

Following injury to the peripheral nerve, pain secondary to low threshold innocuous mechanical stimulation (mechanical allodynia) is commonly observed. Its origins are poorly understood and the pain is often refractory to common analgesic therapeutics. The past two decades has brought major advances in our understanding of the complexity of systems that underlie processing of neuropathic pain (pain arising from damage or disease of the somatosensory system). Beginning with the argument by Robert Galambos in the 1960s that glia played a major regulatory role in neural function, current thinking has evolved to emphasize the major role of glia-mediated neuroinflammation in the development of a chronic pain state. Recent use of unbiased high-throughput genetic and proteomics screening technology in preclinical models of nerve injury associated with pain states, attention has become focused on the role of T cell related genes and adaptive immune pathways (Costigan et al., 2009; Liu et al., 2012). In this perspective, we discuss the organizing principle that following local nerve injury, the ability to evoke a pain state with low-threshold, otherwise innocuous, mechanical stimulation (light touch), represents a local, sex dependent, manifestation of an adaptive immune response mediated by myelin basic protein (MBP).
Adaptive immunity in neuropathic pain: Following peripheral nerve injury, over hours to days, there is a large scale in migration of a variety of immune competent cells into the injury site, the dorsal root ganglion and spinal segments receiving projections from the injured axon. Among these are T-cells, which, though they perform an ongoing immunosurveillance, are among the last immune cell types to infiltrate the damaged nerve, with their presence being seen after a week (Moalem et al., 2004; Cao and DeLeo, 2008; Costigan et al., 2009). Extensive work by multiple laboratories has confirmed the essential role played by T cells in the development of the neuropathic pain phenotype. i) T cell-deficient animals, including CD4 null, recombination activating gene-1 null, interleukin (IL)-17 null mice and athymic nude rats, display significantly diminished neuropathic pain states after a peripheral nerve injury. ii) Adoptive transfer of CD4+ T helper (Th) cells and transgenic animal models generally implicate Th1 and Th17 cells, expressing algesic/pro-inflammatory cytokines in the development of neuropathic pain. Importantly, while adaptive immunity and T cell migration play a pivotal role in the expression of this neuropathic phenotype, the antigens initiating this Th cell activity after nerve injury are poorly characterized. Recent work, reviewed below, indicate the likely role of myelin sheath auto-antigens in initiating this activation of adaptive immune cascade leading to the neuropathic state.
Aβ-pain and myelin sheath auto-antigens: Unlike other pain states, mechanical allodynia engage large-diameter Aβ-afferents, which are activated by low-threshold, typically innocuous, non aversive, mechanical stimuli. Aβ-afferents, are enveloped by Schwann cells, producing a multilamellar myelin sheath that enables neurons to conduct with high velocity. Breakdown in myelin resulting in desheathing of the axon has been considered as a factor in neuropathic pain. Specifically, demyelination is associated with increased expression, trafficking and insertion of voltage gated ion channels in the demyelinated axons, trophic changes in DRG protein expression and transynaptic changes in dorsal horn function. These changes correlate with ectopic Aβ-afferent firing, an enhanced driving of dorsal horn neurons, and an increased response to evoked afferent input (Devor, 2009). It is now appreciated that the myelin sheath disguises major auto-antigens. Among these is MBP.
MBP, a pivotal auto-antigen: MBP is an intrinsically unstructured and positively charged protein responsible for adhesion and compaction of myelin membranes into a sheath. Its sequence contains a number of cryptic T cell epitopes (Boggs, 2006). Several converging lines of work emphasize the importance of these cryptic sequences. i) Liberated by proteolysis, the MBP epitopes, including the 69–86 and 84–104 sequence regions, contribute to autoimmune demyelination in several clinical syndromes, including Guillain-Barré syndrome and multiple sclerosis. ii) In animals, injection of these peptide sequences serve as immunization antigens which model these neurodegenerative conditions. iii) Using the antibody to the degraded MBP69–86 (AB5864, Millipore), we found that these epitopes are released in models of mononeuropathy associated with focal trauma to peripheral nerve (Liu et al., 2012). iv) Injection of certain immunodominant MBP epitopes, including MBP69–86 and MBP84–104, but not MBP2–18, into the intact peripheral nerve, without an adjuvant, is sufficient to initiate robust and long-lasting (up to >30 day) mechanical allodynia (Liu et al., 2012; Ko et al., 2016). v) Using athymic nude rats, we confirmed that the MBP peptides require T cell activity to maintain their algesic action (Liu et al., 2012).
An important observation is that, MBP84–104 injection into the mixed (motor and sensory) sciatic nerve induces mechanical allodynia without affecting heat sensitivity or motor functions (Liu et al., 2012; Ko et al., 2016). This specific effect likely arises from changes in the afferent signaling in mechanoselective, Aβ afferents (Devor, 2009). The absence of a change in thermal escape latencies after intra-sciatic MBP84–104 injection or in major inflammatory cytokine expression compared with the scrambled peptide and failure of systemic COX inhibitor (ketorolac) or sodium channel blocker (lidocaine) to attenuate MBP-induced allodynia (Ko et al., 2016) imply that allodynia is not secondary to a simple sensitization of C polymodal nociceptors. It has been widely appreciated that injury leading to demyelination of Aβ-afferents can result in local changes leading to ectopic activity arising from the injury site, perhaps secondary to local insertion of ion channels as well as changes within the dorsal root ganglion leading to altered ion channels, gap junctions and receptors in the distal cell body and glia. These changes result in an engagement of Aβ-fibers in the pain circuitry.
Sexual dimorphism in mechanical allodynia: Neuroinflammation has been the focus of chronic pain research for several decades, but only recently have sex differences in innate and adaptive immunity been considered distinguishably contributory. Studies on TLRs have shown that after tissue inflammation and nerve injury deficiencies in spinal TLR4 and microglial signaling will reduce the onset of allodynia in males, but not females (Stokes et al., 2013; Sorge et al., 2015). Conversely, following nerve injury, female mice develop pain hypersensitivity via adaptive immune signaling, likely mediated by T lymphocytes. Thus, T cell deficient females, but not males, fail to develop tactile sensitivity. Our studies conducted in females corroborate these findings that hypersensitivity to mechanical stimulation in females involves T cell-mediated signaling and evades conventional neuroinflammation circuitry engaging microglia (Sorge et al., 2015). We suggest that aberrant T cell function relates to changes in large A-afferent function. While myelin disruption occurs in males and females, the female-specific changes may include production, processing, transport or function of the auto-antigens, as well as mechanisms of polarization or integration of myelin-specific T cells into the dorsal horn pain networks, among others. In addition, the presence of suppressive mechanisms preventing T cell integration into pain processing networks in males is conceivable.
We believe that microglia- and T cell-mediated mechanisms of mechanical allodynia are not mutually exclusive and likely contribute to pain circuitry in both sexes to varying degrees, depending on the site of damage, species, genetic background and other factors. In support of this conclusion, loss of adaptive immunity in the T cell-deficient female reveals microglial functionality in the hyperpathic state (Sorge et al., 2015). Conversely, multiple studies that established important T cell roles in neuropathic pain employed male animals. Since MBP84–104 and scrambled peptide induce most of the tested inflammatory cytokines at comparable levels, yet only MBP84–104 produces allodynia, and as this allodynia is refractory to common anti-inflammatory therapy, microglia-independent or -silencing mechanisms by which adaptive cells establish pain circuitry likely exist in females.
Clinical implications: Women more commonly appear afflicted with persistent and chronic pain and autoimmune disorders. The discovery of a sexual dimorphism in immunology of mechanical pain hypersensitivity has far-reaching implications for its basic understanding and for identifying T cell activity as a strategic focus in its therapeutic targeting in females. Accordingly, tactile allodynia in females is susceptible to pharmacological block of both MBP epitope release and immune cell infiltration using matrix metalloproteinase (MMP) inhibitors (Liu et al., 2012) or IL-6 blockage of the algesic MBP activity (Ko et al., 2016). Because T cells gain access into the site of inflammation via the integrin α4β1 binding to the CS1 isoform of fibronectin, interference with this binding using acute CS1 peptide therapy diminishes the peripheral nervous system (PNS) levels of T cell infiltration and mechanical allodynia (Liu et al., 2015). Our data further implies that targeting algesic MBP activity could be considered when broad-spectrum anti-inflammatory (ketolorac), lidocaine and other algesic therapeutics are found to be ineffective (Ko et al., 2016). In a PNS trauma or compression model of sciatic nerve chronic constriction injury, where the algesic MBP peptides are released (Liu et al., 2012), the use of MBP-derived mutant/altered peptide ligand analogues impairing aberrant T cell function attenuates allodynia (Perera et al., 2015).
We further suggest that the algesic MBP epitopes contribute to human pain states associated with whole body autoimmune demyelinating conditions, such as Guillain-Barré syndrome and multiple sclerosis, where these epitopes are reportedly released. Since we find the algesic MBP peptides released in the absence of widespread demyelination (Liu et al., 2012), MBP and other myelin auto-antigens may contribute to pain from mechanical stimulation in subjects suffering from idiopathic neuropathies unaccompanied by evident demyelination. Further, we propose that due to the established MBP homology to human Epstein-Barr virus (EBV), the most common human virus, and to additional subtypes of the herpes simplex virus (HSV) family members, MBP-related algesic pathways may underlie the mechanisms of post-herpetic neuralgia and other idiopathic neuropathies accompanied by mechanical allodynia.
Conclusion: We here argue that mechanical allodynia after physical local nerve injury has mechanistic features of an autoimmune disorder initiated by the liberation of immunodominant, pro-nociceptive epitopes from the myelin sheath of mechanically selective afferents, promoting T cell homing to low-threshold Aβ fibers after PNS injury. The robust and lasting MBP-induced mechanical allodynia may exist without widespread inflammation or involvement of conventional nociceptive circuitry. In this scenario, unmyelinated polymodal C-afferents may be spared from T cell homing, as illustrated in Figure 1. Following the nerve injury-induced release of immunodominant MBP peptides by proteases, such as MMPs, major histocompatibility complex (MHC) class II presents the MBP epitopes on a surface of antigen presenting cells, including Schwann cells (Liu et al., 2012) and the MBP·MHC II complex guides the T cell receptor (TCR) binding (Boggs, 2006). This model does not exclude an important role of additional antigens and factors in T cell recruitment, polarization and activity in pain. In the segmental spinal cord, where Aβ primary afferent axons terminate within an area extending from the inner part of lamina II to lamina V, intra-sciatic administration of the algesic MBP peptides initiates adaptive immune, vitamin D receptor and IL-17 signaling pathways (Liu et al., 2012). Although IL-17 expression is typically restricted to Th17 cells, including that in the damaged nerve, others have shown that the infiltrating spinal cord CD4+ cells after peripheral nerve injury display primarily a Th1 phenotype (Draleau et al., 2014). In light of the ability of myelinating glia to regulate IL-17 expression, the possibility of Schwann cell and oligodendrocyte lineage taking on a role in adaptive pain signaling should be considered. While maintenance of the algesic MBP activity relies on T cell function (Liu et al., 2012), the mechanisms initiating MBP-induced pain preceding T cell recruitment are still under investigation.
Figure 1.
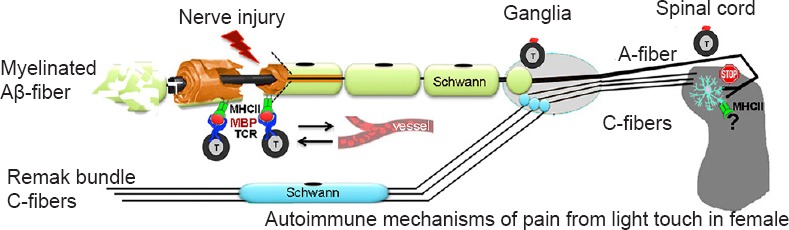
Autoimmune nature of pain from mechanical stimulation in females (a hypothesis diagram).
The myelin sheath of large mechanosensory Aβ-afferents contains auto-antigens, such as myelin basic protein (MBP). After peripheral nervous system (PNS) injury, matrix metalloproteinases (MMPs) and other proteases release the pro-nociceptive, cryptic T cell epitopes on myelin, normally sheltered from immunosurveillance. Schwann cells present MBP epitopes on the surface via major histocompatibility complex (MHC) class II (MHC II), promoting selective T cell homing to A-afferents (but not C-afferents) via T cell receptor (TCR). The repeated release and exposure of MBP epitopes and formation of MBP-specific T cell clones sustain the state of mechanical allodynia. In the spinal cord, activated microglia and adaptive, presumably, T cell signaling, are differentially active in females.
National Institutes of Health R01DE022757 (to VIS, AYS, TLY) supported this work.
References
- Boggs JM. Myelin basic protein: a multifunctional protein. Cell Mol Life Sci. 2006;63:1945–1961. doi: 10.1007/s00018-006-6094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, DeLeo JA. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur J Immunol. 2008;38:448–458. doi: 10.1002/eji.200737485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, Barrett L, Brenner GJ, Vardeh D, Woolf CJ, Fitzgerald M. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. 2009;29:14415–14422. doi: 10.1523/JNEUROSCI.4569-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M. Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp Brain Res. 2009;196:115–128. doi: 10.1007/s00221-009-1724-6. [DOI] [PubMed] [Google Scholar]
- Draleau K, Maddula S, Slaiby A, Nutile-McMenemy N, De Leo J, Cao L. Phenotypic identification of spinal cord-infiltrating CD4 T lymphocytes in a murine model of neuropathic pain. J Pain Relief Suppl. 2014;3:003. doi: 10.4172/2167-0846.S3-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JS, Eddinger K, Angert M, Dolkas J, Chernov AV, Strongin AY, Yaksh TL, Shubayev VI. Spinal activity of IL-6 mediates mechanical allodynia induced by myelin basic protein. Brain Behav Immun. 2016 doi: 10.1016/j.bbi.2016.03.003. doi:10.1016/j.bbi.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Shiryaev SA, Chernov AV, Kim Y, Shubayev I, Remacle AG, Baranovskaya S, Golubkov VS, Strongin AY, Shubayev VI. Immunodominant fragments of myelin basic protein initiate T cell-dependent pain. J Neuroinflammation. 2012;9:119. doi: 10.1186/1742-2094-9-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Dolkas J, Hoang K, Angert M, Chernov AV, Remacle AG, Shiryaev SA, Strongin AY, Nishihara T, Shubayev VI. The alternatively spliced fibronectin CS1 isoform regulates IL-17A levels and mechanical allodynia after peripheral nerve injury. J Neuroinflammation. 2015;12:158. doi: 10.1186/s12974-015-0377-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalem G, Xu K, Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience. 2004;129:767–777. doi: 10.1016/j.neuroscience.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Perera CJ, Duffy SS, Lees JG, Kim CF, Cameron B, Apostolopoulos V, Moalem-Taylor G. Active immunization with myelin-derived altered peptide ligand reduces mechanical pain hypersensitivity following peripheral nerve injury. J Neuroinflammation. 2015;12:28. doi: 10.1186/s12974-015-0253-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 2015;18:1081–1083. doi: 10.1038/nn.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes JA, Cheung J, Eddinger K, Corr M, Yaksh TL. Toll-like receptor signaling adapter proteins governs spread of neuropathic pain and recovery following nerve injury in male mice. J Neuroinflammation. 2013;10:148. doi: 10.1186/1742-2094-10-148. [DOI] [PMC free article] [PubMed] [Google Scholar]