

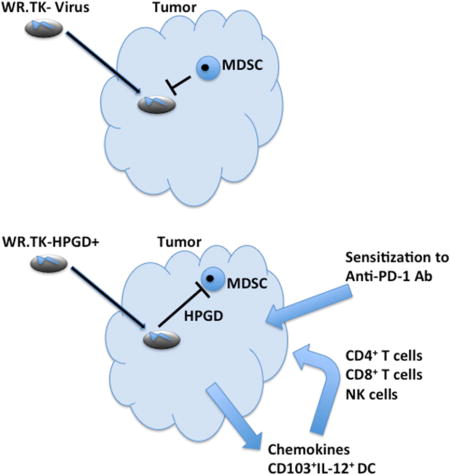
SUMMARY
Immunotherapies are highly promising cancer treatments, but understanding the factors mediating their resistance remains critical. Successes in randomized clinical testing have supported the growing appreciation that oncolytic virotherapies primarily act as immunotherapies. Here we identified prostaglandin E2 (PGE2) in the tumor as a key mediator of resistance to immunotherapies, including oncolytic vaccinia virotherapy. Elevated levels of PGE2 coupled to suppressive chemokine profiles and high levels of granulocytic myeloid derived suppressor cells (MDSC) resulted in loss of immunotherapeutic potential. Viral vectors engineered to target PGE2 were capable of overcoming localized immunosuppression leading to profound changes in the tumor’s immune status. This allowed the viral vectors to raise robust anti-tumor adaptive immune responses and sensitized established and previously resistant tumors to immunotherapies.
Keywords: PGE2, MDSC, oncolytic virus, immune suppression, COX2
Graphical abstract
INTRODUCTION
Recent clinical successes have focused interest on the potential of cancer immunotherapies. However, solid tumors often display the capacity to limit immune induction or to mediate early immune shut-off both locally and systemically. Identifying the key mediators of resistance to immunotherapy will allow the development of more robust treatments with more predictable responses.
Oncolytic viruses (OV) are vectors designed to selectively replicate in and destroy cancer cells, and multiple OVs based on many different viral strains are currently undergoing clinical testing. However, notable among the current clinical generation of oncolytic viral vectors is that those that have succeeded in randomized trials have expressed an immune activating cytokine (GM-CSF)(Andtbacka et al., 2013; Heo et al., 2013). This reinforces a plethora of preclinical data indicating that the immune response can be a key mediator of OV activity (Lichty et al., 2014) and has led to the development of several ingenious strategies to enhance the immune activating potential of OVs (Kottke et al., 2013; Tysome et al., 2012; Zhang et al., 2014). The situation is complex however, as enhanced immune activation frequently reduces oncolytic activity and other reports have demonstrated that certain immune-suppression strategies can also enhance OV activity (Alvarez-Breckenridge et al., 2012; Chen et al., 2013; Lun et al., 2009). A better understanding of the importance of OV-mediated immunotherapeutic activity, how this interacts with oncolytic activity and how OVs can be most beneficially engineered to interact with the host immune response is therefore needed.
Multiple OV strains based on vaccinia have been reported (Kirn et al., 2007; Mastrangelo et al., 1999; Thorne et al., 2007; Zhang et al., 2007), and one of these expressing GM-CSF, Pexa-Vec (JX-594), has produced encouraging responses in randomized clinical testing (Heo et al., 2013; Park et al., 2008). However it is apparent that even in these successful clinical trials, some patients appear resistant to the therapy. It will therefore be critical for the future development of the platform to discover how and why some patient’s tumors do not respond, and to develop approaches to overcome this.
Treating different immunocompetent mouse tumor models with oncolytic vaccinia results in a range of in vivo sensitivities, creating an opportunity to interrogate the causes of resistance and to determine the relative importance of immunotherapeutic and oncolytic mechanisms of tumor killing under different conditions. Strategies to overcome the causes of resistance might then be developed.
RESULTS
Immunocompetent mouse tumor models display differing sensitivities and patterns of response to oncolytic vaccinia therapy
We initially examined a panel of syngeneic immunocompetent mouse tumor models in order to delineate the mediators of resistance or susceptibility to oncolytic vaccinia therapies. The in vitro sensitivities of 14 different mouse tumor cell lines to viral replication and cell killing were compared to in vivo responses with syngeneic tumors formed from a subset of seven of the same cell lines (Figures 1A and 1B). Mice were treated via direct intratumoral injection of a low dose of viral therapy to remove any variability due to differences in systemic delivery. No correlation was seen between viral replication or viral-mediated cell killing in vitro and in vivo anti-tumor effects, indicating factors in addition to direct oncolytic activity may be primarily responsible for optimal therapeutic benefit.
Figure 1.
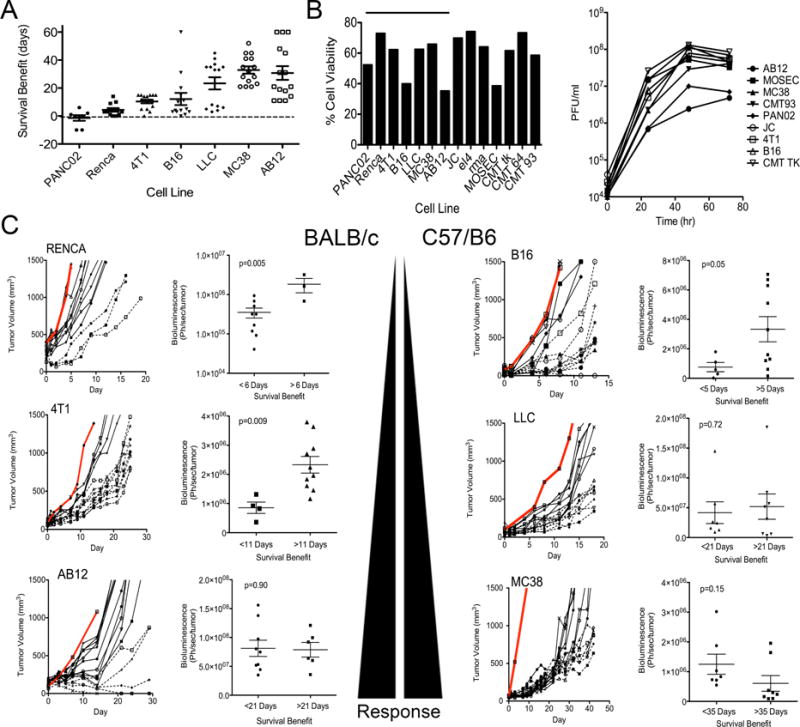
Response to oncolytic vaccinia therapy in different immunocompetent mouse models. (A) Syngeneic tumors were implanted subcutaneously into BALB/c or C57/BL6 mice and treated with a single intratumoral injection of low dose (1×107 PFU) WR.TK- when tumors reached 50–100 mm3. Survival benefit (increased survival compared to PBS treated control mice) was plotted for each tumor model. (B) Cell viability (left), as determined by MTS assay 72 hr after infection with WR.TK- at an MOI of 1.0 was plotted as percentage viability relative to uninfected cells for indicated mouse tumor cell lines and viral replication (right) was followed in the same cell lines after the same treatment by plaque assay on BSC-1 cells. (C) Tumor growth for individual mice treated as in (A) are shown for 6 of the 7 tumor models (PAN02 displayed no survival benefit) and compared to PBS controls (Grey lines). For each tumor model, individual mice are divided into good responders (dashed lines) or poor responders (solid lines) depending on assigned survival benefits. Bioluminescence imaging (BLI) was performed at 24 hr post-treatment to measure viral gene expression from within the tumors and BLI signal for poor and good responders was normalized to tumor volume and plotted (right hand graph for each cell line). Error bars ±SEM. See also Figure S1
Oncolytic vaccinia strain WR.TK-.Luc+ was used during these initial experiments. This virus bears the same thymidine kinase deletion as all three oncolytic vaccinia strains currently in the clinic (Kim et al., 2006; McCart et al., 2001; Worschech et al., 2009; Zeh et al., 2014) and expresses luciferase so that viral gene expression levels could be quantified over time in individual mice (as a surrogate for viral replication and persistence), and compared to subsequent response. It was noted that two distinct kinetic patterns of viral gene expression emerged in vivo (Figure 1C). In the more resistant tumor models (defined as those in which viral therapy increased overall survival by less than 2 weeks, as seen with PANC02, RENCA, 4T1 & B16 (Figure 1A)), the level of viral gene expression measured from within the tumor at 24 hr after delivery correlated closely with subsequent response (Figure 1C). Therefore, within any one of these resistant tumor models greater initial infection and early replication in the tumor (early viral gene expression) results in improved subsequent response. This pattern would be predicted if viral replication was the key mediator of therapeutic effect and suggests that the limited response seen in the more resistant tumor models is primarily due to oncolytic activity.
However, a different pattern was noted in the tumor models that were more susceptible to viral therapy (LLC, MC38 & AB12)(Figures 1A and 1C). In these models, there was no correlation between early viral gene expression in the tumor (at 24 hr post treatment) and subsequent response. Instead, the best responders within each of the sensitive tumor models demonstrated a trend towards a more rapid and robust clearance of the virus, as seen with reduced levels of viral gene expression in the tumor at 4 or 5 days after treatment (Figure S1). This robust viral clearance might indicate that a strong immune response is being induced within the tumor that reinforces any direct oncolytic effects in the more sensitive tumor models.
Several lines of evidence supported the above hypothesis. Firstly, when LLC tumors were implanted into immunodeficient mice, the viral gene expression pattern changed to match that of the poor responders (oncolytic activity only), with viral bioluminescence within the tumor at 24 hr correlating with subsequent response (Figure 2A). Of note, when LLC tumors implanted into immunodeficient mice were treated, most of the tumors displayed a pattern of short-term stable disease (for 10–15 days) followed by progression (Figure 2A), this is despite evidence of ongoing viral replication (luciferase gene expression) in the tumor (Figure S2). However, when the same tumor model was implanted and treated in immunocompetent mice (Figure 1C) 50% of the animals (6 of 12) displayed longer-term (>21 days) stabilization of tumor growth despite the fact that the viral therapy was cleared within 10 days of treatment (Figure S2). Secondly, anti-CD8 antibody was used to deplete CD8+ cells from both sensitive (MC38) and resistant (Renca) tumor models (Figure 2B). Depletion of CD8+ T-cells significantly reduced the vector’s therapeutic activity in the sensitive MC38 tumor model, but had no effect in Renca, further supporting the hypothesis that the resistant tumor models were unable to mount a robust immunotherapeutic response. Of note, the reduced therapeutic activity in MC38 tumors after CD8+ depletion occurred despite increased viral replication (Figure 2C), again highlighting the increased importance of immunotherapeutic over oncolytic activity in these vectors.
Fig 2.
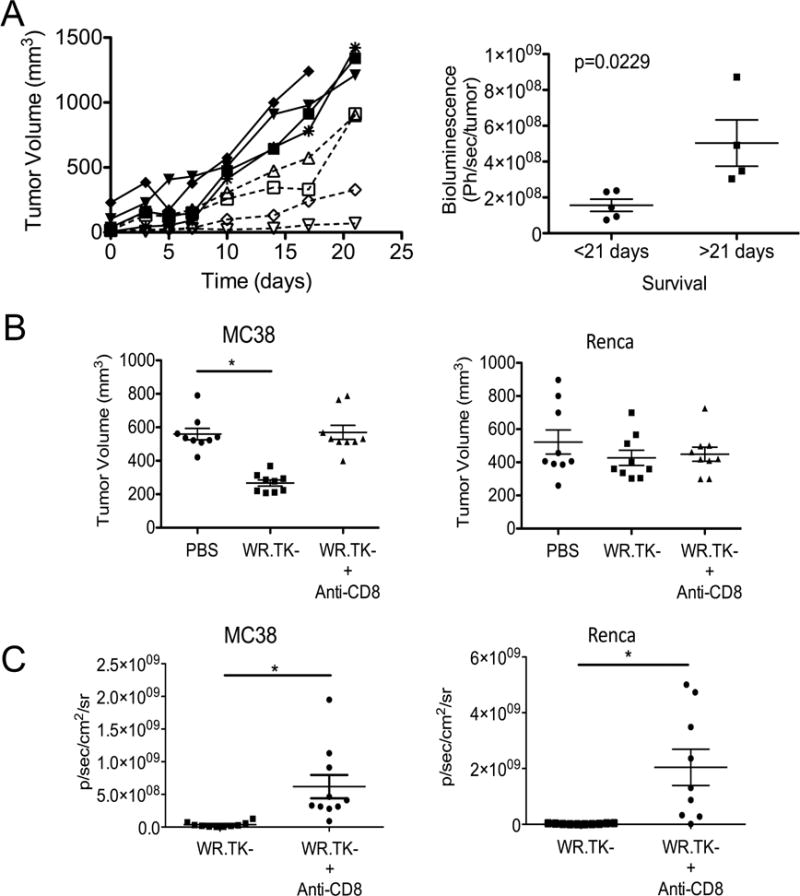
Role of Immune Response in Theraeputic Effect of Oncolytic Vaccinia. (A) LLC tumors were implanted into NOD SCID mice and tumor growth for individual mice is shown over time after treatment with 1×107 PFU WR.TK- (left). These were divided into good (dashed lines) and poor (solid lines) responders and bioluminescence imaging used to determine viral luciferase transgene expression from the tumor at different times after treatment (right). (B) MC38 and Renca cells were implanted subcutaneously into syngeneic mice and treated with WR.TK- (1×107 PFU IT) after antibody depletion of CD8+ cells from the mice. * p<0.05. (C) The viral gene expression from the tumor in the same mice as in (B) was followed by bioluminescence imaging at day 3 after treatment. * p<0.05. Error bars ±SEM. See also Figure S2.
Resistance to viral therapy correlated with MDSC in the tumor environment
It appears therefore that in order to produce a significant therapeutic effect, the viral vector needs to be able to mediate both an oncolytic and immunotherapeutic response, while resistant tumors were able to limit this to an oncolytic-mediated response only. The immune response to viral therapy was therefore examined in more detail in order to define differences between susceptible and resistant tumors.
It was determined that pS6 levels were reduced systemically in myeloid DCs in tumor bearing animals, and that this reduction was more pronounced in the resistant tumor-bearing mouse models (Figure S3A), indicating that a defect in or suppression of the DC response may be important for resistance to immunotherapy in these tumors. However, seeing as oncolytic viral immune activation is likely to primarily occur subsequent to replication in the tumor, the condition of the more localized immune environment within the tumor was examined. Different immune cells are associated with a suppressive phenotype, including MDSC and T-regs, and so the overall level of these different cell types in both the spleen and the tumor of the same seven mouse tumor models were determined prior to therapy. It was observed that the overall level of MDSC found in the tumor for different tumor models correlated very closely with the resistance or sensitivity of that model to subsequent viral therapy (Figure 3A and S3B). No similar correlation was seen with T-reg levels or with either cell type in the spleen (Figure 3A and S3C).
Fig 3.
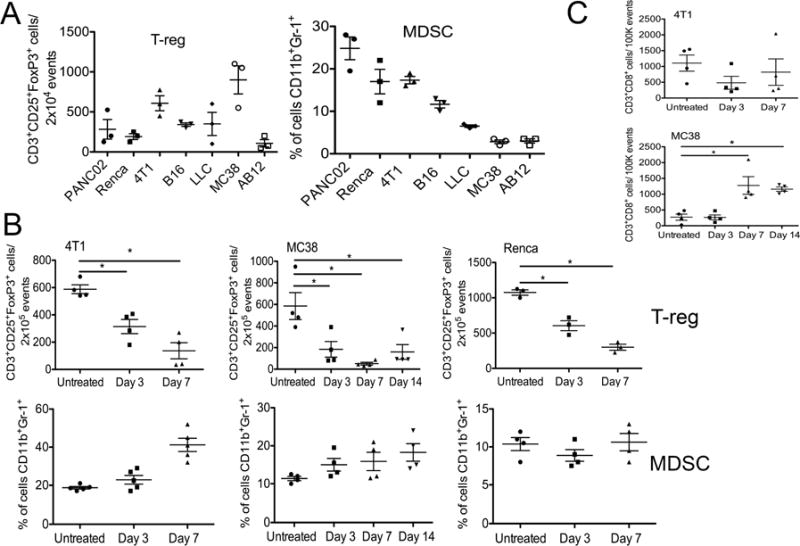
Resistance of different tumor models to oncolytic viral therapy is mediated by localized immune suppression within the tumor. (A) Baseline levels of regulatory T-cells (T-reg) and Monocyte Derived Suppressor Cells (MDSC) in tumors prior to therapy. Syngeneic subcutaneous tumors were formed from different cell lines and mice sacrificed when tumors reached 100 to 200 mm3 and disaggregated. Flow cytometry was used to quantify the relative levels of T-reg (CD3+CD25+FoxP3+CD8−) and MDSC (CD11b+Gr-1+) in the tumors. (B) Effect of viral therapy on suppressive immune cell profile within the tumor. Tumor-bearing mice were treated with a single low dose (1×107 PFU) intratumoral injection of WR.TK- and the levels of T-reg and MDSC in the tumors at different times after treatment were analyzed as in (A). (C) CD3+CD8+CD4− T-cells were also quantified in the tumor as in (A) (*p<0.05). Error bars ±SEM. See also Figure S3 and S4.
We further examined what changes occurred in the tumor after viral therapy and saw that for different tumor models (4T1, MC38 and Renca, Figure 3B), the addition of vaccinia therapy resulted in a rapid loss of T-reg, but that MDSC levels were unaffected (and actually continued to increase over time, as they also did in control groups). It therefore appears that MDSC are unaffected by the presence of oncolytic vaccinia and high levels of MDSC can block the immunotherapeutic activity of these vectors. Of note, in the sensitive MC38 tumor model (with lower background levels of MDSC), the viral therapy was actually found to significantly increase the number of CD8+ T-cells present in the tumor, an indication of immunotherapeutic activity, whereas the more resistant 4T1 tumor model (with higher baseline MDSC levels) did not display any significant increase in CD8+ T-cells in the tumor after treatment (Figure 3C).
We have recently described an oncolytic vaccinia strain, WR.B18R-.IFNβ+, with enhanced immunotherapeutic effects(Kirn et al., 2007; Wang et al., 2011), while oncolytic viral strains expressing GM-CSF (including vaccinia and HSV based strains(Andtbacka et al., 2013; Heo et al., 2013)) have produced the most dramatic clinical responses to date despite GM-CSF being associated with MDSC proliferation(Kohanbash et al., 2013). The effects of these immune-enhanced vectors were therefore also examined to see if they were able to overcome MDSC-mediated immunosuppression in the tumor. It was found that the more immunogenic vaccinia strains (WR.TK-mGMCSF and WR.B18R-mIFNβ+) provided no additional benefit over WR.TK- in a sensitive tumor model (MC38, Figure S4), with WR.TK-mGMCSF even increasing MDSC in the tumor. In the resistant tumor model (4T1, Figure S4) all viral treatments resulted in significant increases in MDSC levels in the tumor. This was less dramatic for the immune-enhanced vectors, and this correlated with a small but not significant increase in CD8+ T-cell infiltration. It therefore appears that the inability of the virus to induce a robust immunotherapeutic effect in tumors with high levels of MDSC is a critical determinant of resistance and cannot be overcome by increasing viral-mediated immune activation.
Targeting of PGE2 can reduce MDSC and re-sensitize resistant tumors to viral therapy
Recent reports have identified COX2-mediated production of the prostaglandin PGE2 as a key determinant of MDSC tumor-infiltration and maintenance of the suppressive phenotype in these cells (Donkor et al., 2009; Fujita et al., 2011; Kalinski, 2012; Obermajer et al., 2011a; Obermajer et al., 2011b; Rodriguez et al., 2005). It was noted that viral therapy did not significantly alter the overall levels of COX2 expression in the tumor (Figure S5A).
We therefore looked to develop approaches to reduce PGE2 levels, including addition of the COX2 inhibitor celecoxib or viral expression of the prostaglandin-inactivating enzyme HPGD (hydroxyprostaglandin dehydrogenase 15-(NAD), 15-PGDH) (Figure S5B, S5C). Initial in vitro experiments determined that even when used at levels known to be toxic in vivo, celecoxib was unable to reduce PGE2 levels by the amounts achieved with HPGD expression (Figure S5D). Oncolytic vaccinia expressing HPGD (WR.TK-HPGD+) was therefore tested in several different mouse tumor models.
It was found that WR.TK-HPGD+ was non-toxic and that the numbers of MDSC cells in Renca (resistant) tumors were rapidly and significantly reduced after treatment with WR.TK-.HPGD+ (Figure 4A). Interestingly, WR.TK-.HPGD+ also induced a more rapid and robust reduction in T-reg numbers in the tumor. Because several sub-sets of MDSC have been defined in mice, we further looked to determine if the monocytic (CD11b+Ly6G−Ly6Chi) and granulocytic (CD11bLy6G+Ly6CLo) MDSC were equally targeted. It was found that HPGD expression selectively depleted the granulocytic MDSC population (Figures 4B and S5E) that is typically found in greater numbers and thought to be terminally differentiated.
Fig 4.
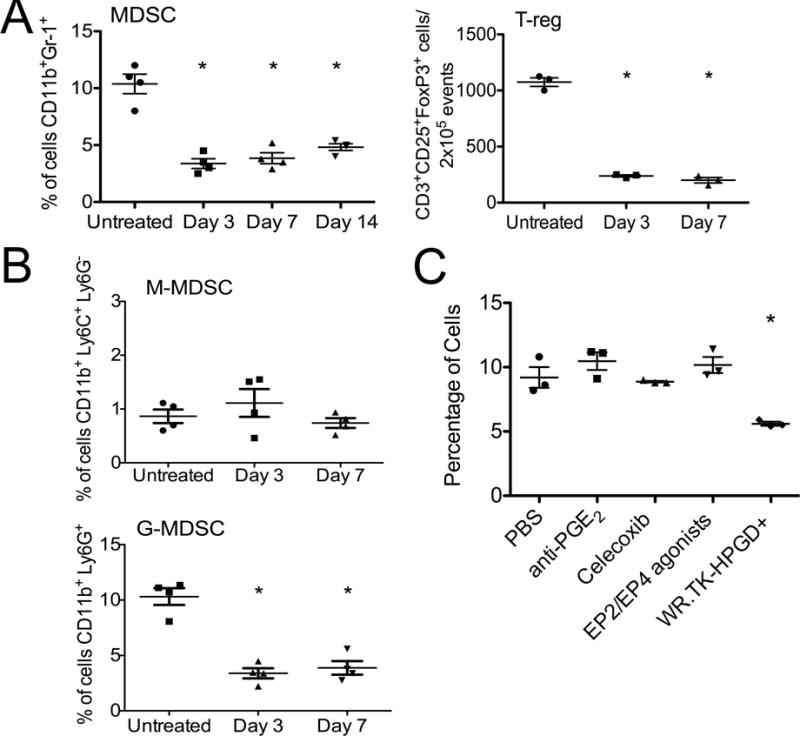
HPGD expression from oncolytic vaccinia reduces MDSC in the tumor. (A) Mice bearing Renca tumors were treated with intratumoral, low dose (1×107 PFU) injection of WR.TK-.HPGD+ and mice sacrificed at indicated times, tumors recovered, disaggregated and analyzed by flow cytometry as in Fig 3. *p<0.05 compared to control. (B) Cells as in (A) were additionally stained for Ly6G and Ly6C to distinguish effects on monocytic MDSC (CD11b+Ly6C+Ly6G−) and granulocytic MDSC (CD11b+Ly6G+) in the tumor. (C) G-MDSC levels in the tumors for mice treated as in (A) and sacrificed after 3 days were compared to mice treated with PBS, anti-PGE2 antibody, celecoxib or EP2/4 agonists. All non-viral treatments were administered daily for 3 days. Error bars ±SEM. See also Figure S5.
The identification of the COX2-PGE2 pathway as a key mediator of immunosuppression has led to a variety of inhibitory approaches being proposed. We therefore compared the in vivo effects of different COX2 or PGE2 inhibitors on the levels of G-MDSC in the tumor, including PGE2 depleting antibody, celecoxib and agonists of the PGE2 receptors EP2 and EP4. The only approach capable of reducing G-MDSC levels was WR.TK-HPGD+ (Figure 4C), indicating that high level HPGD expression from within the tumor is uniquely able to break this immunosuppressive cycle.
These alterations in the tumor microenvironment further correlated with an enhanced therapeutic effect in different mouse tumor models (Figure 5A). Of note, the Renca tumor model that was previously resistant to viral therapy (displaying an ‘oncolytic only’ phenotype and high baseline levels of MDSC) displayed the greatest increase in therapeutic benefit after HPGD transgene expression (Figures 5AB). However MC38 tumors, that had low-level baseline MDSC and already displayed sensitivity to therapy also showed a significant further therapeutic advantage after treatment with WR.TK-.HPGD+.
Fig 5.
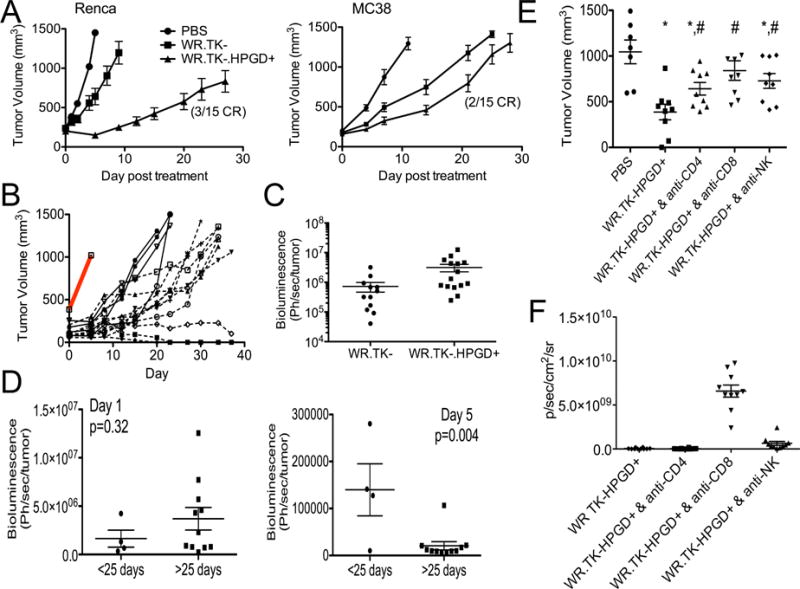
Enhanced therapeutic activity of WR.TK-.HPGD+. (A) Mice bearing subcutaneous RENCA or MC38 tumors were treated with a single intratumoral injection of PBS or 1×107 PFU of WR.TK- or WR.TK-HPGD+ and subsequent tumor growth followed by caliper measurement (n=15 per group). (B) Renca tumor growth in individual mice treated with WR.TK-HPGD+ are plotted, compared to PBS control (grey bar) and divided into good (solid line) and best (dashed line) responders. (C) The viral gene expression (bioluminescence) from the tumor at 24 hr after therapy was compared for mice treated with WR.TK- and WR.TK-HPGD+. (D) The bioluminescence signal (viral gene expression) from the tumor of mice treated with WR.TK-HPGD+ at day 1 and 5 were normalized to tumor volume and shown for both good and best responders. (E) The role of different immune subsets in the increased therapeutic activity of WR.TK-HPGD+ in Renca tumors was examined through depletion of CD4+, CD8+ and NK cells. (*p<0.05 v PBS; #p<0.05 v WR.TK-HPGD+). (F) Viral gene expression from the tumor (bioluminescence imaging) at day 3 after treatment of immune cell depleted mice. Error bars ±SEM. See also Figure S6
The patterns of viral luciferase transgene expression were also compared for WR.TK- and WR.TK-HPGD+) in the previously resistant Renca tumor model (Figures 1B and 5CD). Interestingly, it was initially noted that, unlike many immune enhancing transgenes, HPGD expression did not reduce the initial replicative capability of the virus (Figures 5C and S6). However, the virus was cleared from the tumor slightly faster with HPGD expression than for WR.TK- alone, again indicative of the raising of a robust immune response. It was also seen that whereas WR.TK- treatment displayed the ‘oncolytic only’ phenotype (with higher gene expression at day 1, correlating with greatest subsequent therapeutic benefit)(Figure 1B), WR.TK-.HPGD+ treatment of the same tumor model displayed the ‘oncolytic plus immunotherapeutic’ phenotype, with the best responders displaying a robust and rapid clearance of the virus at day 5 after treatment (Figure 5D). This was further explored through depletion of different immune subsets from the mice prior to treatment with WR.TK-HPGD+ (Figure 5E). It was noted that depletion of CD8+ cells had the most profound effect, resulting in a loss of significance in the therapeutic benefit after treatment. However both CD4+ and NK cells also appear to be important, as their depletion resulted in a significant reduction in therapeutic effect (even though WR.TK-HPGD+ treatment maintained a significant therapeutic benefit). It therefore appears that HPGD expression, in addition to depleting granulocytic MDSC is capable of inducing further immunotherapeutic benefits.
We looked to define in more detail the mechanisms underlying the therapeutic advantage seen with WR.TK-HPGD+. It was noted that at 3 days after treatment, WR.TK- alone was able to modestly increase the levels of several Th1-associated chemokines both systemically and in the tumor (Figures 6AB). The expression of HPGD however produced significant further increases and was also capable of significantly reducing the level of the suppressive chemokine CXCL12 (SDF-1), which is associated with MDSC attraction into the tumor, tumor metastasis and a poor prognosis(Chatterjee et al., 2014; Obermajer et al., 2011b) (Figure 6B). Further analyses looked at the levels of selected cytokines and inflammatory pathways within the tumor. As expected, WR.TK- infection resulted in an increase in the type I IFN response, seen with increased lift-1 and lift-2 expression (Figure 6C). However, this was not increased further with HPGD expression, indicating HPGD does not enhance the innate immune response that might be expected to reduce viral oncolytic effects and mediate premature viral clearance from the tumor. Instead, HPGD expression did significantly increase the level of IFNγ produced within the tumor (Figure 6D). Oncolytic viral infection alone therefore appears capable of inducing an inflammatory response and initial immune activation, whereas HPGD expression may be required to prevent premature shutdown of the immune response prior to full induction of adaptive immunity. This is supported by the observation that at 7 days after treatment WR.TK-HPGD+ had induced a fourfold increase in the number of anti-tumor CTLs in the spleen relative to WR.TK- treatment (Figure 6E).
Fig 6.
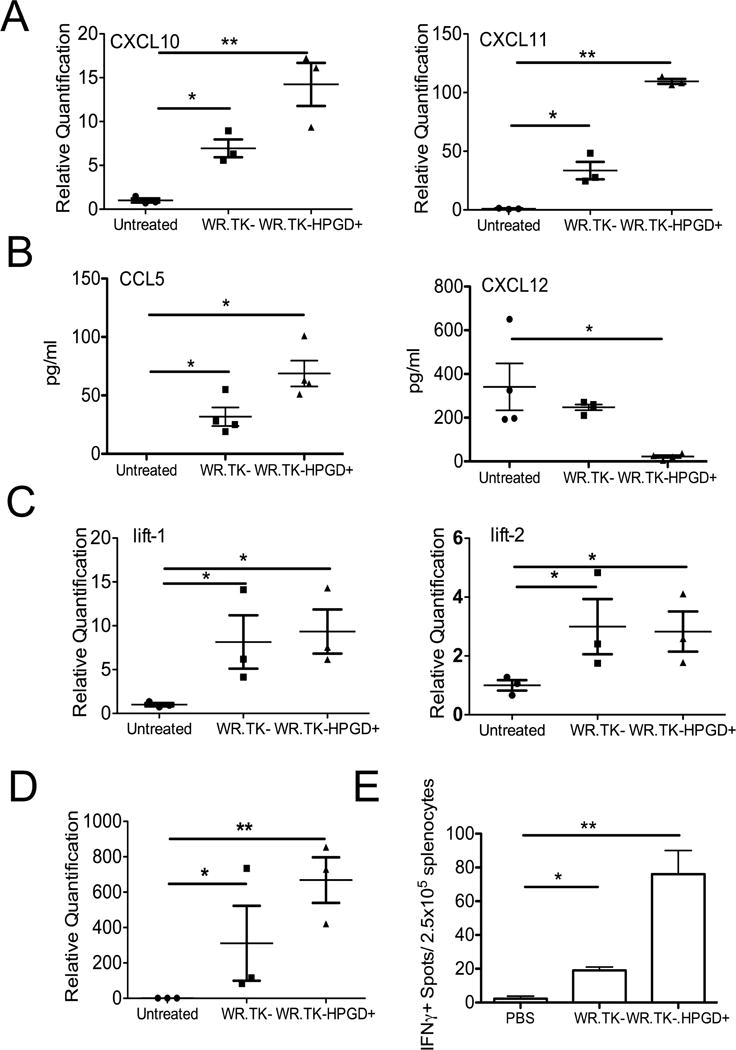
HPGD expression enhances the immune response within and against the tumor. (A) Mice bearing Renca tumors were treated as indicated with 1×107 PFU of different viral strains IT and sacrificed after 3 days. qRT-PCR was used to detect the expression of CXCL10 and CXCL11 in the tumor. (B) ELISA was used to detect the levels of CCL5 and CXCL12 in the serum of mice in (A) at the same times (BLD=Below Limits of Detection). (C) Innate (type I IFN) immune response of mice as in (A) was determined by qRT-PCR of lift-1 and lift-2. (D) Adaptive immune response, as measured by qRT-PCR of IFNγ was determined for mice as in (A). (E) Anti-tumor CTL response was determined in splenocytes collected form RENCA tumor bearing mice 7 days after the indicated treatments. Anti-tumor CTL response was determined by ELISPOT (*p<0.05, **p<0.001). Error bars ±SEM.
WR.TK-HPGD+ can enhance sensitivity of resistant tumors to other immunotherapies
The observed changes in the tumor’s chemokine profile may be responsible for enhancing the CTL response through mediating changes in the immune cell repertoire within the tumor. This might further provide benefits for attracting therapeutic T-cells into the tumor after adoptive T-cell transfer or application of a therapeutic vaccine. This was examined using a bilateral Renca tumor model, whereby one tumor was injected with WR.TK- while the tumor on the opposite flank was injected with WR.TK-HPGD+. It was seen that activated NK T-cells (Cytokine Induced Killer, CIK cells) delivered intravenously, trafficked significantly more efficiently to the HPGD expressing tumor (Figure 7A). This indicates that the expression of HPGD not only limits the suppressive environment within the tumor, but is also capable of enhancing systemic attraction of T-cells.
Fig 7.
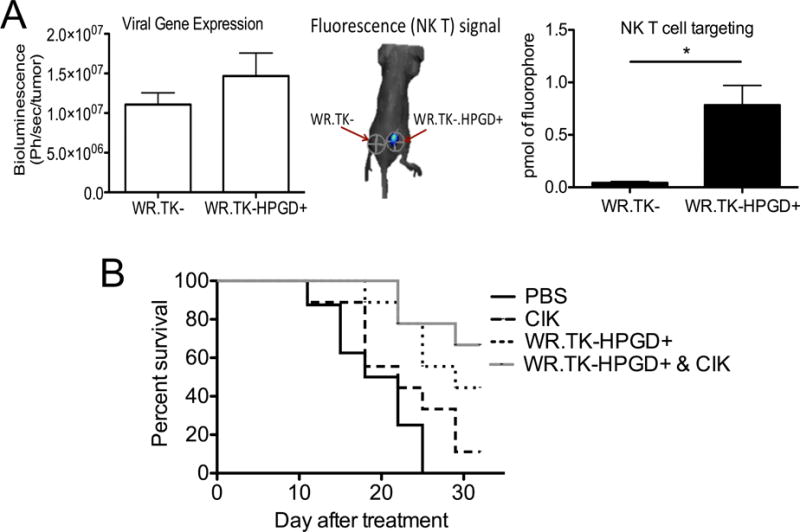
Enhanced Therapeutic Effects of Combination with adoptive T-cell Transfer (A) Mice were implanted bilaterally with Renca tumors. When tumors reached 50–100 mm3, the tumor on one flank was injected with 1×107 PFU of WR.TK- and the tumor on the opposite flank was injected with WR.TK-HPGD+. After 24 hr 1×107 activated and Cy5.5 labeled NK T (CIK) cells were delivered via tail vein injection. 24 hr later mice were imaged for bioluminescence to measure viral gene expression and fluorescence to determine NK T cell trafficking to tumors (*p<0.05). A representative example of fluorescence imaging is shown. (B) BALB/c mice with subcutaneous Renca tumors were treated with PBS, 1×107 PFU of WR.TK-HPGD+ (Intratumoral), 5×106 CIK cells (intravenous) or both therapies on the same day. Animal survival, taken as time to tumor burden reaching 1000 mm3, was determined by caliper measurement. Error bars ±SEM.
It was seen that Cytokine Induced Killer (CIK) cells, used as a model of adoptive T-cell transfer (Figure 7B), only had a small therapeutic effect in Renca tumor models, but combined with WR.TK-HPGD+ to increase survival relative to either treatment used alone.
Perhaps more dramatic was the observed effects of combining WR.TK-HPGD+ with blockade of immune checkpoint inhibitor using anti-PD-1 (Figure 8A). This antibody is known to have no effect in the Renca tumor model (Masters et al., 2014), as was confirmed here, yet combination with WR.TK-HPGD+ produced a large therapeutic advantage, again indicating that WR.TK-HPGD+ has the potential to sensitize otherwise resistant tumors to different immunotherapies. Of particular note, it has recently been reported that aspirin could be used to block Cyclooxygenase activity and sensitize tumors to anti-PD-1 therapy (Zelenay et al., 2015), however this was only possible when the drugs were added prior to formation of established tumors. In the Renca model used here a similar result is seen (Figure 8A), with anti-PD-1 and aspirin added 3 days after tumor implantation producing a small but significant therapeutic benefit, whereas either used alone had no effect, even at this early time. However, if this same combination is added at later time points, after tumor formation (10 days after tumor implantation), there is no therapeutic benefit. However addition of anti-PD-1 in combination with WR.TK-HPGD+ produces highly significant therapeutic effects, even when these are added at late times to established tumors. Also, of note is the observation that adding aspirin to WR.TK- produced no therapeutic benefit, indicating aspirin could not substitute for HPGD expression.
Fig 8.
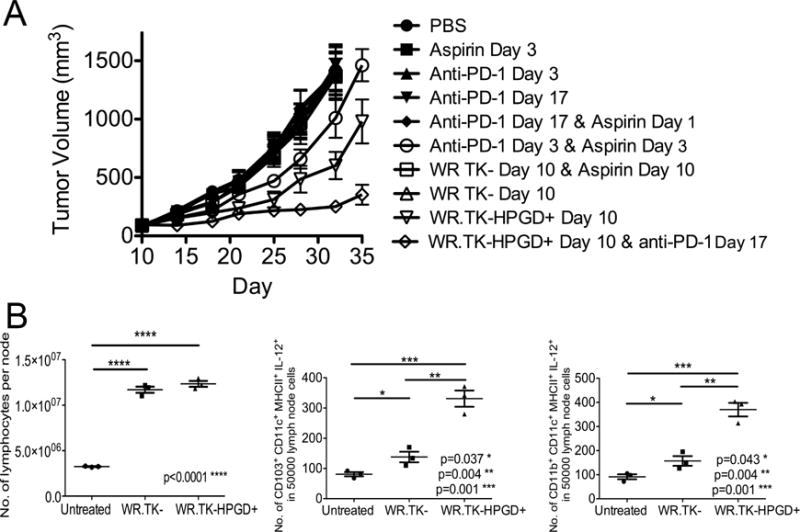
WR.TK-HPGD+ also sensitizes tumors to anti-PD-1 Therapy. (A) Combination of different viruses with anti-PD-1 antibody is compared to combinations with aspirin, and with aspirin + anti-PD-1 after treatment in BALB/c mice bearing subcutaneous Renca tumors (N.B. the indicated days of treatment are days after tumor implantation). Viruses (WR.TK- or WR.TK-HPGD) were given as single IT doses of 1×107 PFU; anti-PD-1 antibody (applied twice weekly for 21 days after start of treatment); and aspirin (600 μg/ml provided continuously in drinking water). (B) Draining lymph nodes were collected 5 days after treatment of mice as in (A) and dissociated for determination of total number of lymphocytes (left), number of CD103+CD11c+MHCII+IL-12+ cells (center), and number of CD11b+CD11c+MHCII+ cells (right) per 5000 events. Error bars ±SEM
The effects of aspirin on sensitizing tumors to anti-PD-1 treatment when added prior to tumor formation were further examined in (Zelenay et al., 2015) using cyclooxygenase deficient Ptgs1−/−Ptgs2−/− mice, and a role for IL-12p40+CD103+ DC was identified in mediating anti-tumor immunity. These cells were depleted when PGE2 was present in wild type mice. Here we found that expression of HPGD from an oncolytic vaccinia could disrupt PGE2 activity sufficiently, even in established tumors, to induce significant numbers of IL12p40+CD103+ DC in the draining lymph nodes of treated tumors (Figure 8B). A small, but significant increase was seen even with WR.TK- treatment alone, but the effects were increased significantly when HPGD was also expressed.
DISCUSSION
A variety of approaches have been proposed to overcome the immunosuppressive microenvironment in large solid tumors, with the COX2-PGE2 pathway as a key mediator of this suppressive activity. This pathway is an attractive target as it has been associated with attracting and maintaining the suppressive phenotype of MDSC. Breaking this cycle might even allow these suppressive cells to differentiate into an immune activating phenotype. Here we demonstrated that an oncolytic vaccinia virus expressing the PGE2 inactivating enzyme HPGD is able to significantly reduce levels of G-MDSC within the tumor. Of several approaches known to target COX2-PGE2, WR.TK-HPGD+ was the only one able to actually reduce MDSC levels.
This appears to be especially important in the context of oncolytic viral therapy as the level of MDSC in the tumor at baseline was inversely related to the sensitivity of the tumor to oncolytic vaccinia therapy. Higher levels of MDSC in the tumor suppressed the immunotherapeutic activity of the virus, limiting its activity to oncolytic-mediated cell killing. Although vaccinia has a known capacity to produce a fast spreading and highly lytic infection in humans or non-human primates (Naik et al., 2006), it is apparent that the immune response plays a more critical role for therapeutic activity, as confirmed here. The determination that local rather than systemic immune suppressive activity is key in preventing viral immunotherapeutic action allows the creation of vectors such as WR.TK-HPGD+ that express transgenes to overcome this suppression locally within the tumor.
Recent clinical success using antibodies that block immune checkpoint inhibitors, such as anti-CTLA-4, anti-PD-1 and anti-PD-L1(Leach et al., 1996; Topalian et al., 2012; Yuan et al., 2008), have revealed the importance of overcoming the tumor’s capacity to prematurely shut down or curtail an immune response. It is apparent that a successful therapeutic strategy would require both activation of the immune response and prevention of its early shut down. As such, it is interesting that the standard oncolytic vaccinia strain (WR.TK-) and several immune-enhanced strains (including WR.TK-mGM-CSF) are capable of inducing inflammation at early times, even in resistant tumor models, but were unable to subsequently prime high level anti-tumor adaptive immunity (in tumors with high baseline G-MDSC).
The expression of HPGD from oncolytic vaccinia however produces a vector that is capable of both immune activation and limiting premature immune shutdown in the tumor. The primary mediator of early immune shut down and suppression after viral therapy appeared to be the granulocytic MDSC lineage within the tumor and that targeting of the prostaglandin PGE2 was shown to be a potent strategy to reduce the levels of these cells. This combination of immune activation after local viral replication in the tumor and overcoming of immune suppressive effects though G-MDSC depletion resulted in greatly increased anti-tumor CTL and significantly enhanced therapeutic effects. The greatest therapeutic advantage occurs in the previously resistant tumors, where the viral gene expression pattern switches to that of an ‘immunotherapeutic’ response. This confirms that previously resistant tumors can be sensitized to viral therapy through HPGD transgene expression. It would be predicted that expression of HPGD from immune-enhanced vectors (such as WR.TK-GMCSF) could further increase their immunotherapeutic potential.
It is also of note that HPGD expression was able to significantly enhance therapeutic activity in already sensitive tumor models (with low baseline G-MDSC levels). In this respect, altered chemokine production patterns and enhanced trafficking of activated T-cells to tumors treated with virus expressing HPGD may play an important therapeutic role.
This multi-faceted targeting of the immunosuppressive microenvironment within the tumor through treatment with WR.TK-HPGD+ was further found to sensitize resistant tumors to other immunotherapies, including adoptive immune cell transfer and immune checkpoint modulation. In particular, it was observed that Renca tumors that are naturally resistant to anti-PD-1 therapy, displayed enhanced sensitivity to anti-PD-1 when anti-PD-1 was applied after the WR.TK-HPGD+. Other approaches that target cyclooxygenase activity (such as with aspirin) have also been shown to sensitize mouse tumors to anti-PD-1 therapy, however this was only possible if both therapies were administered prior to tumor formation. The WR.TK-HPGD+ virus was the only approach found to be effective against pre-established, large solid tumors.
Finally, the description of distinct tumor response phenotypes to oncolytic viral therapy is also a finding that has direct clinical relevance for the application of OVs and potentially other immunotherapies. Recent clinical demonstrations of the ability to image OV expressed reporter transgenes in the clinic, such as the Sodium-Iodide symporter expressed from oncolytic measles virus using SPECT imaging(Penheiter et al., 2011; Russell et al., 2014), opens up the possibility of looking for similar patterns of gene expression and robust early OV clearance in a clinical setting. This might be used to predict clinical responses to oncolytic viral therapies at early time points after treatment, a particular problem for immunotherapies, where tumor swelling often precedes therapeutic response. However other mechanisms of tumor destruction may also play a role, such as viral-mediated vascular collapse in the tumor, which has been implicated in anti-tumor activity of several OVs, including those based on vaccinia (Breitbach et al., 2013; Breitbach et al., 2007). Further, the use of subcutaneous tumor xenografts minimizes the involvement of stromal cells and matrix in the response and tumor-associated fibroblasts have recently been implicated in anti-tumor activity of several OVs, including vaccinia (Ilkow et al., 2015).
This original approach to targeting tumor-induced local defects in the immune system therefore appears to be uniquely capable of targeting the highly suppressive MDSC population in the tumor itself. As a result resistant tumors can be sensitized to the immunotherapeutic effects of the viral therapy itself and to other commonly used immunotherapies, such as immune checkpoint blockade. The broad applicability of this approach would be especially exciting in the development of combination immunotherapies that produce more reliable and robust responses in a variety of solid tumors.
Experimental Procedures
Cell lines, viruses
A variety of mouse tumor cell lines, including AB12, MOSEC, MC38, CMT93, PAN02, JC, 4T1, B16, CMT TK, RENCA and LLC were used in this research, all were obtained from the American Type Culture Collection (Manassas, VA), except for MC38 (gift from Dr. David Bartlett, University of Pittsburgh), and JC, CMT93 and CMT TK (Cancer Research UK tissue culture collection). All were cultured according to vendors’ recommendations. Mouse NK T (CIK) cells were expanded from mouse splenocytes and cultured as previously described (Baker et al., 2001).
The wild-type vaccinia virus WR (Western Reserve) strain was obtained from ATCC (BEI Resources). WR.TK-.Luc+ and WR.B18R-.IFNβ+ were described previously (Kirn et al., 2007). WR.TK-.HPGD+ and WR.TK-.GMCSF+ were constructed for this work, with the pSC65 plasmid (gift from Prof. Bernie Moss, NIH) cloned to express firefly luciferase from the viral pSE/L promoter and mouse HPGD (or mouse GM-CSF) from the p7.5 promoter. This was recombined into the viral thymidine kinase gene (See Figure S5. In addition, vvDD.Luc+ and GFP (WR with deletions in TK and the viral growth factor genes, as described previously) was used as a second model of oncolytic vaccinia.
In vitro cell killing was determined by MTS assay of cell survival relative to uninfected controls and plaque assay on lysed cell samples was performed on BSC-1 cell layers.
Animal models
NOD SCID, C57/BL6 and BALB/c mice (female 6–8 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME). Tumor cells were implanted subcutaneously with 5 × 105 cells injected per mouse. IT (intratumoral) injection treatments began when tumors reached 50–100 mm3 (unless otherwise stated). Treatment doses and timings were as indicated. Tumor size was monitored by caliper measurement unless otherwise indicated and mice sacrificed when tumors reached 1500 mm3.
Other treatments included; Anti-PD-1 blocking antibody (BioXCell, RMP1-4) were diluted in PBS and given IP at 200 mg/mouse twice weekly; mouse CIK cells were given intravenously at 5×106 cell/mouse; anti-PGE2 antibody was given at 20ug/mouse IP daily; Celecoxib (Sigma) was given IP at 25 mg/kg daily; EP2 (AH6809) and EP4 (AH23848) agonsists (both Sigma) were given at 10 mg/kg each daily. Aspirin was given in drinking water ad libitum at doses of 600 μg/ml.
In immune cell depeltion experiments, anti-mouse CD8+ (2.43), anti-mouse CD4+ (GK1.5) or anti-mouse NK1.1 (PK136)(all from BioXCell) were injected intraperitoneally (500mg) on days 1 and 2 after tumor implantaion, with follow up injections of 150 mg every 5 days thereafter.
In some experiments tumor homogenates were collected for cytokine and chemokine quatification (by qRT-PCR). Neutralizing antibody titers in serum were determined as described previously(Sampath et al., 2013).
All experiments were performed according to the University of Pittsburgh Institutional Animal Care and Use Committee approved protocols.
Whole animal imaging
In some experiments whole animal bioluminescence imaging was used to image viral luciferase gene expression. Imaging was on an IVIS 200 (Xenogen, part of PerkinElmer, Waltham, MA) after intraperitoneal injection of luciferin substrate. Bioluminescence signal was quantified and images analyzed using the Living Image software (Xenogen, part of PerkinElmer). In some other experiments, NK-T (CIK)cells were covalently labeled with Cy5.5 NHS Ester (GE Healthcare, Waukesha, WI) and fluorescence signal was imaged in vivo using the FMT2500 fluorescence whole animal imaging system (PerkinElmer, Waltham, MA). In addition, XenoLight RediJect COX2 Probe (PerkinElmer, Waltham, MA) was imaged on the IVIS 200 in vivo.
ELISPOT assay
IFN-γ–producing splenocytes were quantified by ELISPOT assay. Splenocytes were separated from mice after different treatments. Splenocytes were stimulated by lysed tumor cells at 10:1 ratio or UV-inactivated Vaccinia virus at 5:1 ratio and seeded on plates (EMD Millipore, Billerica, MA), coated overnight with 15 ng/mg mIFN-γ antibody AN18 (Mabtech, Inc., Cincinnati, OH). These were incubated for 48 hr before the plates were washed and incubated with a biotinylated secondary antibody R4-6A2-biotin (Mabtech) for 2 hr at room temperature. The plates were then washed, incubated for 1 hr with avidin-peroxidase complex (Vectastatin kit; Vector laboratories, Burlingame, CA), and developed by the addition of AEC (3-amino-9-ethylcarbazole) substrate (Vector Laboratories, Inc., Burlingame, CA) according to manufacturer’s protocol. The spots were counted on a CTL-Immunospot analyzer (Cellular Technologies, Shaker Heights, OH). Spots from unstimulated splenocytes from each group were used to subtract the background.
Elisa and Western blot assay
Tumor homogenates were harvested from mice treated as indicated, and mechanically disaggregated and digested with triple enzyme mixture (Collagenase type IV, DNase type IV, and Hyaluronidase type V (Sigma-Aldrich, St Louis, MO)). Serum was collected through sub-mandibular bleed. In vitro cells infected with virus at MOI=1 for 24 hr or pretreated for 24 hr with 20 nm Celecoxib (BioVision, San Francisco, CA) were pretreated with 20 μm arachidonic acid (Sigma-Aldrich, St. Louis, MO) 4 hr prior to harvest. All experiments were performed in triplicate. Mouse SDF-1(CXCL12), RANTES (CCL5) and I-TAC(CXCL11) ELISA were performed according to the manufacturer’s protocol (Abcam, Cambridge, MA) and PGE2 ELISA were performed according to the manufacturer’s protocol (Cayman, Ann Arbor, Michigan) and optical density was detected with the 3,3,5,5-tetramethylbenzidine (TMB) peroxidase substrate kit (Vector Labs, Burlingame, CA).
For western blot assay, in vitro lysed cell protein was prepared. Mouse HPGD antibody (Abcam,Cambridge, MA), COX2 antibody (BD, Franklin Lakes, New Jersey) and beta-actin antibody (Sigma-Aldrich, St. Louis, MO) were used for Western Blot according to the manufacturer’s protocol.
Flow cytometry
Acquisition was performed on Gallios or Cyan flow cytometers. Data were analyzed using the Cyanor Gallios software. Antibodies included those to CD3, CD4, CD8, CD25, FoxP3, CD11b, Ly6g(Gr-1), Ly6c, CD11b(M1/70), MHC II (M5/114.15.2), CD103(2E7), IL12p40(C17.8) (all BD Bioscience or eBioscience) or CD11c(N418) (BioLegend). Intracellular staining for Foxp3 was done according to protocols in the respective kits. Gating strategies are shown (Figure S3)
In some studies splenocytes were collected and rapidly fixed and permeabilized in order to examine surface markers as well as intracellular stains by Phosflow (including pS6, BD BioSciences).
qRT-PCR
Total RNA was isolated and purified using RNeasy Mini Kit (QIAGEN) from whole-tumor homogenates. cDNA was synthesized using cDNA Synthesis Kit (Quanta BioScience Inc.). An array of gene expression assays were performed using a TaqMan Gene Expression Assay following the manufacturer’s instructions (reference gene: Mouse HPRT) and included IFNγ, CXCL10, CXCL11, lift-1, lift-2.
Statistical Analysis
Standard Student’s t-test (two-tailed) were used, with significance considered to be P < 0.05.
Supplementary Material
SIGNIFICANCE.
Cancer immunotherapies, including oncolytic viruses, offer the potential for curative cancer treatments. However patients often present with a combination of systemic immune defects and localized immunosuppression within the tumor that limit immune activation or mediate premature immune shut-off, leading to resistance. Through identification of critical pathways and immunosuppressive cell lineages mediating resistance to oncolytic viral therapies it was possible to develop viral vectors to overcome them. As a result, previously resistant cancer models became sensitive to oncolytic viral therapy and other immunotherapies. These findings have the potential to significantly enhance the effectiveness of oncolytic viral and other immunotherapies in the clinic and to delineate approaches to overcome resistance to other cancer therapies.
HIGHLIGHTS.
Identification of Granulocytic MDSC as key mediators of resistance to immunotherapy.
Oncolytic virus expressed HPGD targets PGE2 and depletes G-MDSC in the tumor.
Reduction in PGE2 in the tumor alters chemokine profiles and immune cell infiltrate.
Targeting of PGE2 sensitizes established and resistant tumors to imnmunotherapies.
Acknowledgments
Thanks to Doug Marvel and Rachel Sikorski for help with preparation and running of flow cytometry samples and David Bartlett and Bernie Moss for providing cell lines and plasmids as indicated.
This work was funded by grant support from NIH (R01 CA178766 & R01 CA140215), The Lustgarten Foundation and through use of CCSG supported core facilities (P30 CA047904), including animal facilities, In Vivo Imaging Facilities, Flow Cytometry Facility and Luminex Core.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions;
WH ran the majority of the experiments described in this work. PS ran in vitro viral replication and cell killing assays. JJR constructed novel viral vectors, including VV-HPGD. SHT oversaw the work and wrote the manuscript.
Dr. Thorne has a financial interest in Western Oncolytics that has licensed this technology.
Hou et al. identify the prostaglandin PGE2 in the tumor as a key mediator of resistance to immunotherapies, including oncolytic virotherapy. Viral vectors engineered to target PGE2 are capable of overcoming localized immunosuppression to allow for robust anti-tumor adaptive immune responses.
References
- Alvarez-Breckenridge CA, Yu J, Price R, Wojton J, Pradarelli J, Mao H, Wei M, Wang Y, He S, Hardcastle J, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nature medicine. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andtbacka RHI, Collichio FA, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Doleman S, Ye Y, et al. OPTiM: A randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol. 2013;31:LBA9008. [Google Scholar]
- Baker J, Verneris MR, Ito M, Shizuru JA, Negrin RS. Expansion of cytolytic CD8(+) natural killer T cells with limited capacity for graft-versus-host disease induction due to interferon gamma production. Blood. 2001;97:2923–2931. doi: 10.1182/blood.v97.10.2923. [DOI] [PubMed] [Google Scholar]
- Breitbach CJ, Arulanandam R, De Silva N, Thorne SH, Patt R, Daneshmand M, Moon A, Ilkow C, Burke J, Hwang TH, et al. Oncolytic Vaccinia Virus Disrupts Tumor-Associated Vasculature in Humans. Cancer Research. 2013;73:1265–1275. doi: 10.1158/0008-5472.CAN-12-2687. [DOI] [PubMed] [Google Scholar]
- Breitbach CJ, Paterson JM, Lemay CG, Falls TJ, McGuire A, Parato KA, Stojdl DF, Daneshmand M, Speth K, Kirn D, et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther. 2007;15:1686–1693. doi: 10.1038/sj.mt.6300215. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Advances in cancer research. 2014;124:31–82. doi: 10.1016/B978-0-12-411638-2.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Sampath P, Hou W, Thorne SH. Regulating cytokine function enhances safety and activity of genetic cancer therapies. Mol Ther. 2013;21:167–174. doi: 10.1038/mt.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donkor MK, Lahue E, Hoke TA, Shafer LR, Coskun U, Solheim JC, Gulen D, Bishay J, Talmadge JE. Mammary tumor heterogeneity in the expansion of myeloid-derived suppressor cells. International immunopharmacology. 2009;9:937–948. doi: 10.1016/j.intimp.2009.03.021. [DOI] [PubMed] [Google Scholar]
- Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, Ohlfest JR, Okada H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011;71:2664–2674. doi: 10.1158/0008-5472.CAN-10-3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, Cho M, Lim HY, Chung HC, Kim CW, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nature Medicine. 2013;19:329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilkow CS, Marguerie M, Batenchuk C, Mayer J, Ben Neriah D, Cousineau S, Falls T, Jennings VA, Boileau M, Bellamy D, et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat Med. 2015;21:530–536. doi: 10.1038/nm.3848. [DOI] [PubMed] [Google Scholar]
- Kalinski P. Regulation of immune responses by prostaglandin E2. Journal of Immunology. 2012;188:21–28. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Oh JY, Park BH, Lee DE, Kim JS, Park HE, Roh MS, Je JE, Yoon JH, Thorne SH, et al. Systemic Armed Oncolytic and Immunologic Therapy for Cancer with JX-594, a Targeted Poxvirus Expressing GM-CSF. Mol Ther. 2006;14:361–370. doi: 10.1016/j.ymthe.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4:e353. doi: 10.1371/journal.pmed.0040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanbash G, McKaveney K, Sakaki M, Ueda R, Mintz AH, Amankulor N, Fujita M, Ohlfest JR, Okada H. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res. 2013;73:6413–6423. doi: 10.1158/0008-5472.CAN-12-4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T, Boisgerault N, Diaz RM, Donnelly O, Rommelfanger-Konkol D, Pulido J, Thompson J, Mukhopadhyay D, Kaspar R, Coffey M, et al. Detecting and targeting tumor relapse by its resistance to innate effectors at early recurrence. Nat Med. 2013;19:1625–1631. doi: 10.1038/nm.3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14:559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- Lun XQ, Jang JH, Tang N, Deng H, Head R, Bell JC, Stojdl DF, Nutt CL, Senger DL, Forsyth PA, McCart JA. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin Cancer Res. 2009;15:2777–2788. doi: 10.1158/1078-0432.CCR-08-2342. [DOI] [PubMed] [Google Scholar]
- Masters G, Dito G, Penhallow B, Lewin A, Kao H, Jure-Kunkel M. Antitumor activity of anti-PD-1 in combination with tyrosine kinase inhibitors in a preclinical renal cell carcinoma model. AACR, (San Diego, CA) 2014:5016. [Google Scholar]
- Mastrangelo MJ, Maguire HC, Jr, Eisenlohr LC, Laughlin CE, Monken CE, McCue PA, Kovatich AJ, Lattime EC. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6:409–422. doi: 10.1038/sj.cgt.7700066. [DOI] [PubMed] [Google Scholar]
- McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, Moss B, Bartlett DL. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–8757. [PubMed] [Google Scholar]
- Naik AM, Chalikonda S, McCart JA, Xu H, Guo ZS, Langham G, Gardner D, Mocellin S, Lokshin AE, Moss B, et al. Intravenous and isolated limb perfusion delivery of wild type and a tumor-selective replicating mutant vaccinia virus in nonhuman primates. Hum Gene Ther. 2006;17:31–45. doi: 10.1089/hum.2006.17.31. [DOI] [PubMed] [Google Scholar]
- Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011a;118:5498–5505. doi: 10.1182/blood-2011-07-365825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Research. 2011b;71:7463–7470. doi: 10.1158/0008-5472.CAN-11-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, Oh SY, Han SY, Yoon JH, Hong SH, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–542. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- Penheiter AR, Griesmann GE, Federspiel MJ, Dingli D, Russell SJ, Carlson SK. Pinhole micro-SPECT/CT for noninvasive monitoring and quantitation of oncolytic virus dispersion and percent infection in solid tumors. Gene Therapy. 2011 doi: 10.1038/gt.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202:931–939. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SJ, Federspiel MJ, Peng KW, Tong C, Dingli D, Morice WG, Lowe V, O’Connor MK, Kyle RA, Leung N, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clinic proceedings. 2014;89:926–933. doi: 10.1016/j.mayocp.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath P, Li J, Hou W, Chen H, Bartlett DL, Thorne SH. Crosstalk between immune cell and oncolytic vaccinia therapy enhances tumor trafficking and antitumor effects. Mol Ther. 2013;21:620–628. doi: 10.1038/mt.2012.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne SH, Hwang TH, O’Gorman WE, Bartlett DL, Sei S, Kanji F, Brown C, Werier J, Cho JH, Lee DE, et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J Clin Invest. 2007;117:3350–3358. doi: 10.1172/JCI32727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tysome JR, Li X, Wang S, Wang P, Gao D, Du P, Chen D, Gangeswaran R, Chard LS, Yuan M, et al. A novel therapeutic regimen to eradicate established solid tumors with an effective induction of tumor-specific immunity. Clin Cancer Res. 2012;18:6679–6689. doi: 10.1158/1078-0432.CCR-12-0979. [DOI] [PubMed] [Google Scholar]
- Wang LC, Lynn RC, Cheng G, Alexander E, Kapoor V, Moon EK, Sun J, Fridlender ZG, Isaacs SN, Thorne SH, Albelda SM. Treating Tumors With a Vaccinia Virus Expressing IFNbeta Illustrates the Complex Relationships Between Oncolytic Ability and Immunogenicity. Molecular therapy: the journal of the American Society of Gene Therapy. 2011 doi: 10.1038/mt.2011.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worschech A, Chen N, Yu YA, Zhang Q, Pos Z, Weibel S, Raab V, Sabatino M, Monaco A, Liu H, et al. Systemic treatment of xenografts with vaccinia virus GLV-1h68 reveals the immunologic facet of oncolytic therapy. BMC Genomics. 2009;10:301. doi: 10.1186/1471-2164-10-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Gnjatic S, Li H, Powel S, Gallardo HF, Ritter E, Ku GY, Jungbluth AA, Segal NH, Rasalan TS, et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci U S A. 2008;105:20410–20415. doi: 10.1073/pnas.0810114105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeh HJ, Downs-Canner S, McCart JA, Guo ZS, Rao UN, Ramalingam L, Thorne SH, Jones HL, Kalinski P, Wieckowski E, et al. First-in-man Study of Western Reserve Strain Oncolytic Vaccinia Virus: Safety, Systemic Spread, and Antitumor Activity. Mol Ther. 2014 doi: 10.1038/mt.2014.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell. 2015;162:1257–1270. doi: 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tai LH, Ilkow CS, Alkayyal AA, Ananth AA, de Souza CT, Wang J, Sahi S, Ly L, Lefebvre C, et al. Maraba MG1 virus enhances natural killer cell function via conventional dendritic cells to reduce postoperative metastatic disease. Mol Ther. 2014;22:1320–1332. doi: 10.1038/mt.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Yu YA, Wang E, Chen N, Danner RL, Munson PJ, Marincola FM, Szalay AA. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res. 2007;67:10038–10046. doi: 10.1158/0008-5472.CAN-07-0146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.