

Peripheral neuropathies refer to a group of conditions in which the peripheral nervous system (PNS) is damaged. These pathological state are are associated with weakness, pain, and loss of motor and sensory control. More than 100 types of peripheral neuropathies have been identified, with distinct symptoms and prognosis classified according to the type of damage to the nerves. Injury to peripheral nerves results in disabling loss of sensory and motor functions. Damaged axons undergo degeneration distal to the injury and regeneration from the proximal stump, a fundamental process for reinnervation and functional recovery. In contrast, damage to the central nervous system (CNS) is followed by poor regeneration. In the PNS, nerve injury triggers a response known as Wallerian degeneration, characterized by axonal damage due to an increase in axoplasmic calcium, mitochondrial dysfunction and cytoskeleton breakdown (Court and Coleman, 2012). Moreover, Schwann cells (SCs) dedifferentiate to a regenerative cell phenotype, characterized by a proliferative state, the secretion of trophic factors, and the organization into a columnar cell configuration known as bands of Bungner, which guide regenerating axons. Also, SCs participate in myelin and axonal breakdown and secrete cytokines and chemokines to recruit immune cells (i.e., macrophages) into the nerve that eliminate cell debris. In addition, axotomized neurons upregulate regeneration-associated genes (RAGs) to promote axon growth. By contrast, axonal damage in the CNS is followed by limited myelin clearance and activation of astrocytes, which secrete growth-inhibitory molecules that generate an unfavorable environment for axonal regeneration. Therefore, successful axonal repair depends on intrinsic capacities of neurons and the reaction of glial cells and microenvironmental factors that modulate the regeneration process.
The endoplasmic reticulum (ER) is a dynamic interconnected network involved in quality control processes that maintain a functional proteome in the cell. The ER contributes to local calcium homeostasis, lipid synthesis and translation, folding and secretion of proteins, among other metabolic functions. Accumulating evidence indicates that CNS and PNS injury alters ER proteostasis engaging a stress reaction in neurons and glial cells (Li et al., 2013; Hetz and Mollereau, 2014). ER stress activates an adaptive mechanism to cope with protein folding alterations, known as the unfolded protein response (UPR). Under irreversible or chronic ER stress, apoptosis is induced to eliminate compromised cells. The UPR is initiated by selective activation of downstream cascades mediated by three UPR sensors, including IRE1α, PERK, and ATF6 (Hetz et al., 2015). IRE1α is a kinase and endoribonuclease that upon activation processes the mRNA encoding the transcription factor XBP1, shifting the coding reading frame of the mRNA to translate XBP1s, an active transcription factor. XBP1s controls genes involved in protein folding, secretion, lipid synthesis and ER-associated degradation (ERAD), among other functions (Hetz et al., 2015). Activation of PERK leads to the direct phosphorylation of translation initiation factor eIF2α, reducing protein translation into the ER. eIF2α phosphorylation allows the selective translation of the mRNA encoding the transcription factor ATF4, which controls genes involved in antioxidant responses, protein folding, metabolism and autophagy (Hetz et al., 2015). Under chronic ER stress conditions, ATF4 activates a pro-apoptotic program mediated in part by the upregulation of CHOP. Finally, upon ER stress, ATF6 translocates to the Golgi, where it is proteolytically processed to release a cytosolic fragment that regulates genes involved in ERAD. Therefore, the UPR integrates information about the intensity and duration of the stress stimuli to orchestrate adaptive or pro-apoptotic mechanisms, determining cell fate.
ER stress has emerged as an important event driving neurodegeneration in pathological conditions of the CNS and PNS (reviewed in Li et al., 2013; Hetz and Mollereau, 2014). Axonal damage to the PNS triggers the UPR in neurons and glial cells. For example, damage to the sciatic nerve induces the activation of specific components of the UPR in motoneurons, including the expression of XBP1s and ATF4 (Penas et al., 2011). Axonal damage also triggers a robust activation of the UPR in sensory neurons of the dorsal root ganglia (DRG) (Ying et al., 2015) and in dedifferentiated SCs (Mantuano et al., 2011). The upregulation of several ER foldases, including ERp57 and BiP, is observed after peripheral nerve injury. At the functional level, we recently reported that the overexpression of ERp57 in neurons accelerates peripheral nerve regeneration (Castillo et al., 2015). A recent study also demonstrated that injury to the sciatic nerve leads to the upregulation of ER stress markers through the expression of the ATF6 orthologue LUMAN/CREB3 in the axonal compartment. This pathway involves the retrograde transport of the cytosolic domain of LUMAN/CREB3 to the neuronal soma, serving as a locally translated injury signal to regulate axonal growth (Ying et al., 2015). In summary, accumulating evidence demonstrates activation of the UPR after peripheral nerve damage in glial cells and neurons, however functional studies were still missing to define the actual contribution of the UPR to axonal degeneration and regeneration.
We recently investigated the impact of the UPR to peripheral nerve regeneration. Using genetic manipulation, we studied the consequences of targeting ATF4 and XBP1 to assess the impact of the UPR to Wallerian degeneration after sciatic nerve damage. Consistent with previous observations reporting a lack of clear activation of the PERK/ATF4 branch after sciatic nerve injury (Mantuano et al., 2011), we reported that Atf4 deficiency does not alter the course of Wallerian degeneration, regeneration and functional recovery (Oñate et al., 2016). In sharp contrast, deletion of Xbp1 in the nervous system led to decreased myelin clearance, axonal regeneration and macrophage infiltration after mechanical damage (Oñate et al., 2016). Importantly, locomotor recovery in Xbp1 deficient mice was significantly delayed. Furthermore, overexpression of XBP1s in neurons using a transgenic mice increased axonal regeneration and locomotor recovery after injury (Oñate et al., 2016). We moved forward and developed a therapeutic strategy to artificially engage XBP1-dependent gene expression programs to enhance axonal repair. We validated a gene transfer approach to deliver XBP1s into sensory axons using adeno-associated viruses (AAVs). AAV-XBP1s transduced neurons showed an enhancement in the axonal regeneration process (Oñate et al., 2016). Altogether, these results demonstrated a differential contribution of the IRE1α/XBP1 signaling branch of the UPR in the injured PNS.
We speculate that the local activation of UPR stress sensors in the axonal compartment after damage may trigger the retrograde transport of active XBP1s to the cell soma to engage transcriptional programs that contribute to alleviate proteostasis alterations similar to the model described for LUMAN/CREB3 (Ying et al., 2015). In addition to cell-autonomous responses in the damaged neuron, changes in the local environment and surrounded cells may also involve ER stress signals. In fact, we observed that the SCs-dependent upregulation of the chemokine MCP-1 is reduced in XBP1 deficient animals, correlating with lower infiltration of macrophages and delayed clearance of myelin debris (Oñate et al., 2016). It remains to be determined if XBP1s controls the expression of factors involved in axonal regeneration, including neurotrophic factors and RAGs. Of note, we recently reported that XBP1s upregulates the levels of brain-derived neurotrophic factor (BDNF) in the hippocampus, enhancing synaptic function, in addition to improve the learning and memory capacity of mice (Martínez et al., 2016). BDNF is known to modulate neuronal survival and axonal growth, which may contribute to the axonal regeneration process engaged by XBP1s in the PNS. Overall, our study provided the first functional evidence indicating that the UPR, and specifically XBP1s and not ATF4, contributes to Wallerian degeneration through cell intrinsic and cell-nonautonomous effects, modulating axonal regeneration and locomotor recovery after peripheral nerve injury (Figure 1). These observations fully contrast with our previous studies showing that mechanical damage to the CNS in models of spinal cord injury (SCI) engages both the ATF4 and XBP1 pathways, attenuating the adverse effects over motor function (Valenzuela et al., 2012). In the SCI model, activation of the UPR reduced tissue damage possibly by enhancing the survival of oligodendrocytes, suggesting differential effects of UPR between PNS and CNS.
Figure 1.
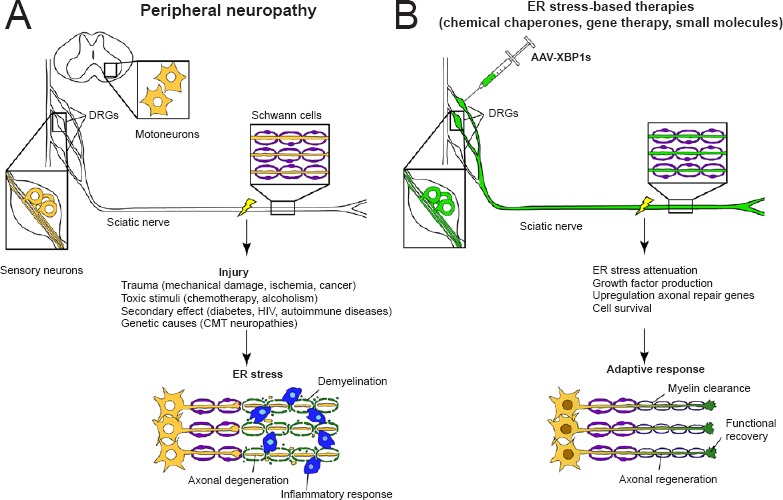
Peripheral neuropathies and endoplasmic reticulum (ER) stress-mediated therapies.
(A) Different neuropathies are associated with ER stress contributing to axonal degeneration. (B) Targeting ER stress may promote axonal regeneration. Strategies to alleviate ER stress may include the use of chemical chaperones and unfolded protein response enhancers (gene therapy or small molecules).
Other studies have reported the possible contribution of the UPR to peripheral nerve pathologies. In models of hereditary demyelinating conditions of the PNS known as Charcot-Marie-Tooth (CMT) diseases, characterized by accumulation of myelin misfolded proteins in the ER of SCs, genetic and pharmacological targeting of the UPR leads to an attenuation of the neurodegenerative process (reviewed in Clayton and Popko, 2016). Peripheral neuropathies are triggered by a large spectrum of conditions, representing a serious public health problem. Only in the United States it is estimated that 20 million people are affected with peripheral neuropathies. Based on the studies discussed here, the next step in the field is to determine if the UPR has therapeutic potentials in other conditions involving peripheral nerve damage including autoimmune diseases, small vessel disease, cancer, kidney disorders, neuromas, viral infections, and diabetes mellitus, or the exposure to environmental toxins and cytotoxic medicines such as chemotherapy (Figure 1). Overall, modulation of axonal regeneration programs by the UPR incorporates novel players in the process of nerve repair after mechanical damage. Since several small molecules and gene therapy strategies are available to target the UPR, manipulation of the ER proteostasis network might emerge as a new avenue to develop interventions that improve axonal regeneration in different degenerative conditions of the nervous system.
This work is funded by FONDAP program 15150012 (to CH and FAC), Millennium Institute, No. P09-015-F, the Frick Foundation 20014-15, ALS Therapy Alliance 2014-F-059, Muscular Dystrophy Association 382453, CONICYT-USA 2013-0003, Michael J Fox Foundation for Parkinson΄s Research – Target Validation grant No. 9277, COPEC-UC Foundation 2013.R.40, Ecos-Conicyt C13S02, FONDECYT No. 1140549, Office of Naval Research-Global (ONR-G) N62909-16-1-2003 and ALSRP Therapeutic Idea Award AL150111 (to CH), Millennium Nucleus-P-07-011-F, FONDECYT, No. 1110987 (to FAC), and PhD fellow supported by CONICYT, No. 21130843 (to MO).
References
- Castillo V, Oñate M, Woehlbier U, Rozas P, Andreu C, Medinas D, Valdés P, Osorio F, Mercado G, Vidal RL, Kerr B, Court FA, Hetz C. Functional role of the disulfide isomerase ERp57 in axonal regeneration. PLoS One. 2015;10:e0136620. doi: 10.1371/journal.pone.0136620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton BL, Popko B. Endoplasmic reticulum stress and the unfolded protein response in disorders of myelinating glia. Brain Res. 2016 doi: 10.1016/j.brainres.2016.03.046. S0006-8993(16)30180-30189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Coleman MP. Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 2012;35:364–372. doi: 10.1016/j.tins.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. 2015;17:829–838. doi: 10.1038/ncb3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- Li S, Yang L, Selzer ME, Hu Y. Neuronal endoplasmic reticulum stress in axon injury and neurodegeneration. Ann Neurol. 2013;74:768–777. doi: 10.1002/ana.24005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantuano E, Henry K, Yamauchi T, Hiramatsu N, Yamauchi K, Orita S, Takahashi K, Lin JH, Gonias SL, Campana WM. The unfolded protein response is a major mechanism by which LRP1 regulates Schwann cell survival after injury. J Neurosci. 2011;31:13376–13385. doi: 10.1523/JNEUROSCI.2850-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez G, Vidal RL, Mardones P, Serrano FG, Ardiles AO, Wirth C, Valdés P, Thielen P, Schneider BL, Kerr B, Valdés JL, Palacios AG, Inestrosa NC, Glimcher LH, Hetz C. Regulation of memory formation by the transcription factor XBP1. Cell Rep. 2016;14:1382–1394. doi: 10.1016/j.celrep.2016.01.028. [DOI] [PubMed] [Google Scholar]
- Oñate M, Catenaccio A, Martínez G, Armentano D, Parsons G, Kerr B, Hetz C, Court FA. Activation of the unfolded protein response promotes axonal regeneration after peripheral nerve injury. Sci Rep. 2016;6:21709. doi: 10.1038/srep21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penas C, Font-Nieves M, Forés J, Petegnief V, Planas A, Navarro X, Casas C. Autophagy, and BiP level decrease are early key events in retrograde degeneration of motoneurons. Cell Death Differ. 2011;18:1617–1627. doi: 10.1038/cdd.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela V, Collyer E, Armentano D, Parsons GB, Court FA, Hetz C. Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death Dis. 2012;3:e272. doi: 10.1038/cddis.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Zhai R, McLean NA, Johnston JM, Misra V, Verge VMK. The unfolded protein response and cholesterol biosynthesis link luman/CREB3 to regenerative axon growth in sensory neurons. J Neurosci. 2015;35:14557–14570. doi: 10.1523/JNEUROSCI.0012-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]