

Abstract
Purpose of review
Highlight recent discoveries about Notch activation and its oncogenic functions in lymphoid malignancies, and discuss the therapeutic potential of Notch inhibition.
Recent findings
NOTCH mutations arise in a broad spectrum of lymphoid malignancies and are increasingly scrutinized as putative therapeutic targets. In T cell acute lymphoblastic leukemia (T-ALL), NOTCH1 mutations affect the extracellular negative regulatory region and lead to constitutive Notch activation, although mutated receptors remain sensitive to Notch ligands. Other NOTCH1 mutations in T-ALL and NOTCH1/2 mutations in multiple B cell malignancies truncate the C-terminal PEST domain, leading to decreased Notch degradation after ligand-mediated activation. Thus, targeting Notch ligand-receptor interactions could provide therapeutic benefits. In addition, we discuss recent reports on clinical testing of Notch inhibitors in T-ALL that influenced contemporary thinking on the challenges of targeting Notch in cancer. We review advances in the laboratory to address these challenges in regards to drug targets, the Notch-driven metabolome, and the sophisticated protein-protein interactions at Notch-dependent super-enhancers that underlie oncogenic Notch functions.
Summary
Notch signaling is a recurrent oncogenic pathway in multiple T and B cell lymphoproliferative disorders. Understanding the complexity and consequences of Notch activation is critical to define optimal therapeutic strategies targeting the Notch pathway.
Keywords: Notch, leukemia, lymphoma, T cell, B cell
Introduction
Notch signaling is a highly conserved signaling pathway with multiple roles in development, tissue homeostasis and disease [1,2]. Notch can act as an oncogene or as a tumor suppressor in different cancers [3]. In lymphoproliferative disorders, data available to date identify Notch as a recurrent oncogene. In fact, human NOTCH was first recognized based on chromosomal translocations generating a constitutively active NOTCH1 allele in T cell acute lymphoblastic leukemia (T-ALL) [3]. These rare translocations were the tip of the iceberg, as frequent activating NOTCH1 mutations were subsequently discovered in T-ALL [4]. Recently, multiple reports described recurrent although less prevalent gain-of-function NOTCH1 and NOTCH2 mutations in B cell malignancies, including chronic lymphocytic leukemia (CLL), splenic marginal zone lymphoma (SMZL), mantle cell lymphoma (MCL), diffuse large B cell lymphoma (DLBCL) and rarely follicular lymphomas (FL) [5–22]. Non-mutational mechanisms of Notch activation may also exist [23,24]. Thus, the overall number of patients with lymphoid malignancies driven by oncogenic Notch signals is high, in sharp contrast with the initial description of infrequent NOTCH1 translocations in a rare disease (T-ALL).
Here, we discuss recent insights into the pathogenesis of oncogenic Notch signaling in T and B cell lymphoproliferative disorders. First, we review the mechanisms of Notch activation by different classes of NOTCH1/2 mutations, highlighting how these mutations remain sensitive to microenvironmental inputs (Figure 1–2). Second, we survey recent efforts to unravel downstream mechanisms of Notch action in T-ALL and other lymphoid malignancies (Figure 3). These considerations identify challenges and opportunities to develop safe and effective strategies of therapeutic Notch inhibition, as well as important areas of future investigation.
Figure 1. Structure of Notch1/2 receptors depicting domain organization and key mutation sites observed in lymphoid malignancies.
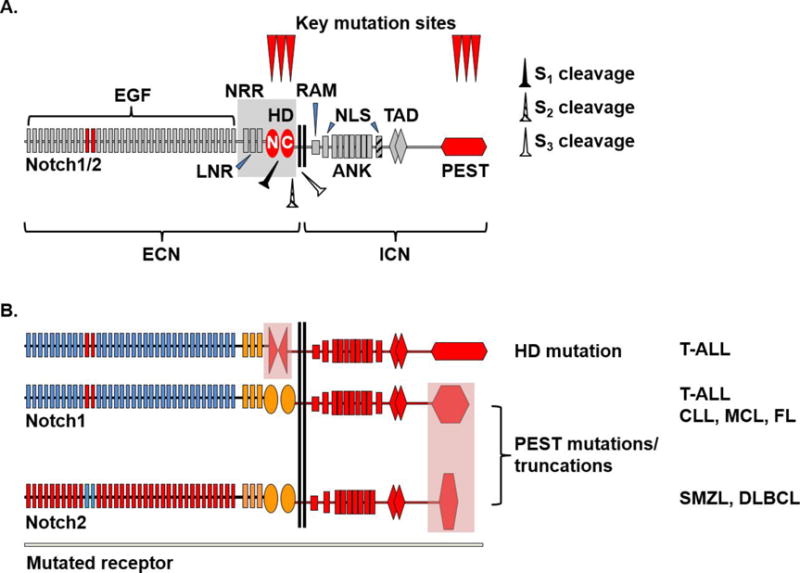
A. Wild-type Notch1/2 receptors depicting extracellular Notch (ECN) and intracellular Notch (ICN) domains. EGF11/12 repeats (red) are important for ligand binding. Sites of proteolytic cleavage are indicated as S1 (furin-like protease), S2 (ADAM10 metalloprotease) and S3 (γ-secretase complex). EGF, epidermal growth factor like domain; LNR, Lin12/Notch repeats; HD, heterodimerization domain; N, N-terminal portion of HD; C, C-terminal portion of HD; NRR, negative regulatory region; RAM, RBPJ-associated molecule domain; NLS, nuclear localization signal; ANK, ankyrin repeats; TAD, transactivation domain; PEST, proline (P), glutamic acid (E), serine (S) and threonine (T)-rich sequence.
B. Mutated Notch1 and Notch2 receptors with corresponding disease associations. Lightly colored areas over HD and PEST domains represent key mutation sites. PEST mutations typically truncate the PEST domain. The most frequent disease associations of individual mutations are shown as follows: T-ALL, T cell acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; FL, follicular lymphoma; SMZL, splenic marginal zone lymphoma; DLBCL, diffuse large B cell lymphoma.
Figure 2. Mechanisms of Notch pathway activation in lymphoid malignancies.

A. Notch1 heterodimerization domain mutations in T-ALL. HD mutations destabilize the receptor and lead to constitutive ligand-independent proteolytic activation (right). HD-mutated Notch1 receptors also remain sensitive to ligand-mediated activation (left). Both ligand-independent and ligand-dependent inputs can contribute to Notch signaling in malignant T cells.
B. Notch1/2 PEST domain mutations in T-ALL and B cell lymphoproliferative disorders. PEST mutations truncate the PEST domain, leading to decreased proteasomal degradation and increased half-life of cleaved ICN1/2. Notch signaling through PEST-mutated receptors requires ligand-dependent activation. The most frequent disease associations are shown as follows: T-ALL, T cell acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; FL, follicular lymphoma; SMZL, splenic marginal zone lymphoma; DLBCL, diffuse large B cell lymphoma.
Figure 3. Emerging insights into transcriptional regulation of Notch target genes.
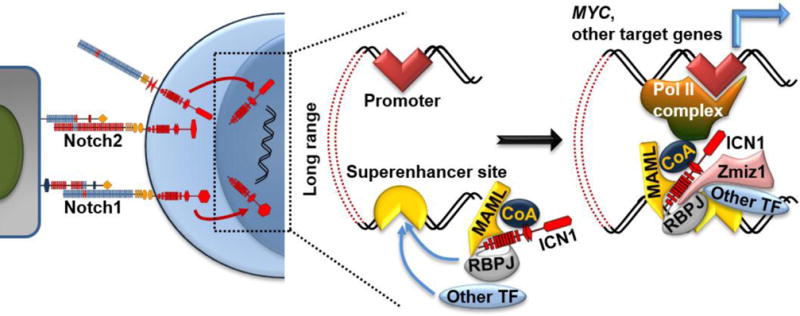
Cleaved ICN translocates to the nucleus and interacts with the DNA-binding transcription factor RBPJ, a member of the Mastermind-like (MAML) family and other transcriptional coactivators (CoA). In T-ALL and possibly in other contexts, activation of key target genes such as MYC involves long-range interactions between the basal promoter and a distant superenhancer. Additional transcription factors (TF) such as ETS1, RUNX1, HEB, E2A, GABPA, and TAL1 converge with ICN to superenhancer sites. The PIAS-like coactivator ZMIZ1 binds ICN1/RBPJ and facilitates the recruitment of ICN to the MYC superenhancer in cancer cells.
Notch signaling pathway
Biochemical aspects of Notch signaling have been reviewed [1]. Notch signaling is a cell-to-cell communication pathway driven by Notch ligand-receptor interactions. Mammals express four Notch receptors (Notch1–4) and five ligands of the Jagged (Jagged1/2) and Delta-like families (Dll1/3/4). Notch receptors are expressed as heterodimers after “S1” cleavage during transit to the cell surface. Ligand-receptor interaction generates a physical force exposing a proximal region of the Notch extracellular domain for cleavage by a disintegrin and metalloprotease (ADAM10). ADAM10-mediated “S2” proteolysis is followed by “S3” cleavage within the transmembrane domain by γ-secretase, releasing intracellular Notch (ICN). ICN migrates into the nucleus where it interacts with the transcription factor RBPJ (also known as RBP-Jk or CSL) and a Mastermind-like family (MAML) transcriptional co-activator. The ICN-RBPJ-MAML complex activates target gene transcription in cooperation with other transcription factors and epigenetic regulators. ICN normally has a short half-life due to rapid proteasomal degradation regulated by the C-terminal ICN PEST domain and other mechanisms.
Oncogenic Notch signaling in T-ALL
Oncogenic Notch activation in T-ALL was first reported to result from rare t(7:9) translocations driving expression of intracellular NOTCH under the control of TCRB regulatory sequences. Experimental models then demonstrated the transforming potential of ICN overexpression in hematopoietic progenitors. In 2004, the Aster and Look laboratories described recurrent NOTCH1 mutations in the majority of human T-ALL, indicating that NOTCH1 is the most frequent oncogene across all T-ALL subtypes [4]. Other large studies confirmed these findings [25–30]. FBXW7 mutations were subsequently discovered to result in decreased degradation of ICN and other oncogenic proteins in T-ALL [31,32]. Altogether, NOTCH1 and FBXW7 mutations were detected in up to 70–80% of patients. In the absence of RAS and PTEN mutations, they are associated with a favorable prognosis and could prove useful as a new molecular prognostication system [26].
NOTCH1 mutations cluster in two areas that dysregulate pathway activation through distinct mechanisms (Figure 1). A first class of missense mutations targets the extracellular negative regulatory region (NRR) containing the heterodimerization (HD) domain and capped by Lin12/Notch repeats (LNR). In the absence of ligand-receptor binding, the NRR buries the S2 cleavage site within a hydrophobic pocket that prevents ADAM10-mediated cleavage and receptor activation [33,34]. The majority of HD mutations affect the NRR hydrophobic core or the HD/LNR interface, leading to receptor destabilization and proteolytic activation even without ligand. Yet, mutated NOTCH receptors remain sensitive to ligand-mediated activation, suggesting that their net in vivo activity cumulates ligand-independent and ligand-dependent activation (Figure 2) [34]. The second class of NOTCH1 mutations truncates the C-terminal PEST domain via non-sense or frameshift events that introduce premature STOP codons (Figure 1–2). PEST truncations and FBXW7 mutations impair ICN proteasomal degradation, increasing its half-life in malignant cells. In some patients, PEST and HD mutations occur in cis, suggesting cooperativity [4].
Oncogenic Notch signaling in CLL and other B cell lymphoproliferative disorders
Gain-of-function NOTCH1/2 mutations were first reported in case series of B cell lymphoproliferative disorders [9,14,19,20], and then in large cohorts [5–8,10–13,15–18,21] (Figure 1). In CLL, NOTCH1 mutations are found in ~10% of patients and are associated with advanced disease, including Richter’s transformation, and with the presence of trisomy 12. Although case series reported an association with worse prognosis, this may not be independent of other prognostic factors. Besides NOTCH1/2 PEST domain truncations, recurrent non-coding mutations in the NOTCH1 3′UTR were recently reported in another ~2% of CLL patients [22]. These mutations introduce of a new splice acceptor site in the 3′UTR, triggering the excision of PEST-coding sequences from the mRNA and effectively truncating the PEST domain. In SMZL, NOTCH2 but not NOTCH1 mutations were reported in 20–25% of cases. In MCL, DLBCL and FL, NOTCH1/2 mutations have also been described, although more limited data are available about their prevalence and significance.
In contrast to T-ALL, gain-of-function NOTCH1 and NOTCH2 mutations in B cell lymphomas nearly exclusively target the PEST domain and not the NRR. Thus, Notch pathway activation in these disorders is predicted to rely on ligand-mediated activation more stringently than in T-ALL (Figure 2). To date, the Notch ligands inducing Notch activation in B cell lymphomas remain unknown. Candidate cellular sources include other hematopoietic cells; endothelial cells, which are capable of inducing Notch signaling in B lymphoma cells [24]; and DLL1/4-expressing fibroblastic cells in secondary lymphoid organs [35]. Aster and colleagues developed an immunohistochemical assay to detect cleaved ICN1 and presented several examples of lymphoproliferative disorders in which active ICN1 was abundant in lymph nodes, but lost abruptly in areas where the tumor extended across the lymph node capsule [23]. These data suggest the presence of functionally important Notch ligands within the lymph node environment, a potential therapeutic target. Moreover, the fraction of lymphomas with detectable ICN1 was much higher than predicted from the prevalence of NOTCH1 mutations, and some lymphomas with no reported mutations (e.g. angioimmunoblastic T cell lymphomas) had abundant ICN1 in a high fraction of tumors [23]. Thus, Notch signaling mediated by Notch ligand-receptor interactions may have pathogenic significance in a broad range of lymphomas.
Limited information is available so far about crosstalk of active Notch signaling and other oncogenic pathways in B cell lymphomas. In CLL, NOTCH1 mutations might be associated with the expression of specific B cell receptor (BCR) subsets [17]. Preclinical data in normal B cells indicate that Notch and BCR signaling can cooperate [36]. Thus, Notch might cooperate with BCR signaling or other microenvironmental inputs in B cell lymphomas. Future studies could identify promising combinations of Notch inhibitors with agents that target downstream BCR signals, such as ibrutinib or idelalisib.
In contrast to its oncogenic role in T-ALL and B cell lymphoproliferative disorders involving mature B cell subsets, Notch signaling was reported to inhibit the growth of precursor B-ALL, where Notch pathway genes tend to be epigenetically silenced [37]. Notch can also act as a growth suppressor in other hematopoietic lineages, such as the myeloid lineage [38–40]. Whether these context-specific suppressive effects happen in other hematological malignancies remains to be determined, but they highlight the versatile effects of Notch signaling in different cancer types [3].
Downstream mediators of the Notch signaling pathway
To date, transcriptional targets of Notch signaling have been studied in T-ALL but not systematically in other lymphoproliferative disorders. Active Notch binds thousands of genomic loci, only a fraction of which appears dynamically regulated [41,42]. In T-ALL, several direct NOTCH1 target genes have been shown to induce and/or maintain disease, such as MYC, HES1, TRIB2, IL7R, HES1, CCND3, and IGF1R (reviewed in [43]). Many of these genes enhance PI3K/AKT/mTOR signaling. Notch-regulated long non-coding RNAs such as LUNAR1 have recently been implicated [44,45]. Understanding the most critical target genes for oncogenesis has gained widespread attention given the “on-target” toxicities and drug resistance seen in early clinical trials of pan-NOTCH inhibitors (γ-secretase inhibitors or GSIs). The hope is to inhibit important Notch target genes that will eliminate malignant cells, but spare normal tissues from the toxicities of pan-Notch inhibition. For example, Hes1 inactivation in mouse T-ALL models induces tumor regression [46,47]. Schnell et al. identified perhexiline as a drug that phenocopies gene expression changes induced by Hes1 inactivation [47]. Perhexiline had antileukemic effects on human T-ALL samples and in a mouse model of Notch-induced T-ALL. Perhexiline, which inhibits mitochondrial carnitine palmitoyltransferase-1, is being used in some countries for cardiac indications. It is encouraging that this drug appears effective and better tolerated than GSIs.
Transcriptional regulation of oncogenic Notch target genes
Notch regulates different target genes in distinct cell types through complex interactions with multiple regulators (reviewed in [48] and [49]), predicting the existence of lineage-specific response elements. Two groups identified a T-lineage specific Notch-dependent MYC enhancer (NDME) ~1.4 Mb downstream of MYC [50,51]. MYC is an important NOTCH target as it controls leukemia-initiating cell activity [52,53], leukemia initiation and maintenance [52,54,55], and can replace Notch1 signals [56] (Chiang M, Pear W, unpublished). Genetic deletion of the NDME in mice [50] or CRISPR/Cas9-mediated excision of the NDME in human T-ALL [57] blocks MYC transcription and leukemia growth. However, in practice, targeting Notch response elements like NDME without pan-Notch inhibition is challenging. One option is to inhibit epigenetic modifiers at Notch response elements, such as BRD4 [51–54]. An alternative strategy is to inhibit protein-protein interactions that build the superenhancer complex at oncogenic Notch target genes. We analyzed publically available ChIP-Seq datasets in human T-ALL [41,58,59] and found several transcription factor peaks close to the NMDE’s RBPJ peak, such as ETS1, RUNX1, HEB, E2A, GABPA, and TAL1 (Chiang, unpublished). HEB and RUNX1 might interact with ICN1 [60]. We recently showed that the PIAS-like coactivator Zmiz1 binds ICN1/RBPJ through a tetratricopeptide repeat (TPR) domain, which enhances NDME functions perhaps by connecting ICN1 to other transcription factors [61,62] (Figure 3). Zmiz1 inactivation induced regression of Notch-dependent mouse T-ALL without toxicities associated with pan-Notch inhibition [61]. Thus, an intriguing possibility is that targeting ICN1 interactions with adjacent transcription factors could strip Notch of its oncogenic functions.
While some protein-protein interactions in chromatin strengthen Notch signals, others weaken them. Cyclin C/CDK inhibits ICN1 activity by phosphorylating ICN1, resulting in ubiquitination and proteosomal degradation via FBXW7 [63]. In ChIP-Seq studies, IKZF1 bound close to ~60% of ICN1/RBPJ sites (including the NDME), suppressing Notch target gene expression [64]. Interestingly, IKZF1 is known to displace ICN1/RBPJ from DNA by competing for shared motifs [65], but more frequently IKZF1 binds next to RBPJ, which may interfere with ICN1 function through protein-protein interactions [64]. The low frequency of inactivating IKZF1 mutations (<5%) in human T-ALL argues against Ikaros having broad functional significance [66]. However, a recent report shows that ICN1 represses IKZF1 expression in human T-ALL and that restoring Ikaros levels induces tumor regression [67]. In human B-ALL, Casein Kinase II (CK2) deactivates IKZF1 [68]. Thus, it is intriguing to consider whether CK2 inhibitors might restore IKZF1 function in T-ALL.
Therapeutic targeting of the NOTCH signaling pathway
Several strategies to block NOTCH signals are predicted to be effective in most NOTCH-dependent cancers, such as NRR-specific antibodies that block NOTCH cleavage, ADAM protease inhibitors, γ-secretase inhibitors (GSI), and MAML-like peptides that disrupt the NOTCH transcriptional complex (reviewed in [43,69]). In contrast, antibodies that block specific NOTCH ligand/receptor interactions are predicted to be more effective in cancers driven by Notch ligands, such as CLL, than cancers that are less ligand-dependent, such as T-ALL. The anti-NOTCH1 antibody OMP-52M51 is currently being tested in CLL and other lymphoid malignancies (NCT01703572). Targeting Notch ligands in CLL and other B cell lymphoproliferative disorders is another attractive approach, although the nature and source of individual Notch ligands in these diseases remain unknown.
As NOTCH was implicated in a growing list of cancers over the past decade, there was increasing excitement that NOTCH inhibitors would be effective anti-cancer treatments. However, initial enthusiasm was tempered by clinical reports of on-target toxicities from pan-NOTCH inhibition and low response rates. More than a dozen clinical trials tested earlier generation GSIs (MK-0752 and R04929097) in patients with mostly solid cancers (reviewed in [69]). Responses were seen in <5% of patients. These initial studies were hampered by dose-limiting GI toxicity, which was attributed to on-target effects of pan-Notch inhibition on the intestinal epithelium. The first clinical trial testing MK-0752 in relapsed/refractory T-ALL was halted due to excessive diarrhea [70], although 1/7 patient had a partial response. Intermittent dosing is more tolerable, but cannot achieve continuous Notch suppression [71]. In a phase I trial with a newer generation GSI, PF-03084014, 1/8 relapsed/refractory T-ALL patient achieved a complete remission [72]. In a preliminary report of a phase I trial (NCT01363817), 25 relapsed/refractory pediatric T-ALL patients were injected weekly with the GSI BMS-906024 with/without dexamethasone at the physician’s discretion [73]. The response rate was encouraging at 32%, perhaps reflecting synergy between GSI and steroids [74]. One patient with ETP-ALL achieved complete remission, received a bone marrow transplant, and has been relapse-free for >19 months [75].
In contrast to other GSIs, PF-03084014 and BMS-906024 were associated with only mild diarrhea [73,76]. Interestingly, NOTCH1 mutation status did not predict response. Thus, it is possible that T-ALLs with wildtype NOTCH1 receptors benefit from anti-NOTCH therapy. This might suggest a role for ligands in triggering oncogenic Notch signals. However, preclinical studies using patient-derived xenografts lack consensus [77–79]. There has been debate about the reasons for the low response rate of T-ALL to GSI so far. This low response rate might reflect technical inability to fully suppress Notch signals because of on-target toxicities. On the other hand, the low response rate might reflect resistance of relapsed/refractory disease.
Resistance to anti-NOTCH agents
Prior therapy may directly result in resistance to NOTCH inhibitors seen in the relapsed/refractory setting. For example, PI3K/MAPK inhibitors can select for resistant, Notch-independent tumors [80]. However, resistance may occur de novo as well, given short, transient responses to upfront intermittent GSI in T-ALL models [81,82]. Several resistance mechanisms are possible (reviewed in [43]). These mechanisms are frequently driven by oncogenic pathways that crosstalk with Notch signals, such as PI3K/mTOR (promoted by PTEN loss [83,84]) and MYC (promoted by FBWX7 loss [31,32], BRD4 [51,54] and ZMIZ1 [61,85]). A recent comprehensive investigation in Notch-induced mouse T-ALL revealed that GSI downregulated glycolysis and glutaminolysis, likely due to broad transcriptional effects on anabolic and catabolic genes [84]. Pten inactivation reversed these changes and promoted GSI resistance in vivo. However, effects were incomplete [84,86], suggesting that multiple pathways operate to promote GSI resistance. Thus, one may need to look beyond GSIs and single sensitizing agents (e.g. PI3K/mTOR [80] or BET inhibitors [54]) and towards combining GSIs with multiple sensitizing agents or other drugs like phenothiazines, which can simultaneously inhibit multiple downstream pathways [87].
Conclusions
Notch pathway activation is emerging as a potential therapeutic target in an expanding range of T and B cell lymphoproliferative disorders. Mutational and non-mutational mechanisms of Notch activation have been described, including mechanisms that rely on ligand-mediated activation. To guide the development of safe and effective therapeutic strategies targeting the Notch pathway, future research should identify Notch ligands and receptors that drive the growth of lymphoid malignancies, evaluate the transcriptional effects of Notch signaling and explore Notch’s crosstalk with other pathways in individual tumor types.
Key bulleted points.
-
-
The Notch pathway has oncogenic effects in multiple T cell and B cell lymphoproliferative disorders.
-
-
In T cell acute lymphoblastic leukemia, a first class of mutated Notch1 receptors targeting the extracellular heterodimerization domain increase Notch signaling intensity through constitutive ligand-independent activation, although the mutated receptors also remain sensitive to ligand-mediated activation.
-
-
In T cell acute lymphoblastic leukemia and multiple B cell malignancies, a second class of mutated Notch1/2 receptors increase the half-life of cleaved intracellular Notch only upon ligand-mediated pathway activation.
-
-
At key oncogenic targets such as MYC, intracellular Notch nucleates a transcriptional activation complex at superenhancer sites in cooperation with other transcription factors and with the PIAS1-like coactivator Zmiz1.
-
-
Understanding the mechanisms of enhanced Notch activity and the downstream consequences of Notch signaling identifies therapeutic challenges and opportunities in T and B cell lymphoproliferative disorders.
Acknowledgments
We thank members of the Maillard and Chiang laboratories for helpful discussions and review of the manuscript.
Financial support and sponsorship
Research on Notch signaling in the Chiang and Maillard laboratories has been supported by the NCI (RO1-CA196604 to M.Y.C.), NIAID (RO1-AI091627 to I.M.) and the Leukemia and Lymphoma Society (CDP 1227-14 and TRP 6462-15 to I.M.). Support for V.R. has been provided by a research training award from the American Society of Hematology and a career development award from the American Society for Blood and Marrow Transplantation.
Footnotes
Conflicts of interest
The authors have no conflict of interest to disclose.
References
- 1.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louvi A, Artavanis-Tsakonas S. Notch and disease: A growing field. Semin Cell Dev Biol. 2012;23:473–480. doi: 10.1016/j.semcdb.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.South AP, Cho RJ, Aster JC. The double-edged sword of Notch signaling in cancer. Semin Cell Dev Biol. 2012 doi: 10.1016/j.semcdb.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 5.Arcaini L, Rossi D, Lucioni M, et al. The NOTCH pathway is recurrently mutated in diffuse large B-cell lymphoma associated with hepatitis C virus infection. Haematologica. 2015;100:246–252. doi: 10.3324/haematol.2014.116855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balatti V, Bottoni A, Palamarchuk A, et al. NOTCH1 mutations in CLL associated with trisomy 12. Blood. 2012;119:329–331. doi: 10.1182/blood-2011-10-386144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baliakas P, Hadzidimitriou A, Sutton LA, et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia. 2015;29:329–336. doi: 10.1038/leu.2014.196. [DOI] [PubMed] [Google Scholar]
- 8.Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208:1389–1401. doi: 10.1084/jem.20110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Ianni M, Baldoni S, Rosati E, et al. A new genetic lesion in B-CLL: a NOTCH1 PEST domain mutation. Br J Haematol. 2009;146:689–691. doi: 10.1111/j.1365-2141.2009.07816.x. [DOI] [PubMed] [Google Scholar]
- 10.Jeromin S, Weissmann S, Haferlach C, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28:108–117. doi: 10.1038/leu.2013.263. [DOI] [PubMed] [Google Scholar]
- 11.Karube K, Martinez D, Royo C, et al. Recurrent mutations of NOTCH genes in follicular lymphoma identify a distinctive subset of tumours. J Pathol. 2014;234:423–430. doi: 10.1002/path.4428. [DOI] [PubMed] [Google Scholar]
- 12.Kiel MJ, Velusamy T, Betz BL, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. The Journal of experimental medicine. 2012;209:1553–1565. doi: 10.1084/jem.20120910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kridel R, Meissner B, Rogic S, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119:1963–1971. doi: 10.1182/blood-2011-11-391474. [DOI] [PubMed] [Google Scholar]
- 14.Lee SY, Kumano K, Nakazaki K, et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer science. 2009;100:920–926. doi: 10.1111/j.1349-7006.2009.01130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oscier DG, Rose-Zerilli MJ, Winkelmann N, et al. The clinical significance of NOTCH1 and SF3B1 mutations in the UK LRF CLL4 trial. Blood. 2013;121:468–475. doi: 10.1182/blood-2012-05-429282. [DOI] [PubMed] [Google Scholar]
- 16.Rossi D, Rasi S, Fabbri G, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119:521–529. doi: 10.1182/blood-2011-09-379966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossi D, Spina V, Bomben R, et al. Association between molecular lesions and specific B-cell receptor subsets in chronic lymphocytic leukemia. Blood. 2013;121:4902–4905. doi: 10.1182/blood-2013-02-486209. [DOI] [PubMed] [Google Scholar]
- 18.Rossi D, Trifonov V, Fangazio M, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209:1537–1551. doi: 10.1084/jem.20120904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sportoletti P, Baldoni S, Cavalli L, et al. NOTCH1 PEST domain mutation is an adverse prognostic factor in B-CLL. Br J Haematol. 2010;151:404–406. doi: 10.1111/j.1365-2141.2010.08368.x. [DOI] [PubMed] [Google Scholar]
- 20.Troen G, Wlodarska I, Warsame A, et al. NOTCH2 mutations in marginal zone lymphoma. Haematologica. 2008;93:1107–1109. doi: 10.3324/haematol.11635. [DOI] [PubMed] [Google Scholar]
- 21.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22*.Puente XS, Bea S, Valdes-Mas R, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526:519–524. doi: 10.1038/nature14666. This paper reports non-coding mutations in CLL, including recurrent mutations in the NOTCH1 3′UTR that lead to aberrant splicing and elimination of PEST-coding sequences. [DOI] [PubMed] [Google Scholar]
- 23**.Kluk MJ, Ashworth T, Wang H, et al. Gauging NOTCH1 Activation in Cancer Using Immunohistochemistry. PloS one. 2013;8:e67306. doi: 10.1371/journal.pone.0067306. Using a sensitive and specific immunohistochemical method to detect cleaved ICN1 as an indication of Notch1 activation in cancer, the authors report a high frequency of Notch activation in several lymphoid malignancies, suggesting that non-mutational mechanisms contribute to Notch activation in these disorders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao Z, Ding BS, Guo P, et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell. 2014;25:350–365. doi: 10.1016/j.ccr.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asnafi V, Buzyn A, Le Noir S, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. 2009;113:3918–3924. doi: 10.1182/blood-2008-10-184069. [DOI] [PubMed] [Google Scholar]
- 26*.Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol. 2013;31:4333–4342. doi: 10.1200/JCO.2012.48.5292. This work reports the development of a new molecular prognostication system in T-ALL including the presence of NOTCH1 or FBXW7 mutations. [DOI] [PubMed] [Google Scholar]
- 27.Mansour MR, Sulis ML, Duke V, et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adults with T-cell acute lymphoblastic leukemia treated on the MRC UKALLXII/ECOG E2993 protocol. J Clin Oncol. 2009;27:4352–4356. doi: 10.1200/JCO.2009.22.0996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jenkinson S, Koo K, Mansour MR, et al. Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia. 2013;27:41–47. doi: 10.1038/leu.2012.176. [DOI] [PubMed] [Google Scholar]
- 29.Clappier E, Collette S, Grardel N, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia. 2010;24:2023–2031. doi: 10.1038/leu.2010.205. [DOI] [PubMed] [Google Scholar]
- 30.Kox C, Zimmermann M, Stanulla M, et al. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia. 2010;24:2005–2013. doi: 10.1038/leu.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson BJ, Buonamici S, Sulis ML, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204:1825–1835. doi: 10.1084/jem.20070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Neil J, Grim J, Strack P, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gordon WR, Roy M, Vardar-Ulu D, et al. Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood. 2009;113:4381–4390. doi: 10.1182/blood-2008-08-174748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malecki MJ, Sanchez-Irizarry C, Mitchell JL, et al. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol. 2006;26:4642–4651. doi: 10.1128/MCB.01655-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fasnacht N, Huang HY, Koch U, et al. Specific fibroblastic niches in secondary lymphoid organs orchestrate distinct Notch-regulated immune responses. Journal of Experimental Medicine. 2014;211:2265–2279. doi: 10.1084/jem.20132528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas M, Calamito M, Srivastava B, et al. Notch activity synergizes with B-cell-receptor and CD40 signaling to enhance B-cell activation. Blood. 2007;109:3342–3350. doi: 10.1182/blood-2006-09-046698. [DOI] [PubMed] [Google Scholar]
- 37.Kuang SQ, Fang Z, Zweidler-McKay PA, et al. Epigenetic inactivation of Notch-Hes pathway in human B-cell acute lymphoblastic leukemia. PLoS One. 2013;8:e61807. doi: 10.1371/journal.pone.0061807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klinakis A, Lobry C, Abdel-Wahab O, et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011;473:230–233. doi: 10.1038/nature09999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kannan S, Sutphin RM, Hall MG, et al. Notch activation inhibits AML growth and survival: a potential therapeutic approach. The Journal of experimental medicine. 2013;210:321–337. doi: 10.1084/jem.20121527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lobry C, Ntziachristos P, Ndiaye-Lobry D, et al. Notch pathway activation targets AML-initiating cell homeostasis and differentiation. The Journal of experimental medicine. 2013;210:301–319. doi: 10.1084/jem.20121484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Wang H, Zang C, Taing L, et al. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:705–710. doi: 10.1073/pnas.1315023111. This paper reports a comprehensive ChIP-Seq study that identifies dynamic NOTCH1-regulated enhancers that are associated with multiple transcription factors and epigenetic modifiers. The ChIP-Seq dataset is a high-quality resource that has been extensively interrogated by several laboratories. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Margolin AA, Palomero T, Sumazin P, et al. ChIP-on-chip significance analysis reveals large-scale binding and regulation by human transcription factor oncogenes. Proc Natl Acad Sci U S A. 2009;106:244–249. doi: 10.1073/pnas.0806445106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tosello V, Ferrando AA. The NOTCH signaling pathway: role in the pathogenesis of T-cell acute lymphoblastic leukemia and implication for therapy. Ther Adv Hematol. 2013;4:199–210. doi: 10.1177/2040620712471368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44**.Trimarchi T, Bilal E, Ntziachristos P, et al. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell. 2014;158:593–606. doi: 10.1016/j.cell.2014.05.049. This is the first, comprehensive screen and analysis of long noncoding RNAs in a Notch-driven cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durinck K, Wallaert A, Van de Walle I, et al. The Notch driven long non-coding RNA repertoire in T-cell acute lymphoblastic leukemia. Haematologica. 2014;99:1808–1816. doi: 10.3324/haematol.2014.115683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wendorff AA, Koch U, Wunderlich FT, et al. Hes1 is a critical but context-dependent mediator of canonical Notch signaling in lymphocyte development and transformation. Immunity. 2010;33:671–684. doi: 10.1016/j.immuni.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 47*.Schnell SA, Ambesi-Impiombato A, Sanchez-Martin M, et al. Therapeutic targeting of HES1 transcriptional programs in T-ALL. Blood. 2015;125:2806–2814. doi: 10.1182/blood-2014-10-608448. This is the first description of a Hes1 pathway inhibitor as well as a genome-wide expression study of downstream targets of Hes1 in Notch-induced T-ALL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H, Zang C, Liu XS, et al. The role of Notch receptors in transcriptional regulation. J Cell Physiol. 2015;230:982–988. doi: 10.1002/jcp.24872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kushwah R, Guezguez B, Lee JB, et al. Pleiotropic roles of Notch signaling in normal, malignant, and developmental hematopoiesis in the human. EMBO Rep. 2014;15:1128–1138. doi: 10.15252/embr.201438842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50**.Herranz D, Ambesi-Impiombato A, Palomero T, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20:1130–1137. doi: 10.1038/nm.3665. Together with Yashiro-Ohtani et al., 2014, this paper identifies a long-range Notch-driven Myc enhancer (NDME) that appears to be specifically active in T-lineage cells and directly implicates this enhancer as an important driver of T-ALL proliferation using NDME-deficient mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51**.Yashiro-Ohtani Y, Wang H, Zang C, et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc Natl Acad Sci U S A. 2014;111:E4946–4953. doi: 10.1073/pnas.1407079111. Together with Herranz et al., 2014, this paper identifies a long-range NOTCH-driven MYC enhancer (NDME) specifically active in T cells. Together with Knoeschel et a., 2014, this paper identifies a second long-range NOTCH-independent, BRD4-dependent MYC enhancer in GSI-resistant T-ALLs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.King B, Trimarchi T, Reavie L, et al. The Ubiquitin Ligase FBXW7 Modulates Leukemia-Initiating Cell Activity by Regulating MYC Stability. Cell. 2013;153:1552–1566. doi: 10.1016/j.cell.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roderick JE, Tesell J, Shultz LD, et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood. 2014;123:1040–1050. doi: 10.1182/blood-2013-08-522698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54**.Knoechel B, Roderick JE, Williamson KE, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364–370. doi: 10.1038/ng.2913. Together with Yashiro-Ohtani et al., 2014, this paper implicates BRD4-driven MYC expression as a resistance mechanism for NOTCH inhibitors, thus providing a rationale for combining phatmacological BET inhibitiors with GSI therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X, Gounari F, Protopopov A, et al. Oncogenesis of T-ALL and nonmalignant consequences of overexpressing intracellular NOTCH1. J Exp Med. 2008;205:2851–2861. doi: 10.1084/jem.20081561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chiang MY, Childs ME, Romany C, et al. C-Myc but not AKT can substitute for Notch in lymphomagenesis. Blood (ASH Annual Meeting Abstracts) 2009:114. [Google Scholar]
- 57.Ohtani Y, Xu S, Petrovic J, et al. Modular Domains within a Super Enhancer Determine Drug Resistance in Leukemia. Blood (ASH Annual Meeting Abstracts) 2015:126. [Google Scholar]
- 58.Palii CG, Perez-Iratxeta C, Yao Z, et al. Differential genomic targeting of the transcription factor TAL1 in alternate haematopoietic lineages. EMBO J. 2011;30:494–509. doi: 10.1038/emboj.2010.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanda T, Lawton LN, Barrasa MI, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22:209–221. doi: 10.1016/j.ccr.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yatim A, Benne C, Sobhian B, et al. NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Mol Cell. 2012;48:445–458. doi: 10.1016/j.molcel.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61**.Pinnell N, Yan R, Cho HJ, et al. The PIAS-like Coactivator Zmiz1 Is a Direct and Selective Cofactor of Notch1 in T Cell Development and Leukemia. Immunity. 2015;43:870–883. doi: 10.1016/j.immuni.2015.10.007. This is the first report of a context-dependent direct Notch1 cofactor that shows proof-of-principle that the oncogenic functions of Notch can be biochemically uncoupled from the essential functions of Notch for homeostasis and tumor supression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Helbig C, Amsen D. Notch Signaling: Piercing a Harness of Simplicity. Immunity. 2015;43:831–833. doi: 10.1016/j.immuni.2015.10.022. [DOI] [PubMed] [Google Scholar]
- 63.Li N, Fassl A, Chick J, et al. Cyclin C is a haploinsufficient tumour suppressor. Nat Cell Biol. 2014;16:1080–1091. doi: 10.1038/ncb3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Geimer Le Lay AS, Oravecz A, Mastio J, et al. The tumor suppressor Ikaros shapes the repertoire of notch target genes in T cells. Sci Signal. 2014;7:ra28. doi: 10.1126/scisignal.2004545. [DOI] [PubMed] [Google Scholar]
- 65.Beverly LJ, Capobianco AJ. Perturbation of Ikaros isoform selection by MLV integration is a cooperative event in Notch(IC)-induced T cell leukemogenesis. Cancer Cell. 2003;3:551–564. doi: 10.1016/s1535-6108(03)00137-5. [DOI] [PubMed] [Google Scholar]
- 66.Marcais A, Jeannet R, Hernandez L, et al. Genetic inactivation of Ikaros is a rare event in human T-ALL. Leuk Res. 2010;34:426–429. doi: 10.1016/j.leukres.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 67*.Witkowski MT, Cimmino L, Hu Y, et al. Activated Notch counteracts Ikaros tumor suppression in mouse and human T-cell acute lymphoblastic leukemia. Leukemia. 2015;29:1301–1311. doi: 10.1038/leu.2015.27. This paper shows that ICN1 suppresses IKZF1 tumor suppressor functions, which could explain why IKZF1 loss-of-function mutations are rare in human T-ALL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Song C, Gowda C, Pan X, et al. Targeting casein kinase II restores Ikaros tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood. 2015;126:1813–1822. doi: 10.1182/blood-2015-06-651505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discov. 2014;13:357–378. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- 70.Deangelo DJ, Stone RM, Silverman LB, et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. Journal of Clinical Oncology, 2006 ASCO Annual Meeting Proceedings Part I. 2009;24:6585. [Google Scholar]
- 71.Krop I, Demuth T, Guthrie T, et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. Journal of Clinical Oncology. 2012;30:2307–2313. doi: 10.1200/JCO.2011.39.1540. [DOI] [PubMed] [Google Scholar]
- 72*.Papayannidis C, DeAngelo DJ, Stock W, et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015;5:e350. doi: 10.1038/bcj.2015.80. This paper is the first published report of a clinical trial of gamma-secretase inhibitors in T-ALL patients. PF-03084014 induced one complete remission with unexpectedly low GI toxicity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zweidler-McKay PA, Deangelo DJ, Douer D, et al. The Safety and Activity of BMS-906024, a Gamma Secretase Inhibitor (GSI) with Anti-Notch Activity, in Patients with Relapsed T-Cell Acute Lymphoblastic Leukemia (T-ALL): Initial Results of a Phase 1 Trial. Blood (ASH Annual Meeting Abstracts) 2014:124. [Google Scholar]
- 74.Real PJ, Tosello V, Palomero T, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15:50–58. doi: 10.1038/nm.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Knoechel B, Bhattt A, DeAngelo DJ, et al. Complete hematologic response of early T-cell progenitor acute lymphoblastic leukemia to the γ-secretase inhibitor BMS-906024: genetic and epigenetic findings in an outlier case. Cold Spring Harb Mol Case Stud. 2015;1 doi: 10.1101/mcs.a000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Messersmith WA, Shapiro GI, Cleary JM, et al. A Phase I, dose-finding study in patients with advanced solid malignancies of the oral gamma-secretase inhibitor PF-03084014. Clin Cancer Res. 2015;21:60–67. doi: 10.1158/1078-0432.CCR-14-0607. [DOI] [PubMed] [Google Scholar]
- 77.Agnusdei V, Minuzzo S, Frasson C, et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia. 2014;28:278–288. doi: 10.1038/leu.2013.183. [DOI] [PubMed] [Google Scholar]
- 78.Minuzzo S, Agnusdei V, Pusceddu I, et al. DLL4 regulates NOTCH signaling and growth of T acute lymphoblastic leukemia cells in NOD/SCID mice. Carcinogenesis. 2015;36:115–121. doi: 10.1093/carcin/bgu223. [DOI] [PubMed] [Google Scholar]
- 79.Armstrong F, Brunet de la Grange P, Gerby B, et al. NOTCH is a key regulator of human T-cell acute leukemia initiating cell activity. Blood. 2009;113:1730–1740. doi: 10.1182/blood-2008-02-138172. [DOI] [PubMed] [Google Scholar]
- 80.Dail M, Wong J, Lawrence J, et al. Loss of oncogenic Notch1 with resistance to a PI3K inhibitor in T-cell leukaemia. Nature. 2014;513:512–516. doi: 10.1038/nature13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cullion K, Draheim KM, Hermance N, et al. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood. 2009;113:6172–6181. doi: 10.1182/blood-2008-02-136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rakowski LA, Lehotzky EA, Chiang MY. Transient Responses to NOTCH and TLX1/HOX11 Inhibition in T-Cell Acute Lymphoblastic Leukemia/Lymphoma. PLoS One. 2011;6:e16761. doi: 10.1371/journal.pone.0016761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Palomero T, Sulis ML, Cortina M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Herranz D, Ambesi-Impiombato A, Sudderth J, et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med. 2015;21:1182–1189. doi: 10.1038/nm.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rakowski LA, Garagiola DD, Li CM, et al. Convergence of the ZMIZ1 and NOTCH1 pathways at C-MYC in acute T lymphoblastic leukemias. Cancer Res. 2013;73:930–941. doi: 10.1158/0008-5472.CAN-12-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Medyouf H, Gao X, Armstrong F, et al. Acute T-cell leukemias remain dependent on Notch signaling despite PTEN and INK4A/ARF loss. Blood. 2010;115:1175–1184. doi: 10.1182/blood-2009-04-214718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gutierrez A, Pan L, Groen RW, et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J Clin Invest. 2014;124:644–655. doi: 10.1172/JCI65093. [DOI] [PMC free article] [PubMed] [Google Scholar]