

Mitochondria, the powerhouses of the cell, are dynamic organelles that constantly move and change their size and morphology to meet energetic demands of the cell (Wai and Langer, 2016). Recent studies demonstrated that energy production is dependent on the ability of mitochondria to undergo cycles of fission and fusion collectively termed “mitochondrial dynamics” (Youle and van der Bliek, 2012). Fidelity of mitochondrial dynamics is especially important in neuronal cells where mitochondria traverse distances over one meter long to deliver energy to the distant parts of the cell for proper synaptic development, maintenance, and activity, and ultimately neuronal survival. Excessive mitochondrial fission has been associated with multiple neurodegenerative diseases including Alzheimer's disease (AD) (Reddy et al., 2011). Based on these studies, modulation of mitochondrial dynamics has been considered as novel therapeutic approach that could be beneficial for multiple human conditions (Reddy, 2014).
Mitochondria vary in size from 0.5 to 10 µm long depending on the tissue and location within the cell. Each mitochondrion has a complicated three-dimensional architecture where the outer membrane surrounds multiple membrane folds or cristae formed by the inner membrane that incorporate components of the electron transport chain (ETC). Fission and fusion machinery depends on the fidelity of multiple proteins including dynamin related protein 1 (Drp1), mitochondrial fission factor, mitochondrial fission protein 1, mitofusin-1 and mitofusin-2, and optical atrophy 1 (Dhingra and Kirshenbaum, 2014). These proteins also regulate the assembly and stability of the ETC inducing the remodeling of mitochondrial cristae and ultimately shaping mitochondrial morphology in response to the energetic demand of the cell. Mitochondrial fission includes multiple steps and is facilitated by phosphorylation of Drp1 at Serine position 616 (S616), which promotes its translocation to the mitochondria. Activated Drp1 assembles on the mitochondrial outer membrane, and then constricts it in the GTP-dependent manner (Chang and Blackstone, 2010). Mitochondrial fission is required for equal segregation of mitochondria during cell division, their enhanced distribution in the cell along microtubules, and to promote degradation of defective mitochondria via mitophagy. However, excessive fission has been linked to apoptosis and neuronal death.
While changes in mitochondrial morphology associated with fission affect the entire organelle, methods utilized to date to monitor mitochondrial dynamics primarily include fluorescence microscopy, which has relatively poor spatial resolution, or/and conventional electron microscopy (EM), which reveals morphological changes in a thin section of the organelle. Moreover, very limited data is available on mitochondrial dynamics in the brain tissue where mitochondrial morphology could be assayed in the context of complex 3D environment.
Formation of mitochondria-on-a-string (MOAS) in animal models of AD and AD patients: To account for a three-dimensional architecture of the brain tissue and organelles, we applied 3-dimensional electron microscopy (3D EM) reconstruction of standard transmission electron microscopy (TEM) images from 10–40 consecutive serial sections (0.1 µm thick) from patients and multiple mouse models of familial AD (FAD) (Zhang et al., 2016). We examined mitochondria in the CA1 hippocampal region from 5 transgenic mouse models carrying human FAD mutations for presenilin 1 (PS1), amyloid precursor protein (APP), and mutant Tau protein. Mutations in PS1 and APP inevitably lead to the development of AD, and the expression of these proteins in mice allows recapitulating some of the mechanisms of human disease. We used non-transgenic (NTG) littermates as control. While examining brain tissue from FAD mice, instead of observing multiple small mitochondria produced by excessive fission previously reported for AD, we identified an unknown mitochondrial phenotype that we termed “MOAS” (Figure 1B, red). MOAS represented elongated interconnected organelles where multiple teardrop shaped mitochondria (~0.5 µm in diameter) were connected by thin double membranes extending up to 5 µm long. This unusual mitochondrial structure could be easily mistaken for multiple individual organelles if the membrane connections are missed (Figure 1A, asterisks). We characterized several MOAS phenotypes where mitochondrial matrix was either present or absent from the membrane connections. The long (> 200 nm) thin double membrane connections seen in MOAS differ significantly in appearance from the short (~100 nm) uniform diameter (50–65 nm) membranes seen in normally dividing mitochondria (~0.3 µm in diameter).
Figure 1.
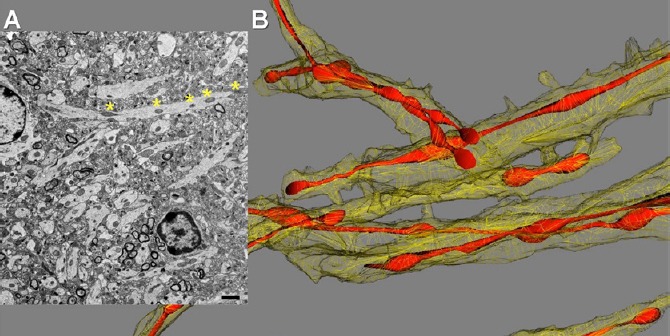
Mitochondrial morphology in the hippocampal brain tissue of APP/PS1 mouse visualized with 2D TEM and 3D EM reconstruction.
(A) Transmission electron microscopy (TEM) of the brain tissue (0.1 μm thick) revealed mitochondria in neuropils as multiple individual teardrop shaped organelles. Asterisks define parts of mitochondria-on-a-string (MOAS) that could be visible only using 3D EM reconstruction. Scale bar: 1 μm. (B) 3D EM reconstruction of the same brain tissue as in A done using 40 consecutive brain slices (0.1 μm thick) revealed a predominant MOAS phenotype.
We found extensive MOAS formation in all FAD mouse models examined. MOAS were most prevalent in mouse models with multiple FAD mutations such as APP/PS1 and 3xTgAD mice. The amount of MOAS was also significantly increased with age. Thus, at 40 and 60 weeks of age the amount of MOAS in brain cells reached 50-70% while almost no MOAS were found in age-matched NTG mice. Thus, the investigation of intact brain tissue of FAD mice using 3D EM reconstruction revealed the presence of mitochondria with novel MOAS phenotype as opposed to excessive fragmentation reported in studies using low-resolution imaging methods.
While animal models represent an informative tool to study AD, they have limitations. We next examined whether MOAS was relevant to human disease. It is well known that postmortem brain tissue undergoes rapid decomposition where mitochondrial morphology is severely affected. To overcome this, we assessed hippocampal, cortical, and cerebellar postmortem tissue (fixed within 5 to 24 hours after death) from cognitively normal individuals and AD patients. We did not observe MOAS in the cerebella, a brain region that is not affected in AD, of control and AD patients where mitochondria were uniformly oval. However, the examination of the serial sections of CA1 hippocampal brain region, which is early and specifically affected in AD, revealed MOAS structures only in AD patients. These structures were similar to the observed in FAD mice suggesting that MOAS formation has relevance to a human disease condition.
Molecular mechanism of MOAS formation: All observations regarding MOAS were done using mouse or human brain tissue. However, brain consists of various neuronal and non-neuronal cells, making the evaluation of the molecular mechanism of MOAS formation difficult. To overcome this, we conducted a detailed examination of the molecular mechanism of MOAS formation in isolated brain mitochondria and primary neurons from wild type (WT) and FAD mice using pharmacological inhibitors, 3D EM reconstruction, super-resolution fluorescence microcopy, western blot analysis, flow cytometry, and time lapse imaging of mitochondrial dynamics in live cells. Our findings indicated that MOAS formation was not associated with a system-wide loss of the expression of fission/fusion proteins or significant changes in mitochondrial mass in the brain of FAD mice. We next examined whether MOAS was associated with the activation of fission machinery, which could be assayed by the levels of activated Drp1 S616. We found a significant increase in Drp1 S616 phosphorylation in whole brain extracts and in the enriched mitochondrial fractions isolated from the brain tissue of FAD mice. These observations were confirmed with three independent methods including Western blot analysis of the brain extracts, flow cytometry determination of the proteins specifically associated with the isolated brain mitochondria, and super-resolution fluorescence microscopy in intact FAD brain tissue. Increased S616 phosphorylation of Drp1, its translocation to hippocampal mitochondria and assembly was observed in animals with most pronounced MOAS phenotype.
After Drp1 assembles on mitochondria, the next step involvesa GTP-dependent membrane fission event. Abnormal energy homeostasis is well documents in AD. Along with hypometabolism, chronic hypoxia and energetic stress are linked to the accumulation of Aβ in brain blood vessels in AD (Zhang and Le, 2010). Since the recruitment and translocation of Drp1 to mitochondria was enhanced in FAD hippocampi, we examined whether formation of MOAS was associated with bioenergetic changes characteristic of AD brain. Indeed, acute energy deprivation induced by hypoxic conditions promoted rapid (within 4 minutes) and excessive (~80% of the neuropils examined in hippocampi) MOAS formation in the brain tissue of young wild type (WT) mice, which was confirmed using 3D EM reconstruction and super-resolution fluorescence microscopy (Figure 2A). Moreover, while Drp1 was discontinuously distributed along WT mitochondria, it formed rings localized to the regions of constriction along hypoxia-induced MOAS suggesting normal Drp1 recruitment but incomplete fission. Furthermore, we have found that chronologically aged WT animals (88 weeks) had increased number of MOAS compared to younger WT mice regardless of hypoxia exposure consistent with age-related decline in brain energetics (Figure 2B). Collectively, these observations suggested that energy depletion induced by hypoxia (Zhang and Le, 2010) or associated with chronological aging (Lopez-Otin et al., 2013) or AD rather than aberrant Drp1 recruitment contributed to MOAS formation.
Figure 2.
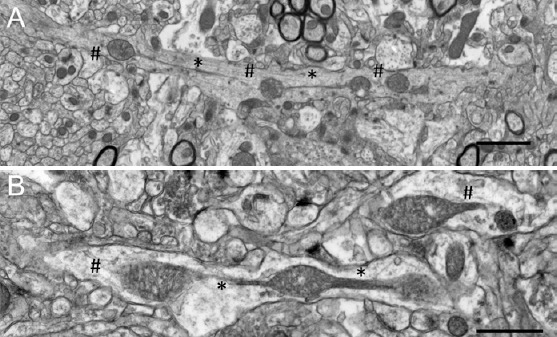
Conditions associated with energy failure including hypoxia or advanced age induce mitochondria-on-a-string (MOAS) formation in the brain tissue of wide-type (WT) mice.
(A) Exposure of a WT mouse 10 weeks of age to CO2 for 5 minutes produced MOAS observed by transmission electron microscopy (TEM) of the hippocampal brain tissue (serial section 0.1 μm thick). Scale bar: 2 μm. (B) MOAS in the hippocampus in a WT mouse 88 weeks of age sacrificed by cervical dislocation. Scale bar: 1 μm. * Depicts MOAS connections visible using 2D TEM; # depicts parts of MOAS that could be accurately defined only using 3D EM reconstruction.
We tested this hypothesis by using time-laps video microscopy to observe mitochondrial dynamics in live primary mouse cortical neurons from WT and FAD mice, and in WT neurons treated with 15-deoxy-Δ12,14-Prostaglandin J2 (PGJ2), a known GTPase inhibitor (Mishra et al., 2010). In WT untreated neurons, mitochondrial fission occurred rapidly, within ~15 seconds, following the earliest indication of MOAS formation. In contrast, in neurons from FAD mice, mitochondria exhibited a marked fission delay (> 2 minutes). Treatment with PGJ2 did not affect mitochondrial movement, but generated persistence (> 5 minutes) of MOAS. These observations support the hypothesis that inhibition of Drp1 GTPase activity contributes to fission arrest and formation of MOAS in healthy neurons.
Our studies reveal a novel mitochondrial phenotype that is relevant to multiple conditions including AD, hypoxia and chronological aging. Our data suggest that MOAS are formed during the final steps of the fission process associated with Drp1 GTP hydrolysis. While we demonstrated that altered energetics, well documented in FAD mice (Trushina et al., 2012) and AD patients (Rabinovici et al., 2010), are sufficient to cause fission arrest, other mechanisms including altered lipid homeostasis, increased production of PGJ2 by activated microglia and altered axonal trafficking associated with AD could contribute to MOAS formation. It also remains to be determined whether MOAS are beneficial or devastating to the cell. Mitochondrial fission is essential for quality control ensuring the removal of damaged organelles via mitophagy (Youle and van der Bliek, 2012) while highly fused mitochondria are resistant to degradation and certain forms of stress. While the details of molecular mechanisms, especially with respect to the involvement of other fission/fusion proteins, remain to be determined, it is feasible that MOAS formation allows mitochondria to sustain cell viability during energy deprivation as in case of AD or hypoxia contributing residual functions towards extending a protective energetic margin that plays a role in neuronal survival.
Taken together, our data reveals greater complexity of mitochondrial fission/fusion that includes sustained transition state dynamics. The occurrence of delayed fission during normal aging, hypoxia or neurodegenerative diseases provides compelling evidence for fundamental compensatory adaptation of mitochondria to bioenergetic stress. The discovery of novel mitochondrial phenotype in the brain tissue accurately detected only using 3D EM reconstruction argues for a major role of mitochondrial dynamics in regulating neuronal survival and emphasizes the importance of utilizing advanced 3D tools to study complex tissues and organelles.
I thank Trace Christensen for help with Figures and together with Drs. Sergey Trushin and Jeffrey L. Salisbury for comments on the paper. My apologies to all authors whose work could not be cited here due to page limitations. This work was partially supported by grants from NIEHS R01ES020715, ADDF 291204, BrightFocus A2011084, GHR Foundation, and NCATS UL1 TR000135.
References
- Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann N Y Acad Sci. 2010;1201:34–39. doi: 10.1111/j.1749-6632.2010.05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra R, Kirshenbaum LA. Regulation of mitochondrial dynamics and cell fate. Circ J. 2014;78:803–810. doi: 10.1253/circj.cj-14-0240. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N, Kar R, Singha PK, Venkatachalam MA, McEwen DG, Saikumar P. Inhibition of mitochondrial division through covalent modification of Drp1 protein by 15 deoxy-Delta(12, 14)-prostaglandin J2. Biochem Biophys Res Commun. 2010;395:17–24. doi: 10.1016/j.bbrc.2010.03.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Furst AJ, Alkalay A, Racine CA, O’Neil JP, Janabi M, Baker SL, Agarwal N, Bonasera SJ, Mormino EC, Weiner MW, Gorno-Tempini ML, Rosen HJ, Miller BL, Jagust WJ. Increased metabolic vulnerability in early-onset Alzheimer's disease is not related to amyloid burden. Brain. 2010;133:512–528. doi: 10.1093/brain/awp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J Alzheimers Dis. 2014;40:245–256. doi: 10.3233/JAD-132060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, Mao P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res Rev. 2011;67:103–118. doi: 10.1016/j.brainresrev.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, Nemutlu E, Zhang S, Christensen T, Camp J, Mesa J, Siddiqui A, Tamura Y, Sesaki H, Wengenack TM, Dzeja PP, Poduslo JF. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer's disease. PLoS One. 2012;7:e32737. doi: 10.1371/journal.pone.0032737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Trushin S, Christensen TA, Bachmeier BV, Gateno B, Schroeder A, Yao J, Itoh K, Sesaki H, Poon WW, Gylys KH, Patterson ER, Parisi JE, Diaz Brinton R, Salisbury JL, Trushina E. Altered brain energetics induces mitochondrial fission arrest in Alzheimer's disease. Sci Rep. 2016;6:18725. doi: 10.1038/srep18725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Le W. Pathological role of hypoxia in Alzheimer's disease. Exp Neurol. 2010;223:299–303. doi: 10.1016/j.expneurol.2009.07.033. [DOI] [PubMed] [Google Scholar]