

Neurovascular unit (NVU): Brain microvasculature has a close structural and functional relationship with brain parenchyma, an aspect governed by the NVU (Hermann and ElAli, 2012). The concept of it was introduced by the Stroke Progress Review Group who defined it as triad of neurons, glia (astrocytes and microglia), and adjacent vasculature (arterioles and capillaries) [The Stroke Progress Review Group (SPRG) 2002. Stroke Priorities for the 21st Century. http://www.ninds.nih.gov/about_ninds/groups/stroke_prg/sprg_overview.pdf]. More precisely, the NVU constitutes an integrative biological system comprising tightly sealed endothelial cells, forming the blood-brain barrier (BBB), that interact with extracellular matrix proteins, pericytes, astrocyte endfeet, microglia and neurons (Herman and ElAli, 2012). The NVU is a site of dynamic and complex biochemical and cellular interactions that work in concert to enable proper brain homeostasis and functioning (Hermann and ElAli, 2012). It maintains an optimal biochemical microenvironment suitable for neuronal function and viability by controlling neurovascular coupling, adjusting regional cerebral blood flow (rCBF) and regulating BBB function (Hermann and ElAli, 2012). Besides, the NVU constitutes an important checkpoint that shapes the adaptive and innate immune responses of the brain (Lampron et al., 2013). More precisely, pericytes and perivascular macrophages have an antigen presentation potential in adaptive immunity, whereas endothelial cells and microglia are richly endowed with innate immunity (Lampron et al., 2013).
Ischemic stroke: The homeostasis of the NVU is altered in several neurological diseases, namely stroke (Hermann and ElAli, 2012). Stroke is one of the leading causes of death and disability in the industrialized world. Ischemic stroke accounts up to 85% of stroke cases (Dirnagl, 2012). Ischemic stroke is caused by the sudden occlusion of a cerebral blood vessel, interrupting cerebral microcirculation in a specific region of the brain, thus initiating the ischemic cascade that leads to neuronal death (Dirnagl, 2012). Ischemic stroke challenges NVU function by causing BBB breakdown, BBB transport deregulation, extracellular matrix protein degradation, pericyte and astrocyte endfeet detachment in the acute phase (minutes to hours after ischemic stroke onset), thus jointly contributing to the secondary progression of brain injury, by increasing brain edema and exacerbating the inflammatory response in the sub-acute phase (hours to days after ischemic stroke onset) (Dirnagl, 2012). The severity of these early events reduces the capacity of neurons to recover in the chronic phase (days to weeks after ischemic stroke onset), thus significantly worsening stroke outcomes (Dirnagl, 2012). Importantly, ischemic stroke results in two major zones of injury, the core, which is constituted by dead tissues caused by necrosis, and the penumbra, which is constituted by damaged tissues undergoing programmed cell death (Lo, 2008). The slow progression of cell death in the penumbra implies that therapeutic salvage is possible. However, although significant progress has been made in stroke prevention and supportive care, still no disease-modifying therapy that aims at protecting injured brain tissue from death and promoting its recovery in the acute and sub-acute phases of ischemic stroke exists. Today, recombinant tissue-plasminogen activator (rtPA)-induced thrombolysis remains the only approach that is used in clinics to recanalize the obstructed artery. Paradoxically, and although stroke is a cerebrovascular disease, most efforts in the field focused on strategies that aimed essentially at targeting neurons per se, omitting somehow the central implication of vascular injury in ischemic stroke pathobiology. The failure of acute and sub-acute ischemic stroke therapies was attributed to the fact that clinical trials did not succeed to properly target the ischemic penumbra in patients (Lo, 2008). Initial experimental studies presumed that injured tissues within the ischemic penumbra would inevitably evolve towards a complete irreversible infarct (Lo, 2008). However, in the last decade, the overwhelming experimental findings suggest that the injured NVU integrates a continuum of complex molecular and cellular responses, which directly affect the balance between injury and repair within the penumbra (Lo, 2008). Understanding these signaling responses may reinvigorate the hope for making breakthroughs in stroke therapies in the near future.
NVU signaling following ischemic stroke: Several reports are suggesting that the biochemical and cellular signaling responses orchestrated at the NVU following injury decisively influence the pathobiology of ischemic stroke. These signaling responses translate an adaptation attempt from the injured brain to counter and face the stress signals triggered by ischemic injury, during which NVU structural, biochemical and cellular integrity are reshaped (Hermann and ElAli, 2012). This process is thought to aim essentially at promoting tissue repair within the ischemic penumbra. However, depending on injury severity, the signaling responses that aim at reshaping NVU structural, biochemical and cellular integrity are not always successful and may lead to NVU loss-of-function, or even prevent successful brain pharmacotherapies (Hermann and ElAli, 2012). The signaling responses orchestrated at the NVU following ischemic stroke are complex, as they integrate several pleiotropic molecular and cellular signals (Hermann and ElAli, 2012). Therefore, in this report we will exemplify these signals by referring to specific studies. For instance, matrix metalloproteinases (MMPs) have been demonstrated to play a multifaceted role at the NVU following ischemic stroke. In the acute phase, MMPs activation contributes to NVU dysfunction by destabilizing BBB structure, whereas in the chronic phase their activity is required to achieve successful neurovascular remodeling and plasticity (Zhao et al., 2006). In parallel, ischemic stroke upregulates the expression of ATP-binding cassette transporter B1 (ABCB1) (i.e., multidrug resistance protein-1; Mdr-1), which eliminate a wide range of molecules and xenobiotics across the BBB (ElAli and Hermann, 2010). Although this adaptive response orchestrated at the NVU aims essentially at eliminating toxic metabolites generated in the brain during the ischemic insult and limiting the entry of toxic molecules into the brain, it impedes the entry of neuroprotective drugs into the ischemic brain (ElAli and Hermann, 2010). In addition, it has been demonstrated that even after a successful cerebral reperfusion, the oxidative and nitrative stress associated to ischemic stroke triggers pericyte contraction thus inducing brain microvasculature constriction, which consequently alters cerebral blood reflow in the injured region (Yemisci et al., 2009). Strategies that aimed at preventing pericyte contraction enhanced brain microvasculature patency and promoted neuronal recovery (Yemisci et al., 2009). On the other hand, pericytes play a crucial role in maintaining brain microvasculature stability. Furthermore, strategies that aim at enhancing pericyte coverage and interaction with brain endothelial cells have been demonstrated to be efficacious in restoring the homeostasis of brain microenvironment in a way that promotes neuronal recovery (Zechariah et al., 2013). Additionally, a recent report showed that following ischemic stroke pericytes acquire a multipotent activity and differentiate into microglial cells, which are an important cellular component of the NVU. For instance, microglia has been shown to play a double role in ischemic stroke. In the sub-acute phase of ischemic stroke microglia adopts a protective phenotype that resembles the phenotype “alternatively” activated by interleukin-4 (IL-4) or interleukin-13 (IL-13), which has been shown to be involved in tissue repair, healing and remodeling (Hu et al., 2012). However, over time microglial cell phenotype shifts toward a destructive phenotype that resembles the phenotype “classically” activated via toll-like receptors (TLRs) or by interferon-γ (INF-γ), which have been shown to be involved in the inflammatory responses that exacerbate ischemic injury (Hu et al., 2012). Several experimental findings have revealed a previously unrecognized role of high mobility group box-1 (HMGB1), which plays an important role in the inflammatory response, in ischemic stroke pathophysiology (Hayakawa et al., 2010). The early release of HMGB1 by ischemic neurons into the extracellular space following ischemic injury may aggravate ischemic injury at the penumbra mainly by exacerbating the inflammatory response, and triggering BBB breakdown caused by astrocyte endfeet swelling, pericyte contraction and endothelial tight junction destabilization (Hayakawa et al., 2010). However, it has been reported that in the chronic phase of ischemic stroke HMGB1 plays a beneficial role by promoting neurovascular recovery (Hayakawa et al., 2010). Recently, it has been shown that reactive astrocytes in the peri-infarct areas increase the expression of pentraxin 3 (PTX3), a prototypical member of the long pentraxin family, thus resulting in BBB preservation by decreasing vascular endothelial growth factor (VEGF)-induced brain endothelial permeability (Shindo et al., 2016). The authors concluded that this response orchestrated by astrocytes at the NVU constitutes an intrinsic compensatory mechanism that aims at maintaining BBB function after ischemic stroke (Shindo et al., 2016).
NVU restitution and loss-of-function balance: The NVU serves as the minimal functional unit in the brain, thereby the subtle balance that has been proposed to exist between the two opposite biological signals within the ischemic penumbra directly affects NVU reshaping following ischemic stroke (Lo, 2008), which itself influences ischemic injury development within the penumbra on the short- and long-term. These signals include the repair signals that promote NVU restitution, and the dysfunction signals that lead to NVU loss-of-function (Figure 1). Depending on the nature and severity of ischemic injury, three scenarios could take place; (a) the repair signals dominate and shift the balance towards NVU restitution, (b) the dysfunction signals dominate and shift the balance towards NVU loss-of-function and in some cases (c) both signals are equally evoked and mutually neutralized giving rise to a virtually silent NVU reshaping, which, due to the repetitive chronic stress that accumulates over time, could evolve and lead to a progressive NVU loss-of-function, such as observed in vascular dementia (Moskowitz et al., 2010). This subtle balance between the repair and dysfunction signals constitutes a new framework to address how NVU signaling influences ischemic stroke pathobiology at the penumbra, which may allow the development of novel therapeutic strategies that combine the reinforcement of the repair signals, and minimize the consequences of dysfunction signal, thus shifting the balance towards NVU restitution and consequently neurorestoration. Therefore, future studies should emphasize on deciphering the spatiotemporal regulation of the signaling responses orchestrated at the NVU following ischemic stroke in order to get better insights into disease pathobiology, which may allow the development of novel efficacious therapeutic interventions. In addition, instead of focusing on directly targeting neurons, stroke therapies should promote the overall function of the NVU.
Figure 1.
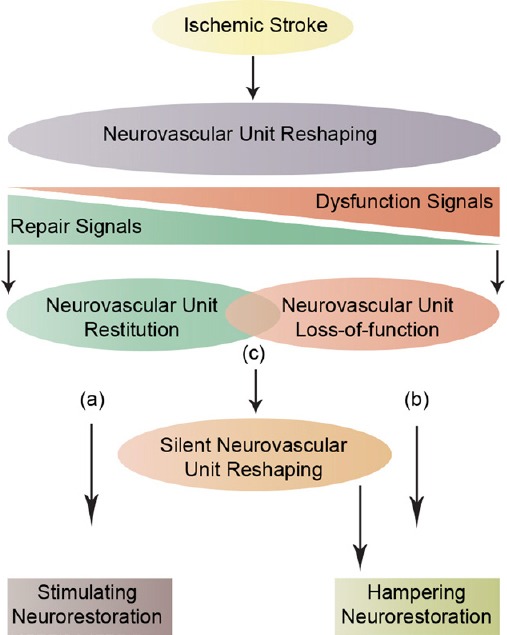
Neurovascular unit (NVU) signaling.
A schematic illustration that represents the subtle balance between two opposite biological signals: the repair signals that lead to NVU restitution and the dysfunction signals that lead to NVU loss-of-function. Three possible scenarios could take place; (a) the repair signals dominate, thus shifting the balance towards NVU restitution and consequently stimulating neurorestoration, (b) the dysfunction signals dominate, thus sifting the balance towards NVU loss-of-function and consequently hampering neurorestoration, and (c) both signals are equally evoked and mutually neutralized, thus giving rise to a silent reshaping that can evolve over time to a total NVU loss-of-function and consequently hampering neurorestoration.
This work was supported by establishment grants from the Foundation du CHU de Québec (2331), and Faculty of Medicine, Laval University.
References
- Dirnagl U. Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann N Y Acad Sci. 2012;1268:21–25. doi: 10.1111/j.1749-6632.2012.06691.x. [DOI] [PubMed] [Google Scholar]
- ElAli A, Hermann DM. Apolipoprotein E controls ATP-binding cassette transporters in the ischemic brain. Sci Signal. 2010;3:ra72. doi: 10.1126/scisignal.2001213. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Qiu J, Lo EH. Biphasic actions of HMGB1 signaling in inflammation and recovery after stroke. Ann N Y Acad Sci. 2010;1207:50–57. doi: 10.1111/j.1749-6632.2010.05728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann DM, ElAli A. The abluminal endothelial membrane in neurovascular remodeling in health and disease. Sci Signal. 2012;5:re4. doi: 10.1126/scisignal.2002886. [DOI] [PubMed] [Google Scholar]
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- Lampron A, ElAli A, Rivest S. Innate immunity in the CNS: redefining the relationship between the CNS and its environment. Neuron. 2013;78:214–232. doi: 10.1016/j.neuron.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo A, Maki T, Mandeville ET, Liang AC, Egawa N, Itoh K, Itoh N, Borlongan M, Holder JC, Chuang TT, McNeish JD, Tomimoto H, Lok J, Lo EH, Arai K. Astrocyte-derived pentraxin 3 supports blood-brain barrier integrity under acute phase of stroke. Stroke. 2016;47:1094–1100. doi: 10.1161/STROKEAHA.115.012133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- Zechariah A, ElAli A, Doeppner TR, Jin F, Hasan MR, Helfrich I, Mies G, Hermann DM. Vascular endothelial growth factor promotes pericyte coverage of brain capillaries, improves cerebral blood flow during subsequent focal cerebral ischemia, and preserves the metabolic penumbra. Stroke. 2013;44:1690–1697. doi: 10.1161/STROKEAHA.111.000240. [DOI] [PubMed] [Google Scholar]
- Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]