems, including, high expression level, rapid growth and low cost.10,22 However, despite the many advantages of E. coli expression system, high-level expression is not easily achieved. The most common preventing factors in the high level protein expression by E. coli include codon bias, gene product toxicity, insolubility, mRNA secondary structure and mRNA instability.23-26 In addition, there are several reports that proposed the use of synthetic genes to increase expression efficiency.27 Considering the factors involved in the heterologous protein expression, in this study optimized synthetic hEs gene based on the Ikemura classification was applied to amplify hEs gene, followed by cloning and producing in E. coli BL21 (DE3).28
Another factor that limits the using of E. coli expression system is the lack of posttranslational modification. To overcome this problem, expression of recombinant protein in periplasmic space is one of the solutions. Secretory production of recombinant proteins into the periplasm has the following advantages: (a) providing purer product because of having fewer impurities than the cytoplasm (b) providing a more oxidative space than cytoplasm which results in facilitating the disulfide bond formation (c) lesser protease concentration which results in decreasing the degradation of heterologous proteins and the possibility of proper folding of proteins. Wild type signal peptides have been used successfully in E .coli for protein translocation into the periplasm.29,30 There are several reports on the cytoplasmic expression of the hEs gene.4,5,14,15 However, there is no article on the periplasmic expression of rhEs protein.
Thus, considering the structure of rhEs that has two disulfide bonds, periplasmic secretion strategy has been used for expression of rhEs via fusing with pelB signal peptide. The expression vector pET26b(+) harbors a pelB signal peptide at N-terminal of protein that transports the expressed protein into the periplasmic space.31 Also, the powerful promoter of pET26b(+) vector allows for over-expression of the cloned gene.19,32
Periplasmic secretion of rhEs protein was attained because of the attendance of pelB signal peptide, which directly leaded the interest protein to periplasmic region.
But the produced soluble rhEs level was very low in comparison with Du et al. and Xu et al findings which expressed rhEs in the cytoplasm.5,15Du et al. studied the effects of glutathione S-transferase (GST) and NusA protein on the solubility of rhEs in E. coli. They revealed that NusA protein increased the production of soluble rhEs (18mg/L); but GST did not effect on the amount of produced rhEs.5 Also, Xu et al produced soluble rhEs in E. coli employing DnaK-DnaJ-GrpE as molecular chaperone. The obtained yield of soluble rhEs was about 16mg/L.15 This difference in the rhEs concentration can be due to the following reasons: (1) expression in the lab scale condition instead of fermenter, (2) choosing pelB as a signal peptide (3) culture condition (4) cultivation medium and (5) smaller space of periplasm than cytoplasmic space. Releasing periplasmic proteins are done with mechanical and physical cell disruption methods. Gao et al. reported that the physical cell disruption methods are safe and effective method than mechanical methods. In mechanical method, the selectivity of releasing proteins is low and purification process is difficult.33 Thus, in this study osmotic shock method was adopted for the releasing rhEs from the periplasm in accordance with Cao et al. report.19 The advantages of this method are low-cost, quickness and high selectivity in the protein releasing process.33
Conclusion
Based on these findings, it is concluded that the designed construct, which pelB signal peptide fused with rhEs, could express rhEs protein into periplasmic space of E. coli. Considering the low level expression of the target protein, optimization of culture condition variables and other effective factors seems to be needed.
Acknowledgments
We thank Dr. Sarvin Sanyi for assistance in editing this manuscript prior to submission. This research was supported by Tuberculosis and Lung Disease Research Center of Tabriz University of Medical Sciences (Grant number 5/76/520).
Ethical Issues
Not applicable.
Conflict of Interest
Authors declare no conflict of interest in this study.
References
- 1.O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS. et al. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(2):277–85. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 2.Fu Y, Wu X, Han Q, Liang Y, He Y, Luo Y. Sulfate stabilizes the folding intermediate more than the native structure of endostatin. Arch Biochem Biophys. 2008;471(2):232–9. doi: 10.1016/j.abb.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Folkman J. Antiangiogenesis in cancer therapy--endostatin and its mechanisms of action. Exp Cell Res. 2006;312(5):594–607. doi: 10.1016/j.yexcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Dhanabal M, Volk R, Ramchandran R, Simons M, Sukhatme VP. Cloning, expression, and in vitro activity of human endostatin. Biochem Biophys Res Commun. 1999;258(2):345–52. doi: 10.1006/bbrc.1999.0595. [DOI] [PubMed] [Google Scholar]
- 5.Du C, Yi X, Zhang Y. Expression and purification of soluble recombinant human endostatin in Escherichia coli. Biotechnol Bioproc Engine. 2010;15(2):229–35. doi: 10.1007/s12257-009-0100-5. [DOI] [Google Scholar]
- 6.Xu YF, Zhu LP, Hu B, Rong ZG, Zhu HW, Xu JM. et al. A quantitative method for measuring the antitumor potency of recombinant human endostatin in vivo. Eur J Pharmacol. 2007;564(1-3):1–6. doi: 10.1016/j.ejphar.2007.01.086. [DOI] [PubMed] [Google Scholar]
- 7.Terpe K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2006;72(2):211–22. doi: 10.1007/s00253-006-0465-8. [DOI] [PubMed] [Google Scholar]
- 8.Muntari B, Amid A, Mel M, Jami MS, Salleh HM. Recombinant bromelain production in escherichia coli: Process optimization in shake flask culture by response surface methodology. AMB Express. 2012;2:12. doi: 10.1186/2191-0855-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baneyx F. Recombinant protein expression in escherichia coli. Curr Opin Biotechnol. 1999;10(5):411–21. doi: 10.1016/s0958-1669(99)00003-8. [DOI] [PubMed] [Google Scholar]
- 10.Lilie H, Schwarz E, Rudolph R. Advances in refolding of proteins produced in e. Coli. Curr Opin Biotechnol. 1998;9(5):497–501. doi: 10.1016/s0958-1669(98)80035-9. [DOI] [PubMed] [Google Scholar]
- 11.Su Z, Wu X, Feng Y, Ding C, Xiao Y, Cai L. et al. High level expression of human endostatin in pichia pastoris using a synthetic gene construct. Appl Microbiol Biotechnol. 2007;73(6):1355–62. doi: 10.1007/s00253-006-0604-2. [DOI] [PubMed] [Google Scholar]
- 12.Balderas Hernandez VE, Paz Maldonado LM, Medina Rivero E, Barba de la Rosa AP , Jimenez-Bremont JF, Ordonez Acevedo LG. et al. Periplasmic expression and recovery of human interferon gamma in escherichia coli. Protein Expr Purif. 2008;59(1):169–74. doi: 10.1016/j.pep.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 13.Huang X, Wong MK, Zhao Q, Zhu Z, Wang KZ, Huang N. et al. Soluble recombinant endostatin purified from escherichia coli: Antiangiogenic activity and antitumor effect. Cancer Res. 2001;61(2):478–81. [PubMed] [Google Scholar]
- 14.Wei DM, Gao Y, Cao XR, Zhu NC, Liang JF, Xie WP. et al. Soluble multimer of recombinant endostatin expressed in e. Coli has anti-angiogenesis activity. Biochem Biophys Res Commun. 2006;345(4):1398–404. doi: 10.1016/j.bbrc.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 15.Xu HM, Zhang GY, Ji XD, Cao L, Shu L, Hua ZC. Expression of soluble, biologically active recombinant human endostatin in escherichia coli. Protein Expr Purif. 2005;41(2):252–8. doi: 10.1016/j.pep.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 16.Xu R, Du P, Fan JJ, Zhang Q, Li TP, Gan RB. High-level expression and secretion of recombinant mouse endostatin by escherichia coli. Protein Expr Purif. 2002;24(3):453–9. doi: 10.1006/prep.2001.1585. [DOI] [PubMed] [Google Scholar]
- 17.Yari K, Afzali S, Mozafari H, Mansouri K, Mostafaie A. Molecular cloning, expression and purification of recombinant soluble mouse endostatin as an anti-angiogenic protein in escherichia coli. Mol Biol Rep. 2013;40(2):1027–33. doi: 10.1007/s11033-012-2144-4. [DOI] [PubMed] [Google Scholar]
- 18.Cao S, Zhang Y, Liu F, Wang Q, Zhang Q, Liu Q. et al. Secretory expression and purification of recombinant escherichia coli heat-labile enterotoxin b subunit and its applications on intranasal vaccination of hantavirus. Mol Biotechnol. 2009;41(2):91–8. doi: 10.1007/s12033-008-9101-4. [DOI] [PubMed] [Google Scholar]
- 19.Cao W, Li H, Zhang J, Li D, Acheampong DO, Chen Z. et al. Periplasmic expression optimization of vegfr2 d3 adopting response surface methodology: Antiangiogenic activity study. Protein Expr Purif. 2013;90(2):55–66. doi: 10.1016/j.pep.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 20.Morowvat MH, Babaeipour V, Rajabi-Memari H, Vahidi H, Maghsoudi N. Overexpression of recombinant human beta interferon (rhinf-beta) in periplasmic space of escherichia coli. Iran J Pharm Res. 2014;13(Suppl):151–60. [PMC free article] [PubMed] [Google Scholar]
- 21.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage t4. Nature. 1970;227(5259):680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Paek SY, Kim YS, Choi SG. The orientation-dependent expression of angiostatin-endostatin hybrid proteins and their characterization for the synergistic effects of antiangiogenesis. J Microbiol Biotechnol. 2010;20(10):1430–5. doi: 10.4014/jmb.1004.04040. [DOI] [PubMed] [Google Scholar]
- 23. Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22(7):346–53. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Hernández VEB, Maldonado LMP, Rivero EM, de la Rosa APB, Acevedo LGO, Rodríguez ADL. Optimization of human interferon gamma production in Escherichia coli by response surface methodology. Biotechnol Bioproc Engine. 2008;13(1):7–13. doi: 10.1007/s12257-007-0126-5. [DOI] [Google Scholar]
- 25.Kane JF. Effects of rare codon clusters on high-level expression of heterologous proteins in escherichia coli. Curr Opin Biotechnol. 1995;6(5):494–500. doi: 10.1016/0958-1669(95)80082-4. [DOI] [PubMed] [Google Scholar]
- 26. Wu X, Jornvall H, Berndt KD, Oppermann U. Codon optimization reveals critical factors for high level expression of two rare codon genes in escherichia coli: Rna stability and secondary structure but not trna abundance. Biochem Biophys Res Commun. 2004;313(1):89–96. doi: 10.1016/j.bbrc.2003.11.091. [DOI] [PubMed] [Google Scholar]
- 27.Maldonado LM, Hernandez VE, Rivero EM, Barba de la Rosa AP , Flores JL, Acevedo LG. et al. Optimization of culture conditions for a synthetic gene expression in escherichia coli using response surface methodology: The case of human interferon beta. Biomol Eng. 2007;24(2):217–22. doi: 10.1016/j.bioeng.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 28. Ikemura T. Correlation between the abundance of escherichia coli transfer rnas and the occurrence of the respective codons in its protein genes: A proposal for a synonymous codon choice that is optimal for the e. Coli translational system. J Mol Biol. 1981;151(3):389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 29. Harrison ST. Bacterial cell disruption: A key unit operation in the recovery of intracellular products. Biotechnol Adv. 1991;9(2):217–40. doi: 10.1016/0734-9750(91)90005-g. [DOI] [PubMed] [Google Scholar]
- 30.Makrides SC. Strategies for achieving high-level expression of genes in escherichia coli. Microbiol Rev. 1996;60(3):512–38. doi: 10.1128/mr.60.3.512-538.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asghari S, Shekari Khaniani M, Darabi M, Mansoori Derakhshan S. Cloning of soluble human stem cell factor in pet-26b(+) vector. Adv Pharm Bull. 2014;4(1):91–5. doi: 10.5681/apb.2014.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang ZW, Huang W-B, Chao Y-P. Efficient production of recombinant proteins in escherichia coli using an improved l-arabinose-inducible T7 expression system. Process Biochem. 2005;40(9):3137–42. doi: 10.1016/j.procbio.2005.03.013. [DOI] [Google Scholar]
- 33.Gao H, Liu M, Liu J, Dai H, Zhou X, Liu X. et al. Medium optimization for the production of avermectin b1a by streptomyces avermitilis 14-12a using response surface methodology. Bioresour Technol. 2009;100(17):4012–6. doi: 10.1016/j.biortech.2009.03.013. [DOI] [PubMed] [Google Scholar]