

Abstract
Six new chamigrane sesquiterpenes, merulinols A‒F (1‒6), and four known metabolites (7‒10) were isolated from the culture of the basidiomycetous fungus XG8D, a mangrove-derived endophyte. Their structures were elucidated mainly by 1D and 2D NMR, while the structures of 1 and 2 were further confirmed by single-crystal X-ray diffraction analysis. The in vitro cytotoxicity of all compounds was evaluated against three human cancer cell lines, MCF-7, Hep-G2, and KATO-3. Compounds 3 and 4 selectively displayed cytotoxicity against KATO-3 cells with IC50 values of 35.0 and 25.3 μM, respectively.
Keywords: chamigrane, sesquiterpene, endophytic fungus, cytotoxic
1. Introduction
Marine-associated microorganisms are recognized as a promising source of chemically diverse, structurally unique, and biologically active secondary metabolites [1,2]. Among them, the fungi derived from mangrove plants are of special interest and continue to attract considerable attention due to the extreme environmental conditions [3,4,5,6,7,8]. Our research group has, therefore, focused on the exploration of bioactive natural products from these types of fungi collected from mangrove areas in Thailand [9,10]. The isolation and characterization of unique sesquiterpene endoperoxides of the chamigrane type was previously reported from the ethyl acetate (EtOAc) extract of the marine-derived fungal strain XG8D, grown in corn-steep liquor-containing medium [11,12]. This fungus was obtained from the leaves of the mangrove plant Xylocarpus granatum. Chamigrane sesquiterpenoids, mostly halogenated, are generally produced by the red alga in the genus Laurencia (Family Rhodomelaceae) [13,14,15,16,17], with those from fungi being rare [18]. The only precedents include merulins A–D from the basidiomycetous endophytic fungus XG8D [11,12], steperoxide A from the higher fungus Steccherinum ochraceum [19], talaperoxides A–D from the mangrove endophytic fungus Talaromyces flavus [20], and acaciicolinolides A–C and acaciicolinols A–L from Pseudolagarobasidium acaciicola, which is also a mangrove-derived endophytic fungus [21].
Moreover, some studies have shown that the production of secondary metabolites might be highly dependent on fermentation conditions and modes [22,23,24]. In particular, endophytic fungi seem to be affected to a greater extent by variations in the culture media, which leads to changes in their metabolite profiles [25]. This prompted us to embark on a study of the effect of the culture medium on the metabolite production of the endophytic fungus strain XG8D. As a result, the primary screening of the cultivation conditions for this strain indicated that alteration of the culture medium greatly affected the chemical profile of this fungus. In the present study, the chemical investigation of the XG8D strain grown in Sabouraud dextrose broth (SDB) led to the isolation of six new chamigrane sesquiterpenes, not of the endoperoxide type, namely merulinols A–G (1–6), and four known compounds, acaciicolinols C (7), K (8), F (9) and D (10) (Figure 1). Details of the isolation, structure elucidation and in vitro cytotoxic activity of these metabolites are reported herein.
Figure 1.

Structures of metabolites 1–10 isolated from the basidiomycetous fungus XG8D.
2. Results and Discussion
Merulinol A (1) was obtained as colorless crystals and was determined to have a molecular formula of C14H22O4 by the HR-ESIMS ion at m/z 277.1419 [M + Na]+, implying four degrees of unsaturation. Combined analysis of the 1H and 13C NMR (Table 1, Figures S1–S3) plus the HSQC (Figure S4) spectroscopic data revealed the presence of three singlet methyls including one downfield signal at δH 1.68, five sp3 methylenes, one oxymethine and five quaternary carbons, including one ketone carbon (δC 212.0). These data accounted for all the NMR resonances of 1 and one of three unsaturations, indicating that 1 was a tricyclic compound. Analysis of the 1H–1H COSY spectrum (Figure 2 and Figure S3) established three partial structures of H2-1/H-2, H2-4/ H2-5, and H2-9/H2-10. The C-10 methylene and C-6 quaternary carbon were connected to C-11 by the HMBC correlations from H2-10, H3-12, and H3-13 to C-11, as well as from H3-12 and H3-13 to C-6 (Figure 2 and Figure S5). Further HMBC cross-peaks from a downfield singlet methyl at δH 1.68 to the C-8 ketone carbon, C-7 oxygenated quaternary carbon and C-6 led to the attachment of this methyl group (C-14) on C-7 and the connection of C-6 and C-8 on C-7. Two other partial structures, H2-1/H-2 and H2-4/ H2-5, were placed between C-6 and C-3 from the HMBC correlations of H2-1, H-2, H2-4, H2-5 to C-6 and from H-2 to C-3, and so established a 6,6-bicyclic skeleton via a C-6 spiro carbon. Moreover, the 13C resonance of C-3 at δC 94.5 implied that C-3 was a hemiketal moiety. According to the molecular formula C14H22O4, and the degree of unsaturation of 1, C-3 was thus linked, through an ether bond, to C-7 to establish a tricyclic structure as shown. The proposed structure of 1 and its relative configuration were further confirmed by single-crystal X-ray diffraction analysis using Mo Kα radiation, and a perspective ORTEP plot is depicted in Figure 3.
Table 1.
1H (400 MHz) and 13C NMR (100 MHz) data of compounds 1‒3.
| Position | 1 a | 2 b | 3 a | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | |
| 1 | 32.4, CH2 | 1.99, m | 29.2, CH2 | 2.19, d (20.0) | 26.9, CH2 | 2.15, m |
| 2.11, dt (14.4, 2.8) | 2.34, d (20.0) | |||||
| 2 | 69.4, CH | 3.85, dd (9.6, 2.0) | 124.0, CH | 5.16, br s | 144.1, CH | 7.18, br s |
| 3 | 94.5, qC | 137.7, qC | 130.0, qC | |||
| 4 | 29.5, CH2 | 1.66, m | 25.1, CH2 | 1.89, m | 23.3, CH2 | 2.24, m |
| 1.94, m | 2.44, m | |||||
| 5 | 25.7, CH2 | 1.64, m | 25.8, CH2 | 2.05, m | 29.1, CH2 | 1.63, m |
| 1.70, m | ||||||
| 6 | 43.5, qC | 44.3, qC | 41.3, qC | |||
| 7 | 86.0, qC | 78.3, qC | 44.9, CH | 1.65, m | ||
| 8 | 212.0, qC | 76.9, CH | 3.56, br s | 72.5, CH | 3.33, td (12.0, 4.0) | |
| 9 | 35.1, CH2 | 2.31, m | 26.9, CH2 | 1.38, dq (12.0, 4.0) | 31.6, CH2 | 1.48,m |
| 2.75, m | 2.10, m | 1.79, dq (12.0, 4.0) | ||||
| 10 | 37.3, CH2 | 1.61, m | 34.7, CH2 | 1.00, m | 36.6, CH2 | 1.23, dt (12.0, 4.0) |
| 1.92, m | 1.86, m | 1.60, m | ||||
| 11 | 37.1, qC | 38.3, qC | 37.3, qC | |||
| 12 | 27.3, CH3 | 0.94, s | 30.5, CH3 | 0.85, s | 27.9, CH3 | 0.78, s |
| 13 | 25.1, CH | 1.24, s | 26.3, CH3 | 1.17, s | 22.5, CH3 | 0.99, s |
| 14 | 25.8, CH3 | 1.68, s | 25.2, CH3 | 1.33, s | 12.8, CH3 | 1.01, d (6.8) |
| 15 | 66.9, CH2 | 3.89, br s | 171.3, qC | |||
| 7-OH | 3.08, s | |||||
a NMR data were measured in CDCl3; b NMR data were measured in acetone-d6.
Figure 2.

1H‒1H COSY and selected HMBC correlations of 1, 5, and 6.
Figure 3.
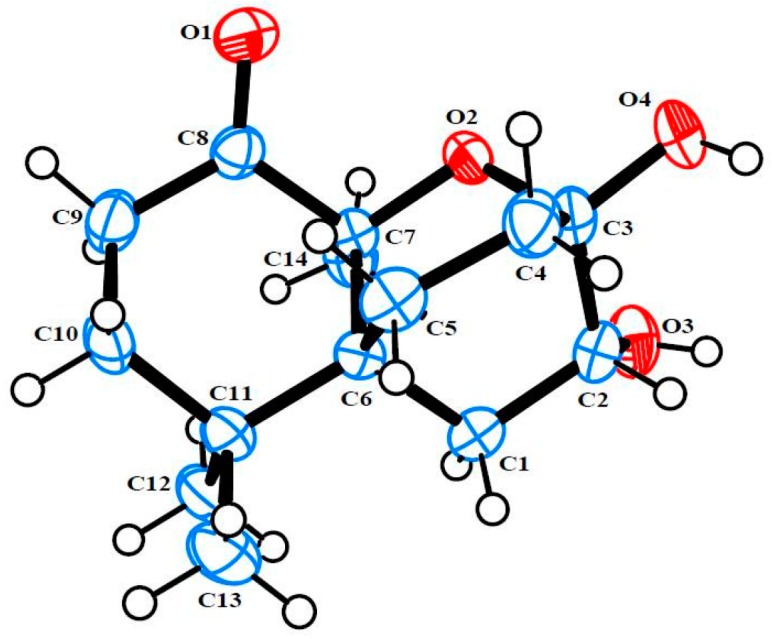
ORTEP plot of 1.
Merulinol B (2) was also obtained as colorless crystals. HR-ESIMS analysis of the molecular ion (m/z 277.1771 [M + Na]+) suggested the molecular formula of C15H26O3, which was indicative of three degrees of unsaturation. Analysis of 1H and 13C NMR (Table 1, Figures S6–S8) plus the HSQC (Figure S9) spectroscopic data indicated the presence of three singlet methyls, six sp3 methylenes (one oxymethylene), one oxymethine, one olefinic methine and four quaternary carbons (one oxygenated and one olefinic carbon). It was revealed that compound 2 had similar 1H and 13C NMR spectra to those of acaciicolinol C (7) [21], except the C-15 carboxylic group in 7 was replaced by a hydroxymethyl in 2. Moreover, an exchangeable proton at δH 3.08 (s) was assigned to 7-OH by its correlations to C-6 and C-7 (Figure S10). Finally, the relative configuration of 2 was assigned by NOESY correlation (Figure S11) and single-crystal X-ray diffraction analysis using Mo Kα radiation, and a perspective ORTEP plot is depicted in Figure 4.
Figure 4.

ORTEP plot of 2.
Merulinol C (3), obtained as a pale yellow gum, was assigned a molecular formula of C15H24O3 by HR-ESIMS (m/z 275.1612 [M + Na]+), implying four degrees of unsaturation. The 1H and 13C NMR spectroscopic data of 3 (Table 1, Figures S12–S15) closely resembled those of 2. The obvious difference in the 1H NMR spectra between 2 and 3 was the presence of a doublet methyl signal at δH 1.01 (J = 6.8 Hz) in 3, replacing the singlet methyl signal (C-14) in 2. In addition, the 13C NMR signal of an oxyquaternary carbon (δC 78.3, C-7) in 2 was absent in 3, while signals for a methine group (δH 1.65 m, δC 44.9) were observed in 3. The location of the doublet methyl at C-14 was confirmed by its 1H–1H COSY correlation with H-7 (Figure S14) and HMBC cross-peaks from H3-14 to C-8, C-7 and C-6 (Figure S16). The NOESY correlations of H3-13/H3-14, H3-13/H2-1 and H3-14/H2-1 (Figure 5 and Figure S17) supported that 3 had the same relative configuration as 2. Additionally, the NOE correlation between H3-14 and H-8, as well as the lack of a correlation between H-7 and H-8, revealed the β-orientation of 8-OH.
Figure 5.

Selected NOESY correlations of 3 and 4.
Merulinol D (4) was obtained as a pale yellow gum. Its molecular formula was established as C15H24O3 by HR-ESIMS (m/z 275.1616 [M + Na]+), being the same as that of 3. Its 1H and 13C NMR spectra (Table 2, Figures S18 and S19) displayed resonances that were nearly identical with those of 3. Interpretation of 2D NMR data (Figures S20–S23) established the same planar structure as 3. The NOESY correlations of 4 indicated that 4 had the same configuration as 3, except for that of C-8. The observed NOE correlation between H-7 and H-8 (Figure 5) suggested the α-orientation of HO-8. Therefore, compound 4 was assigned as the C-8 epimer of 3. This assignment was supported by the opposite signs of their specific rotations ( +6.4 for 3 vs. −47.2 for 4).
Table 2.
1H (400 MHz) and 13C NMR (100 MHz) data of compounds 4‒6 in CDCl3.
| Position | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | |
| 1 | 29.9, CH2 | 2.17, m | 30.1, CH2 | 2.27, d (20.0) | 38.9, CH2 | 2.70, d (14.4) |
| 2.40, m | 2.43, d (20.0) | 2.79, d (14.4) | ||||
| 2 | 142.7, CH | 7.12, br s | 142.4, CH | 7.26, br s | 215.8, qC | |
| 3 | 129.1, qC | 130.6, qC | 75.9, qC | |||
| 4 | 22.6, CH2 | 2.27, m | 22.3, CH2 | 2.10, m | 33.3, CH2 | 1.76, m |
| 2.35, m | 2.58, d (18.0) | 2.24, m | ||||
| 5 | 27.5, CH2 | 1.50, m | 30.7, CH2 | 1.88, m | 23.5, CH2 | 1.54, m |
| 1.82, m | 1.80, m | |||||
| 6 | 39.5, qC | 43.0, qC | 55.2, qC | |||
| 7 | 36.8, CH | 1.95, m | 143.8, qC | 81.8, qC | ||
| 8 | 70.8, CH | 3.91, dd (12.0, 6.8) | 135.9, qC | 213.8, qC | ||
| 9 | 27.7, CH2 | 1.63,m | 192.3, qC | 33.7, CH2 | 2.41,m | |
| 2.74, m | ||||||
| 10 | 35.6, CH2 | 1.48, m | 47.1, CH2 | 2.22, d (18.0) | 36.5, CH2 | 1.61, m |
| 2.78, d (18.0) | 1.91, m | |||||
| 11 | 36.6, qC | 40.8, qC | 39.0, qC | |||
| 12 | 25.7, CH3 | 0.84, s | 23.8, CH3 | 1.01, s | 25.0, CH3 | 1.28, s |
| 13 | 26.3, CH3 | 0.95, s | 24.8, CH3 | 1.05, s | 28.6, CH3 | 1.00, s |
| 14 | 11.4, CH3 | 0.98, d (6.8) | 15.1, CH3 | 1.85, s | 25.4, CH3 | 1.42, s |
| 15 | 171.4, qC | 170.9, qC | 67.1, CH2 | 3.48, d (11.6) | ||
| 3.85, d (11.6) | ||||||
| 7-OH | 4.15, br s | |||||
Merulinol E (5) was obtained as a yellow gum. The molecular formula was determined to be C15H20O4 by the HR-ESIMS ion at m/z 265.1431 [M + H]+, implying six degrees of unsaturation. The 1H and 13C NMR (Table 2, Figures S24–S26) plus the HSQC (Figure S27) data indicated the structure of 5 contained three singlet methyls, four methylenes, one olefinic methine, five quaternary carbons (including two olefinic) and two carbonyls. The two carbonyl signals at δC 192.3 and 170.9 were ascribed to one α,β-unsaturated ketone and one carboxylic acid carbonyl, respectively. Comparison of the NMR data of 5 with those of 3 and 4 revealed that 5 had the same B ring with an α,β-unsaturated carboxylic acid moiety as found in both 3 and 4. Analysis of the HMBC spectrum indicated that an α,β-unsaturated ketone was located at C-7 to C-9 due to the correlations of H3-14/C-7, H3-14/C-8 and H2-10/C-9 (Figure 2 and Figure S28). The C-8 resonance (δC 135.9) was deshielded, which coupled with the HRESIMS data led to the attachment of the hydroxy group on this olefinic carbon. Indeed, the NMR data of 5 were similar to those of acaciicolinol L [21], except the C-15 hydroxymethyl in acaciicolinol L was replaced by a carboxylic moiety in 5. The NOESY data (Figure S29) verified that 5 shared the same configuration as merulinols A‒E. Consequently, the structure of 5 was established as shown.
Merulinol F (6), also obtained as a pale yellow gum, had a molecular formula of C15H24O5 from the HR-ESIMS ion at m/z 307.1516 [M + Na]+. The 1H NMR spectrum (Figure S30) showed signals of three singlet methyls, five methylenes and one hydroxymethyl. The 13C NMR spectrum (Figure S31) exhibited two ketone resonances at δC 213.8 and 215.8. The NMR data of 6 (Figures S30–S34) were similar to those of acaciicolinol K (9) [21], except that the C-9–C-10 double bond in 9 was replaced by a single bond. This was confirmed by the 1H‒1H COSY correlation of H2-9 and H2-10 and the HMBC correlations from H2-9 to a ketone at δC 213.8 (C-8) (Figure 2 and Figures S32 and S34). Based on these spectroscopic data, the structure of 6 was determined as shown. However, its configuration at C-3 and C-7 could not be conclusively established from the NOESY data.
The in vitro cytotoxic activities of the isolated compounds 1‒10 against the MCF-7, Hep-G2, and KATO-3 cell lines were tested using MTT assay to approximate the number of viable cells, while doxorubicin was used as a positive control. Compounds 3 and 4 exhibited activities against KATO-3 cells with IC50 values of 35.0 ± 1.20 and 25.3 ± 0.82 μM, respectively, but against the other two cell lines and all the other compounds, a growth inhibition against the three cell lines of less than 50% at a dose of 50 μM was observed and thus they were considered inactive.
3. Experimental Section
3.1. General Experimental Procedures
Melting points were determined on a melting point M565 apparatus and are uncorrected. Optical rotations were measured on a JASCO P-1010 polarimeter (JASCO Corporation, Tokyo, Japan). UV spectra were recorded on a Spekol 1200 Analytic Jena spectrophotometer. IR spectra were recorded on a Bruker ALPHA FT-IR spectrometer (Bruker, MA, USA). NMR spectra were acquired on a Varian Mercury-400 Plus NMR spectrometer (Varian, CA, USA). High-resolution ESI mass spectroscopy was measured on a Bruker micrOTOF (Bruker). Silica gel (230–400 mesh, Merck, Darmstadt, Germany) and Sephadex LH20 (Amersham Biosciences, NJ, USA) were used for open-column chromatography. Analytical thin layer chromatography (TLC) was performed using precoated silica gel 60 GF254 plates (Merck). Single-crystal X-ray diffraction analysis was performed on a Bruker APEX II diffractometer (Bruker).
3.2. Fungal Material
The endophytic fungus XG8D used in this study was isolated from the healthy leaves of Xylocarpus granatum collected from Samutsakorn province, Thailand, in July 2008. The fungus was identified to the family Meruliaceae (order Polyporales, subclass Incertaesedis, class Agaricomycetes, phylum Basidiomycota) by analysis of the 28S rDNA and ITS data (GenBank accession Nos. HM060640 and HM060641).
3.3. Fermentation, Extraction, and Isolation
The fungus XG8D was cultured on potato dextrose agar at room temperature for 10 days. Then, five pieces (0.5 cm2 × 0.5 cm2) of agar plugs were inoculated in a 1000 mL Erlenmeyer flask containing 200 mL of Sabouraud dextrose broth (SDB) at 30 °C for 21 days. The fungal culture (10 L) was filtered to remove mycelia, and the culture broth was extracted twice with ethyl acetate (EtOAc). The combined EtOAc extract was concentrated under reduced pressure to afford a dark brown residue (12.60 g).
The EtOAc extract was subjected to column chromatography (CC) over Sephadex LH20 and eluted with MeOH to obtain five fractions (A–E). Fraction E (4.68 g) was fractionated by CC over silica gel eluting with a 1:2 to 1:0 gradient of EtOAc-n-hexane to give 11 fractions (E1–E11). Fraction E4 (379.4 mg) was separated by CC using MeOH-CH2Cl2 (1:19) to afford nine subfractions (E4.1–E4.9). Subfraction E4.8 was further purified using the same procedure as fraction E4 to provide 3 (5.2 mg), while subfraction E4.9 was eluted with acetone-CH2Cl2 (1:2) on silica gel column to afford 4 (2.8 mg). Fraction E5 (256.0 mg) was subjected to CC over silica gel using EtOAc-CH2Cl2 (3:2) to give seven subfractions (E5.1–E5.7). Subfraction E5.3 was purified by CC over Sephadex LH20 eluted with MeOH to provide 5 (3.7 mg). Fraction E8 (198.2 mg) was chromatographed on silica gel (acetone-n-hexane, 1:3) to afford six subfractions (E8.1–E8.6). Subfraction E8.4 gave 6 (5.2 mg) and 8 (25.2 mg) upon CC over silica gel (acetone-CH2Cl2, 2:8). Subfraction E8.6 (52.6 mg) was further purified by a silica gel CC using EtOAc-CH2Cl2 (1:1) to yield a pale yellow solid, followed by recrystallization with MeOH to give 7 (16.2 mg). Subfraction E8.5 (75.3 mg) was subjected to CC over silica gel using MeOH-CH2Cl2 (1:19) to obtain five fractions (E8.5.1–E8.5.5). After solvent evaporation, a light yellow solid was formed from fraction E8.5.5, which was further recrystallized with MeOH to afford 2 (9.8 mg). Fraction E9 was purified by silica gel CC using 2:3 EtOAc-benzene elution followed by recrystallization from MeOH to yield 1 (2.4 mg). Fraction E10 (239.4 mg) was subjected to CC over silica gel using MeOH-CH2Cl2 (1:19, then 1:9) to provide 14 subfractions (E10.1–E10.14). Subfractions E10.11 (12.8 mg) and E10.14 (10.1 mg) were further purified by Sephadex LH20 CC eluting with MeOH to afford 9 (5.6 mg) and 10 (3.3 mg), respectively.
Merulinol A (1): colorless crystals; mp 195–199 °C, +18.4 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 218 (2.71), 279 (2.31) nm; IR (neat) νmax 3413, 2969, 1718, 1460, 1375, 1217, 1049 cm–1; 1H and 13C NMR data (Table 1); HR-ESIMS m/z 277.1419 [M + Na]+ (calcd. for C14H22O4Na, m/z 277.1416 [M + Na]+).
Merulinol B (2): colorless crystals; mp 126–129 °C, ‒16.8 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 213 (2.25), 239 (2.25) nm; IR (neat) νmax 3359, 2924, 2853, 1738, 1658, 1632, 1468, 1367, 1216, 1054 cm–1; 1H and 13C NMR data (Table 1); HR-ESIMS m/z 277.1771 [M + Na]+ (calcd. for C15H26O3Na, m/z 277.1780 [M + Na]+).
Merulinol C (3): Pale yellow gum; +5.6 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 225 (3.06) nm; IR (neat) νmax 3441, 2925, 2855, 1737, 1685, 1457, 1367, 1217, 1024 cm–1; 1H and 13C NMR data (Table 1); HR-ESIMS m/z 275.1612 [M + Na]+ (calcd. for C15H24O3Na, m/z 275.1623 [M + Na]+).
Merulinol D (4): Pale yellow gum; ‒47.2 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 227 (2.98) nm; IR (neat) νmax 3435, 2925, 2856, 1732, 1687, 1455, 1360, 1223, 1021 cm–1; 1H and 13C NMR data (Table 2); HR-ESIMS m/z 275.1616 [M + Na]+ (calcd. for C15H24O3Na, m/z 275.1623 [M + Na]+).
Merulinol E (5): Pale yellow gum; +127.2 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 218 (2.82), 280 (2.82) nm; IR (neat) νmax 3361, 2923, 2852, 1738, 1658, 1468, 1366, 1217 cm–1; 1H and 13C NMR data (Table 2); HR-ESIMS m/z 265.1431 [M + H]+ (calcd. for C15H21O4, m/z 265.1440 [M + H]+).
Merulinol F (6): Pale yellow gum; ‒138.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 215 (2.49), 291 (2.05) nm; IR (neat) νmax 3388, 2927, 2855, 1706, 1659, 1367, 1230 cm–1; 1H and 13C NMR data (Table 2); HR-ESIMS m/z 307.1516 [M + Na]+ (calcd. for C15H24O5Na, m/z 307.1521 [M + Na]+).
3.4. Single X-ray Crystallograpic Analysis of Merulinols A (1) and B (2)
All crystallographic data were collected at 293 K on a Bruker APEX II diffractometer with Mo Kα radiation (λ = 0.71073). The structures were solved by direct methods using SHELXS-97 and refined by full-matrix least-squares on all F2 data using SHELXS-97 to final R values [26,27]. All hydrogen atoms were added at calculated positions and refined using a rigid model. Crystallographic data for 1 and 2 have been deposited with the Cambridge Crystallographic Data Centre with the deposition numbers CCDC 1404677 and 832629, respectively [28].
Crystal Data for 1: colorless crystal, C14H22O4, Mr = 254.32, orthorhombic, a = 9.0809(17) Å, b = 10.223(2) Å, c = 13.933(4) Å, space group P212121, Z = 4, Dx = 1.306 Mg/m3, and V = 1293.5(5) Å3, μ (Mo Kα) = 0.09 mm‒1, and F(000) = 552. Crystal dimensions: 0.40 mm × 0.25 mm × 0.20 mm. Independent reflections: 1901 (Rint = 0.022). The final R1 values were 0.042, wR2 = 0.103 (I > 2σ(I)).
Crystal Data for 2: colorless crystal, C30H48O8, Mr = 536.68, orthorhombic, a = 11.3273(7) Å, b = 11.8750(7) Å, c = 20.5308(13) Å, space group P212121, Z = 4, Dx = 1.291 Mg/m3, and V = 2761.6(3) Å3, μ (Mo Kα) = 0.09 mm‒1, and F(000) = 1168. Crystal dimensions: 0.40 mm× 0.30 mm× 0.18 mm. Independent reflections: 5564 (Rint = 0.112). The final R1 values were 0.095, wR2 = 0.268 (I > 2σ(I)).
3.5. Cell Culture
Human breast (MCF-7), liver (Hep-G2), and gastric (KATO-3) cancer cell lines were cultured in RPMI-1640 (HiMedia Laboratories, Mumbai, India) supplemented with 100 U/mL of penicillin, 100 μg/mL of streptomycin and 10% fetal bovine serum (FBS, Thermo Scientific, Waltham, MA, USA). The cells were incubated in a humidified atmosphere of 5% CO2 at 37 °C and sub-cultured every three days.
3.6. Cytotoxicity Assay
Cytotoxicity of isolated compounds against the MCF-7, Hep-G2 and KATO-3 cancer cells was assessed using the MTT (3-[4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium] bromide) assay as previously described [29]. Briefly, freshly trypsinized cell suspensions were seeded into 96-well culture plates at 1 × 104 cells/well in the presence or absence of the test compound. After incubation at 37 °C for 72 h, 10 μL of MTT solution (5 mg in PBS 1 mL) was added to each well for 4 h. The cell-free supernatant was then removed, and DMSO was added to dissolve the formazan crystals. Absorbance values were measured with a microplate reader at 540 nm. Experiments were operated in triplicate, and data are described as mean ± SD of three independent experiments. Doxorubicin was used as a positive control.
4. Conclusions
As a result, six new fungal metabolites in the class of chamigrane sesquiterpenes (1‒6), together with four known compounds (7‒10), were isolated from the basidiomycetous endophytic fungus strain XG8D which was isolated from a Thai mangrove plant. Among the isolated compounds, merulinol A (1) is a nor-chamigrane with a novel tricyclic ring system, whereas compounds 2‒10 are 6/6 spirobicyclic charmigrane sesquiterpenes. Compounds 3 and 4 selectively exhibited cytotoxicity toward gastric KATO-3 cells with IC50 values of 35.0 ± 1.20 and 25.3 ± 0.82 μM, respectively.
Acknowledgments
This work was supported by the Thailand Research Fund (Grant No. RSA5880050). We thank Sujitra Hantanong and Supichar Chokpaiboon, Program in Biotechnology, Faculty of Science, Chulalongkorn University, for fungal cultivation. The authors also wish to thank Robert Douglas John Butcher, the Publication Counseling Unit, for English language editing.
Supplementary Materials
The following are available online at www.mdpi.com/1660-3397/14/7/132/s1, NMR spectra of compounds 1–6 and CIF files for compounds 1 and 2.
Author Contributions
Khanitha Pudhom was the principal investigator, who proposed ideas for the present work, managed and supervised the whole research work, prepared and corrected the manuscript, and contributed to the structure elucidation of the new compounds. Siwattra Choodej performed all experiments for compounds 1‒10, including fermentation, isolation, structure elucidation, biological assay and manuscript preparation. Thapong Teerawatananond performed the single-crystal X-ray crystallographic experiment and analyzed the data. Tohru Mitsunaga contributed material and analysis tools.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Rateb M.E., Ebel R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011;28:290–344. doi: 10.1039/c0np00061b. [DOI] [PubMed] [Google Scholar]
- 2.Chen G., Wang H.F., Pei Y.H. Secondary metabolites from marine-derived microorganisms. J. Asian Nat. Prod. Res. 2014;16:105–122. doi: 10.1080/10286020.2013.855202. [DOI] [PubMed] [Google Scholar]
- 3.Anada K., Sridhar K.R. Diversity of endophytic fungi in the roots of mangrove species on the west coast of India. Can. J. Microbiol. 2002;48:871–878. doi: 10.1139/w02-080. [DOI] [PubMed] [Google Scholar]
- 4.Debbab A., Aly A.H., Proksch P. Mangrove derived fungal endophytes—A chemical and biological perception. Fungal Divers. 2013;61:1–27. doi: 10.1007/s13225-013-0243-8. [DOI] [Google Scholar]
- 5.Zhou Z.F., Kurtán T., Yang X.H., Mándi A., Geng M.Y., Ye B.P., Taglialatela-Scafati O., Guo Y.W. Penibruguieramine A, a novel pyrrolizidine alkaloid from the endophytic fungus Penicillium sp. GD6 associated with Chinese Mangrove Bruguiera gymnorrhiza. Org. Lett. 2014;16:1390–1393. doi: 10.1021/ol5001523. [DOI] [PubMed] [Google Scholar]
- 6.Meng L.H., Li X.M., Lv C.T., Huang C.G., Wang B.G. Brocazines A–F, cytotoxic bisthiodiketopiperazine derivatives from Penicillium brocae MA-231, an endophytic fungus derived from the marine mangrove plant Avicennia marina. J. Nat. Prod. 2014;77:1921–1927. doi: 10.1021/np500382k. [DOI] [PubMed] [Google Scholar]
- 7.Peng J., Lin T., Wang W., Xin Z., Zhu T., Gu Q., Li D. Antiviral alkaloids produced by the mangrove-derived fungus Cladosporium sp. PJX-41. J. Nat. Prod. 2013;76:1133–1140. doi: 10.1021/np400200k. [DOI] [PubMed] [Google Scholar]
- 8.Zang L.Y., Wei W., Guo Y., Wang T., Jiao R.H., Ng S.W., Tan R.X., Ge H.M. Sesquiterpenoids from the mangrove-derived endophytic fungus Diaporthe sp. J. Nat. Prod. 2012;75:1744–1749. doi: 10.1021/np3004112. [DOI] [PubMed] [Google Scholar]
- 9.Pudhom K., Teerawatananond T., Chookpaiboon S. Spirobisnaphthalenes from the mangrove-derived fungus Rhytidhysteron sp. AS21B. Mar. Drugs. 2014;12:1271–1280. doi: 10.3390/md12031271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pudhom K., Teerawatananond T. Rhytidenones A–F, spirobisnaphthalenes from Rhytidhysteron sp. AS21B, an endophytic fungus. J. Nat. Prod. 2014;77:1962–1966. doi: 10.1021/np500068y. [DOI] [PubMed] [Google Scholar]
- 11.Chokpaiboon S., Sommit D., Teerawatnanond T., Muangsin N., Bunyapaiboonsri T., Pudhom K. Cytotoxic nor-chamigrane and chamigrane endoperoxides from a basidiomycetous fungus. J. Nat. Prod. 2010;73:1005–1007. doi: 10.1021/np100103j. [DOI] [PubMed] [Google Scholar]
- 12.Chokpaiboon S., Sommit D., Bunyapaiboonsri T., Matsubara K., Pudhom K. Antiangiogenic effect of chamigrane endoperoxides from a Thai mangrove-derived fungus. J. Nat. Prod. 2011;74:2290–2294. doi: 10.1021/np200491g. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki M., Daitoh M., Vairappan C.S., Abe T., Masuda M. Novel halogenated metabolites from the Malaysian Laurencia pannosa. J. Nat. Prod. 2001;64:597–602. doi: 10.1021/np0006317. [DOI] [PubMed] [Google Scholar]
- 14.Brito I., Cueto M., Díaz-Marrero A.R., Darias J., Martín A.S. Oxachamigrenes, new halogenated sesquiterpenes from Laurencia obtusa. J. Nat. Prod. 2002;65:946–948. doi: 10.1021/np010580t. [DOI] [PubMed] [Google Scholar]
- 15.Davyt D., Fernandez R., Suescun L., Mombrú A.W., Saldaña J., Domínguez L., Coll J., Fujii M.T., Manta E. New sesquiterpene derivatives from the red alga Laurencia scoparia. Isolation, structure determination, and anthelmintic activity. J. Nat. Prod. 2001;64:1552–1555. doi: 10.1021/np0102307. [DOI] [PubMed] [Google Scholar]
- 16.Kimura J., Kamada N., Tsujimoto Y. Fourteen chamigrane derivatives from a red alga, Laurencia nidifica. Bull. Chem. Soc. Jpn. 1999;72:289–292. doi: 10.1246/bcsj.72.289. [DOI] [Google Scholar]
- 17.Al-Massarani S.M. Phytochemical and biological properties of sesquiterpene constituents from the marine red seaweed Laurencia: A review. Nat. Prod. Chem. Res. 2014;2:1000147. [Google Scholar]
- 18.Ebel R. Terpenes from marine-derived fungi. Mar. Drugs. 2010;8:2340–2368. doi: 10.3390/md8082340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu D.Z., Dong Z.J., Wang F., Liu J.K. Two novel norsesquiterpene peroxides from basidiomycete Steccherinum ochraceum. Tetrahedron Lett. 2010;51:3152–3153. doi: 10.1016/j.tetlet.2010.04.048. [DOI] [Google Scholar]
- 20.Li H., Huang H., Shao C., Huang H., Jiang J., Zhu X., Liu Y., Liu L., Lu Y., Li M., et al. Cytotoxic norsesquiterpene peroxides from the endophytic fungus Talaromyces flavus isolated from the mangrove plant Sonneratia apetala. J. Nat. Prod. 2011;74:1230–1235. doi: 10.1021/np200164k. [DOI] [PubMed] [Google Scholar]
- 21.Wibowo M., Prachyawarakorn V., Aree T., Mahidol C., Ruchirawat S., Kittakoop P. Cytotoxic sesquiterpenes from the endophytic fungus Pseudolagrobasidium acaciicola. Phytochemistry. 2016;122:126–138. doi: 10.1016/j.phytochem.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 22.Lawrence R.N. Rediscovering natural product biodiversity. Drug Discov. Today. 1999;4:449–451. doi: 10.1016/S1359-6446(99)01405-1. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y., Li X.M., Shang Z., Li C.S., Ji N.Y., Wang B.G. Meroterpenoid and diphenyl ether derivatives from Penicillium sp. MA-37, a fungus isolated from marine mangrove rhizospheric soil. J. Nat. Prod. 2012;75:1888–1895. doi: 10.1021/np300377b. [DOI] [PubMed] [Google Scholar]
- 24.Zhou X.M., Zheng C.J., Chen G.Y., Song X.P., Han C.R., Li G.N., Fu Y.H., Chen W.H., Niu Z.G. Bioactive anthraquinone derivatives from the mangrove-derived fungus Stemphylium sp. 33231. J. Nat. Prod. 2014;74:2021–2028. doi: 10.1021/np500340y. [DOI] [PubMed] [Google Scholar]
- 25.Hewage R.T., Aree T., Mahidol C., Ruchirawat S., Kittakoop P. One strain-many compounds (OSMAC) method for production of polyketides, azaphilones and an isochromanone using the endophytic fungus Dothideomycete sp. Phytochemistry. 2014;108:87–94. doi: 10.1016/j.phytochem.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 26.Sheldrick G.M. SHELX-97, Program for Crystal Structure Solution. University of Göttingen; Göttingen Germany: 1997. [Google Scholar]
- 27.Sheldrick G.M. SHELX-97, Program for Crystal Structure Refinement. University of Göttingen; Göttingen, Germany: 1997. [Google Scholar]
- 28.Cambridge Crystallographic Data Centre. [(accessed on 8 July 2016)]. Available online: http://www.ccdc.cam.ac.uk/data_request/cif.
- 29.Xiao D., Zhu S.P., Gu Z.L. Quercetin induced apoptosis in human leukemia HL-60 cells. Acta Pharmacol. Sin. 1997;18:280–283. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.