

Abstract
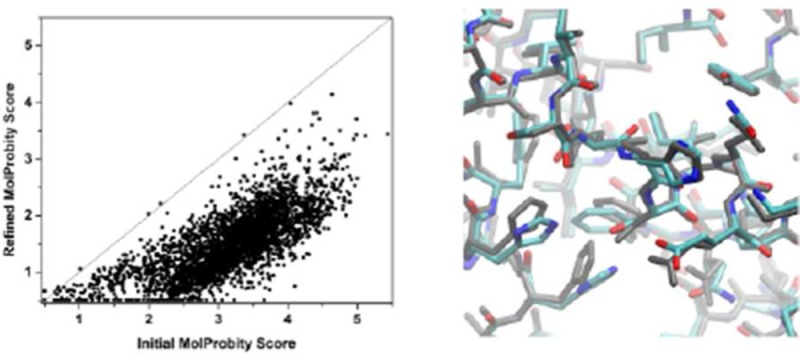
A method for the local refinement of protein structures that targets improvements in local stereochemistry while preserving the overall fold is presented. The method uses force field-based minimization and sampling via molecular dynamics simulations with a modified force field to bring bonds, angles, and torsion angles into an acceptable range for high-resolution protein structures. The method is implemented in the locPREFMD web server and was tested on computational models submitted to CASP11. Using MolProbity scores as the main assessment criterion, the locPREFMD method significantly improves the stereochemical quality of given input models close to the quality expected for experimental structures while maintaining the Cα coordinates of the initial model.
Introduction
Protein structures are the key link between genes and their function without which a full mechanistic understanding of biology could not be developed. High-resolution protein structures are also an essential starting point for rational drug design. Experimental efforts such as X-ray crystallography1 and NMR (nuclear magnetic resonance) spectroscopy,2 and recently also increasingly high-resolution cryo-electron microscopy,3 have provided a wealth of structures. This is evident from the rapid growth of the PDB (Protein Data Bank),4 where those structures are deposited. However, there is little hope that experimental structures will become available for all or even most of the rapidly increasing number of known genes.
Computational methods have long been established as an alternative to predict protein structures based on a given amino acid sequence.5 Usually this is accomplished by exploiting available structural information for related sequences,6 whereas de novo physics-based protein folding methods are also becoming increasingly successful.7,8 Using elaborate state-of-the-art protocols, it is now possible to generate models without experimental data as input for a majority of sequences that closely resemble the overall fold of the native structure.9 Computational methods also play a key role in generating structures based on experimental data. With the exception of very high-resolution X-ray structures, the experimental data does not provide true atomic resolution and at least some degree of modeling under the constraints of the experimental data is almost always necessary to generate fully atomistic models.
The inherent quality of computer-generated protein structure models can be assessed with a number of well-established structure validation tools. Early tools include VERIFY3D,10 PROSA,11 and PROCHECK.12 A more recent quality assessment suite is MolProbity.13 These programs analyze whether a given model conforms to expected structural qualities of proteins as observed in known high-resolution protein structures. Such quality assessment focuses, in particular, on whether bond lengths, angles, and torsion angles lie within statistically expected ranges, whether ring side chains preserve planarity, whether atomic packing avoids both clashes and excessive internal cavities, and whether hydrophobic and polar residues are distributed as expected. When protein structures are generated via computational methods, the resulting models often deviate substantially from these criteria. To some degree, deviations from ideality are expected in real structures under biological conditions where the overall conformational free energy is minimized and thermal fluctuations are present. However, the majority of apparent structural violations in computational models are more likely attributed to artifacts in the model generation protocol and/or imperfect energy functions. Since computational models also deviate overall from the “true” native structure, based on backbone Cα atoms, one may assume that stringent high-resolution structure validation assessment criteria cannot be fulfilled unless the overall fold also matches the native structure. To explore this question, publicly available assessment results from the latest round of CASP, CASP11,14 were analyzed. Figure 1 compares the MolProbity scores,13 assessing the local structural quality, with the GDT-TS (Global Distance Test) score,15 measuring the similarity to the native, experimentally determined structure, for computational models submitted to CASP11. A weak degree of correlation appears to exist, but there are a large number of models with excellent MolProbity scores but very low GDT-TS values, while most of the models with very high GDT-TS values actually have fairly poor MolProbity scores (above 2). Therefore, it should be possible to significantly improve local structural quality largely independently of how close the overall fold of a given model is from the native structure.
Figure 1.

MolProbity score13 vs GDT-TS scores (relative to experimental native structure based on Cα coordinates) for all models submitted to CASP11 after separation into domains using data provided by the CASP assessors on the CASP Web site.14
In the past, several efforts have been made to develop protocols for improving the local structural quality. A straightforward strategy may involve simple all-atom minimization using atomistic force fields with constraints to preserve the overall fold.16,17 More elaborate algorithms have employed side chain repacking with or without minimization and/or molecular dynamics to target the refinement of PDB structures in the Autofix method18 and the refinement of computational models in GalaxyRefine.19 In another study, a simulated annealing protocol was developed using a modified potential energy function combined with a statistical torsion potential to improve the quality of NMR structures.20 The proposed methods have demonstrated success in achieving improved local structure quality as measured by MolProbity scores, but a broad application to a wide variety of models has either not been attempted or resulted in mixed success.19
Here, a more elaborate protocol is presented that combines minimization, sampling via restrained molecular dynamics, and targeted rebuilding of problematic residues to significantly improve the local structural quality of virtually any given protein model. Previously, an initial implementation of this protocol was tested in the limited context of the final stage in a protein refinement protocol.21 Here, an improved version is presented and tested in an expanded context to a wide range of models. Briefly, it appears to be possible, with only moderate computational effort, to significantly improve the MolProbity score to values below 2 for almost any model and below 1 for models where the initial MolProbity score is below 2. The improvement in MolProbity scores is significantly greater than what has been achieved with other previously published methods. In the following, the methodology is described in more detail before validation results are presented and discussed.
Methodology
Refinement Protocol
The local structure refinement protocol (termed locPREFMD; local Protein structure REFinement via Molecular Dynamics) consists of a series of empirically optimized steps where all-atom force field-based minimization and sampling and targeted rebuilding of problematic regions are applied. At the end, an ensemble of conformations is generated, and the structure with the lowest MolProbity score and closest to the initial conformation based on the Cα root-mean-square deviation (RMSD) is selected. The detailed protocol is shown in Figure 2. The protocol described here is similar to but improved over what was applied previously just in the context of the Feig group refinement pipeline.21
Figure 2.

Flowchart of the local structure refinement protocol described in detail in the methods section. Force field-based minimization and molecular dynamics (MD) steps are shown as red boxes, and blue boxes describe targeted rebuilding steps. Values given in gray indicate force constants (for minimization and MD steps) and temperatures (for MD steps). Values highlighted in blue were used in an alternate protocol to allow Cα atoms to deviate from the initial positions. The end points of different variants (locPREFMD, locPREFMin, locPREFMin0) are also indicated.
To be able to handle a diverse set of input models, including models that may be missing atoms or are compromised in other ways, the protocol begins by adding missing atoms using the complete.pl tool from the MMTSB Tool Set.22 This tool generates fully atomistic models with as little as only Cα atoms while preserving the atoms that are present in the input file in several stages. First, missing backbone atoms are reconstructed from Cα positions using a reconstruction procedure developed by us earlier.23 Then, side chains are completed. If at least Cβ positions are available, a residue is converted to the SICHO (SIdeCHain-Only) coarse-grained model and subsequently rebuilt to atomistic detail using a previously developed method.23 If a side chain is missing completely, the tool SCWRL24,25 is used for reconstruction. Finally, models are submitted to the CHARMM all-atom modeling program26 to add missing hydrogen atoms and complete (zwitterionic) termini. This step resolves gross clashes where two atoms are within 0.1 Å of each other, for example, when two different atoms have the same coordinates in the input file. Nearly overlapping atoms are treated as missing atoms that are subsequently rebuilt. Finally, as part of generating a complete initial model, leucine side chains are inspected to ensure that the naming of Cδ carbons, CD1 and CD2, follows the standard pro-chiral convention.
The complete atomistic model is then inspected for the presence of cis peptide bonds. If any cis bonds are found in residues that are neither proline nor precede proline, the backbone is rebuilt in the trans conformation. Dihedral constraints are generated to maintain the backbone ω torsion angle for all nonproline residues that are not preceding proline near 180° using a force constant of 10 kcal/mol/degree2 in all of the subsequent minimization and sampling steps. The constraint is applied because initial models may be energetically very unfavorable and can flip to the cis conformation due to high initial strain energies. The rationale for automatically converting cis peptide bonds is that although they may occur in real structures,27 it is much more likely that they are artifacts when they are found in computational models. In fact, an inspection of the CASP11 predictions used here as test sets found that about 0.4% of residues that are neither proline nor preceded proline had a cis backbone.
The model is subsequently minimized using an all-atom force field for the first time using a relatively weak restraint (0.1 kcal/mol/Å2) on Cα atoms with respect to the initial model. The minimized structure is then inspected for possible ring penetrations where a bond in another residue crosses the ring plane of histidine, phenylalanine, tyrosine, or tryptophan. Such artifacts may already be present in the initial model or occur when two side chains are overlapping closely initially and are not separated properly in the initial minimization step. If such a problem is found, the side chains of the two involved residues are rebuilt based on the Cα and Cβ positions only using rotamers that avoid overlap followed by a brief minimization. Typically, this resolves the issue. If the initial models have very tightly packed side chains and it is difficult to place selected side chains in a way that overlap is avoided, the backbone needs to be allowed to move to create space. Therefore, this step is iterated with successively decreasing restraints on the Cα atoms until all of the ring penetrations are resolved. Ring penetrations occur less frequently than cis backbone bonds but are also present in some CASP models based on our analysis, and they often do not resolve easily via simple molecular dynamics simulations.28
The model is then minimized twice with increasing restraints (0.2 and 0.5 kcal/mol/Å2) to relax the structure further while bringing the Cα positions back to the initial model in case they moved during the first two steps. Afterward, the model is examined with MolProbity13 for the existence of unfavorable side chain rotamers. If present, those side chains are rebuilt again followed by another minimization step.
The structure is minimized again with stronger restraints on Cα atoms (1.0 kcal/mol/Å2) to generate starting models for four short molecular dynamics (MD) simulations that all start from the minimized structure. The first and second simulations (MD1, MD2) are run at 20 and 100 K, respectively, using restraints of 1.0 kcal/mol/Å2; the third and fourth simulations (MD3, MD4) are run at 150 K using restraints of 0.5 and 2.0 kcal/mol/Å2. The variations in temperature and restraint strength increase the chances of reaching improved models during the MD step. Higher temperatures than 150 K lead to distortions due to thermal fluctuations, whereas weaker restraints increase the likelihood of departure from the initial model. From the MD simulations, snapshots are collected that are then minimized again to remove thermal noise. For every snapshot in the final ensemble, MolProbity scores and the RMSD from the initial model (iRMSD) for Cα atoms are determined. The final model is then selected based on the lowest MolProbity score, and if multiple models with the same score are found, the model with the lowest iRMSD value is chosen.
To test how different stages of the locPREFMD protocol contribute to the final improvement in quality, a partial protocol was also explored where only the first minimization step (locPREFMin0) or all of the minimization steps until the MD step (locPREFMin) were applied (Figure 2).
The main goal of the locPREFMD protocol is to improve MolProbity scores while, at the same time, preserving the initial Cα coordinates as closely as possible. An alternative protocol was also explored where one of the MD runs (MD4 at 150 K) sampled conformations without restraints on the Cα atoms, and the final minimization of all snapshots from MD1–4 used only very weak Cα restraints (0.01 kcal/mol/Å2). This protocol was tested to examine to what degree better MolProbity scores can be achieved when allowing for larger deviations from the initial Cα trace.
All-Atom Minimization and Molecular Dynamics
In all minimization and molecular dynamics runs, a modified version of the CHARMM36 all-atom force field29 was used. In order to generate structures that satisfy the stringent requirements of the quality assessment tools, certain bond and angle term force constants were increased (Tables S1 and S2), improper torsions were added to enforce planarity of histidine, tyrosine, and phenylalanine (Table S3), and the CMAP potential was modified to increase penalties for ϕ/ψ angles outside the preferred Ramachandran map areas (Figure S1). The modifications were introduced empirically to minimize the MolProbity scores of the final models.21 It should be emphasized that the resulting modified potential would not be appropriate for running unrestrained simulations and is only meant as a knowledge-based correction to encode idealized structural properties of experimental structures of proteins.
Solvation effects were accounted for implicitly by using a distance dependent dielectric function (ε = 4r). Electrostatic and Lennard-Jones interactions were cut off at 18 Å using a switching function between 16 and 18 Å. All of the minimization runs involved 50 steps of steepest descent minimization followed by 500 steps of adopted-basis Newton–Raphson minimization. The molecular dynamics (MD) runs were carried out for 5000 steps, saving snapshots every 500 steps. The Berendsen thermostat was used in the MD simulations to maintain a constant temperature using a coupling constant of 0.1 ps–1. SHAKE30 was applied to constrain heavy atom-hydrogen distances so that an integration time step of 2 fs could be used.
Test Sets
The locPREFMD protocol was tested on three test sets based on models submitted for CASP11 targets. In the first set, termed “regular”, all (8098) model 1 submissions with complete backbone and side chains for 99 targets in the regular 3D structure prediction category were used. A few models with missing side chains and/or C, N, or O backbone atoms were excluded. Although locPREFMD can handle initial models that are missing side chains and/or non-Cα backbone atoms, MolProbity cannot calculate meaningful initial scores for such models complicating the analysis presented here. In the second set, termed “server”, all (3495) model 1 submissions for 97 targets in the server category were used. The server submissions are part of the “regular” test set, but they were analyzed separately to gauge the potential impact of the locPREFMD protocol on fully automatic prediction pipelines. Finally, the third test, termed “refined”, consisted of all (1305) model 1 submissions for 32 targets in the refinement category. All of the models were downloaded from the CASP Web site14 and used as is without further modification as input to locPREFMD. Since CASP11 covers a wide variety of computational methods and the CASP11 targets cover a wide variety of structural types of proteins, these three test sets should sufficiently validate this method for its intended application.
Web Server
The locPREFMD method is available as a web service.31 Users can submit a PDB structure and will receive a refined model via email after a few minutes.
Results
Local Refinement of Protein Models with locPREFMD
The locPREFMD protocol shown in Figure 2 was applied to three large test sets consisting of model predictions from the last round of CASP11. The main criterion considered here is the improvement in MolProbity scores as a measure of local structural quality that combines a variety of aspects of protein structure quality. Experimental structures are expected to have MolProbity scores below 2, while scores near 1 would be desirable for high-quality structures. The range of MolProbity scores for the submitted models in the “regular” and “server” test sets covers the entire range from 0.5 (the best possible score) to near 6 with the majority of scores well above 2. Therefore, most of these models would be considered to have poor local structural quality. In the models submitted for the “refinement” category, the distribution of MolProbity scores is shifted to smaller values with the majority between 1 and 2. Application of the locPREFMD protocol significantly reduced the MolProbity scores across all test sets with virtually every model being improved to at least some degree (Figure 3). After refinement, almost all “server” and “regular” predictions had MolProbity scores below 3 with the majority below 2, while most “refinement” predictions were improved to scores of 1 or below. Because the initial models submitted to CASP generally did not contain hydrogen atoms and the MolProbity program suite uses its own program to add hydrogen atoms before calculating the scores, hydrogen atoms were removed from the refined models before submitting them to MolProbity so that the results reported here match the published CASP analysis of the submitted models. However, an alternate protocol where hydrogens are added to the initial CASP models using an in-house protocol (with CHARMM) before submitting them to MolProbity and comparing with the full all-atom models that result from locPREFMD gives essentially identical results (data not shown).
Figure 3.

MolProbity scores of initial CASP submissions vs models refined with locPREFMD for the refinement (A), server (B), and regular (C) prediction test sets.
Average MolProbity scores before and after refinement are given in Table 1. For the “regular” and “server” prediction sets, average scores improved from around 3 to 1.5 while the “refinement” predictions were improved on average from 2 to 0.9. At the same time, average GDT-TS and GDT-HA scores remained virtually unchanged indicating that the local structure quality could be improved without affecting Cα positions, thereby preserving the overall fold.
Table 1. Average MolProbity Scores, RMSD from the Initial Model, and GDT-TS/HA Scores before and after Refinement Using Different Variations of the locPREFMD Protocol Applied to the Refinement, Server, and Regular Prediction test setsa.
| test set | protocol | MolProbity | iRMSD Cα [Å] | GDT-TS | GDT-HA |
|---|---|---|---|---|---|
| refinement predictions | initial | 2.00 | 0 | 70.02 | 50.78 |
| locPREFMin0 | 1.15 | 0.13 | 70.18 | 51.04 | |
| locPREFMin | 1.14 | 0.15 | 70.18 | 51.04 | |
| locPREFMD | 0.91 | 0.25 | 70.19 | 51.02 | |
| locPREFMD/2 | 0.86 | 0.32 | 70.17 | 50.96 | |
| server predictions | initial | 3.16 | 0 | 44.50 | 31.85 |
| locPREFMin0 | 1.98 | 0.24 | 44.47 | 31.85 | |
| locPREFMin | 1.93 | 0.25 | 44.50 | 31.88 | |
| locPREFMD | 1.51 | 0.44 | 44.43 | 31.73 | |
| locPREFMD/nofix | 1.16 | 2.03 | 41.55 | 28.20 | |
| regular predictions | initial | 2.90 | 0 | 36.24 | 25.45 |
| locPREFMin0 | 1.82 | 0.20 | 36.28 | 25.53 | |
| locPREFMin | 1.78 | 0.21 | 36.28 | 25.52 | |
| locPREFMD | 1.41 | 0.39 | 36.22 | 25.41 |
While the focus here is primarily on the overall MolProbity scores, other quality measures were also analyzed (Table 2). locPREFMD is especially effective in reducing clashes (as measured by the MolProbity clashscore, see Table 2), but the refined models also have reduced fractions of rotamer and backbone torsion (Ramachandran) outliers. VERIFY3D scores were improved for the “refinement” test set but remained unchanged for the “server” and “regular” test sets. Since this score focuses more on the overall packing of side chains, which the locPREFMD refinement protocol does not target, this would be expected. However, PROCHECK measures that again emphasize local stereochemistry also exhibited significant improvements after models were submitted to locPREFMD. PROCHECK’s overall G-factor is a log-odds score of bonds, angles, and torsion angles with respect to observed distributions.34 G-factors of favorable structure should be at least above −0.5 and ideally near zero. After application of the locPREFMD protocol, G-factors improved substantially, especially for the server models, reaching average values between −0.2 and zero.
Table 2. Average Quality Assessment Measures before and after Refinement Using locPREFMD from MolProbity (MP),13 VERIFY3D,10 and PROCHECK (PC)12 for the Refinement, Server, and Regular Test Sets Considered Here.
| refinement |
servers |
regular |
||||
|---|---|---|---|---|---|---|
| quality measure | initial | locPREFMD | initial | locPREFMD | initial | locPREFMD |
| MP totalscore | 2.00 | 0.91 | 3.16 | 1.51 | 2.90 | 1.41 |
| MP clashscore | 23.27 | 0.11 | 59.54 | 0.96 | 51.17 | 0.80 |
| MP rotamer outliers [%] | 3.64 | 1.11 | 5.72 | 3.53 | 6.05 | 3.11 |
| MP Ramach. outliers [%] | 1.97 | 1.08 | 4.51 | 2.47 | 4.63 | 2.35 |
| VERIFY3D | 0.36 | 0.43 | 0.33 | 0.33 | 0.32 | 0.32 |
| PC G-factor | –0.19 | –0.06 | –0.48 | –0.20 | –0.33 | –0.18 |
| PC mainchain bonds [%] | 89.51 | 99.96 | 90.97 | 99.90 | 89.86 | 99.92 |
| PC mainchain angles [%] | 86.80 | 94.43 | 85.18 | 93.15 | 84.55 | 93.47 |
| PC side chain planarity [%] | 86.83 | 96.43 | 94.08 | 95.05 | 89.73 | 95.46 |
Minimization vs Molecular Dynamics
While MolProbity scores were improved substantially with locPREFMD, just simple minimization (locPREFMin0 and locPREFMin protocols, see Methodology section) also led to significantly improved MolProbity scores (Table 1). Average scores after just one round of initial minimization were improved by about one unit for all test sets. Subsequent minimization runs only offered marginal returns as the difference between the locPREFMin0 and locPREFMin protocols is at most 0.05 score units. However, additional MD sampling resulted in a further decrease of 0.3–0.4 units as a consequence of additional sampling and the ability to select the lowest score from an ensemble of models. While the low cost of just a simple minimization (<1 min) may be attractive, the additional cost of running short MD simulations (5–20 min depending on the target) appears to be worthwhile.
MolProbity Scores and Refinement vs GDT
The distribution of MolProbity scores as a function of GDT-TS in the initial models showed only a moderate trend of decreasing MolProbity scores as a function of GDT-TS (Figure 1). After refinement with locPREFMD, this trend is more pronounced (Figure 4A). Models with GDT-TS scores above 50 exhibit scores that are almost entirely limited to the 0.5–2 interval with the majority of scores reaching values between 0.5 and 1 for the models with the highest GDT-TS scores (>80). On the other hand, models with poor MolProbity scores are most prominent for models with the very lowest GDT-TS scores (<20). However, as in the distribution of the initial models, there is a significant fraction of models with very low MolProbity scores even for models with very low GDT-TS scores. This confirms that high local structural quality can be achieved for models with entirely incorrect folds. However, models that reproduce the native fold closely can generally be refined toward higher local structural quality.
Figure 4.

MolProbity scores of models refined with locPREFMD (A) and improvement in MolProbity scores after refinement with locPREFMD (B) as a function of GDT-TS of the initial models from the native structure based on Cα coordinates. Results for refinement and regular prediction test sets are colored in red and black, respectively.
Figure 4B shows the improvement in MolProbity scores after application of locPREFMD. The majority of models were improved by 1.5–2 units. A strong trend of the degree of refinement as a function of GDT-TS is not apparent. This indicates that the effectiveness of locPREFMD does not depend on how close the initial model is to the native structure.
Refinement without Constraining Cα Atoms
While the main application of locPREFMD is the improvement of the local structural quality of models without affecting the Cα positions, preserving the backbone in poor initial models may significantly hinder effective refinement. Therefore, an alternative protocol was also tested where Cα atoms were allowed to move during the final MD simulations to achieve lower MolProbity scores (see Methodology section). This altered protocol was tested for the “server” test set. Significantly lower MolProbity scores can indeed be achieved if Cα atoms are allowed to move (on average 1.2 vs 1.5), but at the same time, GDT-TS and GDT-HA scores decreased significantly (Table 2), which is probably not acceptable for most applications.
Figure 5 shows how much refined models deviated from the initial models, measured by the Cα RMSD between initial and refined models. While the restraints in the locPREFMD protocol keep the refined models very closely to the initial models, large deviations are observed if Cα atoms are not restrained. In both cases, the final MolProbity score is correlated with the deviation of the final model from the initial model since poor initial models could not be refined without deviating more significantly from the initial model than good initial models.
Figure 5.
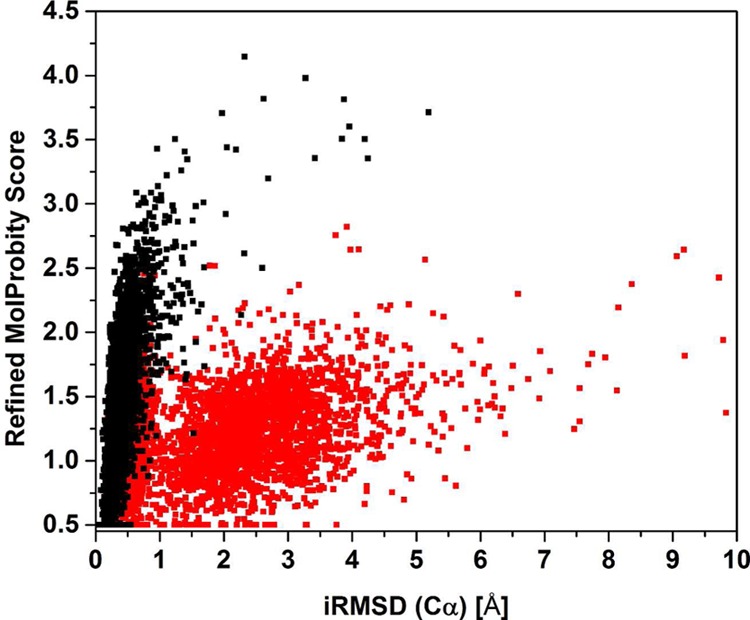
MolProbity scores of models refined with locPREFMD vs Cα RMSD from the initial model for the server prediction test set using the regular protocol that restrains Cα positions to the initial model (black) and a modified protocol where larger deviations of Cα positions are allowed (red, see Methodology section and Figure 2 for details).
Refinement for Initial Models Developed with Different Methods
Previous efforts to improve the local structural quality have reported difficulties in refining models generated by certain methods. In particular, the Seok group reported that refinement of models generated with ROSETTA using GalaxyRefine was challenging.19Figure 3 suggests that locPREFMD was also not able to refine the MolProbity scores in a few cases. Therefore, further analysis was carried out to examine the degree of refinement for models generated by different groups during CASP11. Table 3 compares average MolProbity scores for selected, highly performing server and refinement prediction. In all cases, locPREFMD was able to significantly improve initial models (including models that already had very low initial MolProbity scores presumably as a result of targeted efforts to achieve high local structural quality such as predictions from the Seok, LEE, Baker, and RFMQA groups in the refinement category). Therefore, the locPREFMD protocol should be applicable to improve models generated with essentially any method during CASP11.
Table 3. Average MolProbity Scores before and after Refinement with locPREFMD for Server and Refinement Predictions from Selected Top-Performing Prediction Groups.
| group | initial MolProbity score | refined MolProbity score |
|---|---|---|
| Zhang-Server (iTASSER) | 2.96 | 1.71 |
| ROSETTA server | 2.03 | 0.96 |
| RaptorX server | 2.16 | 1.18 |
| HHPredX server | 4.17 | 1.98 |
| MULTICOM-CONSTRUCT server | 2.83 | 1.11 |
| TASSER-VMT server | 3.96 | 1.88 |
| nns server | 2.93 | 1.09 |
| FFAS-3D server | 3.35 | 1.57 |
| FEIG refinement | 1.84 | 0.76 |
| Schroderlab refinement | 1.65 | 0.69 |
| Kiharalab refinement | 1.61 | 0.88 |
| PRINCETON_TIGRESS refinement | 2.25 | 0.88 |
| Seok refinement | 1.02 | 0.77 |
| KnowMIN_server refinement | 2.20 | 0.95 |
| LEE refinement | 0.91 | 0.71 |
| BAKER-REFINESERVER | 1.30 | 0.81 |
| PRINCETON_MD_REFINE | 2.53 | 1.02 |
| FUSION refinement | 1.53 | 0.83 |
| RFMQA refinement | 1.02 | 0.83 |
| MULTICOM-REFINE refinement | 1.63 | 0.86 |
Repeated Application of locPREFMD
While a single round of locPREFMD offered significant improvements in MolProbity scores, it was also tested whether a second round of locPREFMD could offer additional improvements. This test was focused on the “refinement” test set where the goal is to maximize both accuracy and quality for predictions that already exhibit high initial structural accuracy. locPREFMD was repeated only for models that did not already achieve a “perfect” score of 0.5 after the first round. Figure 6 and the average values reported in Table 1 indicate that a second round of locPREFMD can further improve MolProbity scores, although by a much smaller degree than after the first round, while still preserving the original GDT-TS and GDT-HA values. The repeated application of locPREFMD could be improved further by only selecting refined models from the second (or further rounds) if the MolProbity score is lower than in initial rounds. Such a scheme may be attractive for a focused high-resolution modeling of selected structures, but the additional costs of running several rounds of locPREFMD may not be justified in the context of high-throughput automatic structure prediction.
Figure 6.

MolProbity scores of models in the refinement test set after one round of locPREFMD vs models generated after repeating the locPREFMD protocol for a second time.
Discussion and Conclusions
Force field-based minimization and molecular dynamics via the locPREFMD method was successfully applied to significantly improve the local structural quality of a wide variety of computational models. An earlier version of the protocol described here was able to improve the structural quality of the Feig group predictions in the refinement category during CASP11,21 but because those models were among the best-submitted models, it was not clear that the method would be equally applicable to a broader set of models. Here, it is shown based on extensive test sets from CASP11 submissions that locPREFMD can refine virtually any model irrespective of the method that was used to generate the initial model. MolProbity scores were improved on average by about 1.5 units for server and regular prediction models and, slightly less, by about 1.2 units for models submitted in the refinement category. The degree of refinement is more substantial than what has been reported previously using other protocols such as GalaxyRefine.19 locPREFMD is also more universally applicable since improvements were possible irrespective of how the initial model was generated compared to, again, GalaxyRefine, which faced challenges in refining models from certain methods, such as ROSETTA.19
The significant extent of refinement achieved here is a result of combining force field-based sampling with an empirically modified potential function that favors idealized protein stereochemistry. While the modified potential would not be appropriate for unrestrained molecular dynamics simulations, it focuses sampling on the most likely bonding geometries and speeds up convergence to conformations that are both energetically and structurally sound. The use of restraints and targeted rebuilding of problematic residues further contributes to speeding up convergence. A related approach was also taken recently in an improved protocol for the refinement of NMR structures,20 where a statistical potential was combined with flat-bottom distance potentials. But, the approach taken here went further by modifying specific bond and angle terms as well using a customized CMAP potential35−37 to limit the sampling of regions of the Ramachandran map that are considered outliers.
While MolProbity is used here to guide the refinement and to select the best models from the final ensemble, a more efficient protocol could, in principle, use the MolProbity score directly as part of the energy function during minimization and sampling. Currently, this is not a practical proposition because the only available MolProbity implementation from the Richardson group takes on the order of seconds for a single conformation and does not provide derivatives. However, the development of faster code for calculating MolProbity scores may be possible in the future.
The protocol proposed here involves moderate computational costs of between 5 and 20 min depending on the size of the input model and could easily be incorporated as the final stage of prediction pipelines. The locPREFMD protocol would be especially useful for improving server-generated models since an analysis of CASP submissions indicates that many servers generate models with relatively poor local stereochemistry. locPREFMD could significantly improve those models, although it was more difficult to refine MolProbity scores from certain servers (for example, the Zhang-Server or HHPredX) well below 2 than for others. This is puzzling given that those models are among the best in terms of proximity to the native state (based on GDT scores). To understand this observation better, the server models were analyzed in terms of Cα–Cα distances for neighboring residues. Ideally, this distance should be distributed narrowly around almost exactly 3.8 Å. In models generated by the Zhang-Server, the Cα–Cα distance distribution is systematically shifted to larger values with a peak near 3.85 Å (Figure S2). The Cα–Cα distribution in HHPredX models is centered near 3.80 Å, but it is very broad with a significant number of Cα–Cα pairs that are relatively far from the ideal value (Figure S2). Since Cα positions are restrained in locPREFMD, nonideal distance distributions in initial models from these servers likely hinder local structure quality improvement. In two other methods, FFAS-3D and MULTICOM-CONSTRUCT, the Cα–Cα distributions are shifted to shorter distances (near 3.75 Å; Figure S2), although this appears to be slightly less problematic for generating high quality structures with our protocol. The origin of the shifted Cα–Cα distributions in some server models is unclear, but if they are a result of tuning predictions toward maximal GDT scores based on Cα atoms, our analysis would suggest that such optimization may not be fully compatible with achieving high local structural quality that satisfies crystallographic standards.
For initial models with high structural quality, locPREFMD is able to achieve further refinement. In many cases, optimal scores of 0.5 are reached, and it also appears to be possible that repeated application of locPREFMD can successively improve models to very low scores. This raises the question of what scores are good enough. Crystallographers are often satisfied if MolProbity scores approach a value near 1 as they realize that real structures do not strictly conform to the idealized criteria derived from statistics of what constitutes a “correct” structure. Therefore, once MolProbity scores for a given model approach 1, there may be little sense in further improvement. With that criteria, locPREFMD, on average, achieves optimal structures for the refinement test set and is not too far from optimality for the server and regular test sets, even although a significant number of input models were structurally highly problematic.
Improvements of the local structural quality using the protocol described here are expected to enhance the utility of computer-generated models when used as docking targets or as starting structures for computer simulations, where not just the Cα backbone but also realistic side chain conformations are important. Improved stereochemistry may also be helpful when initial homology models are subjected to additional global refinement, for example, via molecular dynamics simulations, to bring such models closer to the native state.17,21
Acknowledgments
M.F. thanks Dr. Bercem Dutagaci, Dr. Grzegorz Nawrocki, and Ms. Asli Yildirim for help in testing the locPREFMD web service. Funding from National Institute of Health Grant R01 GM084953 is acknowledged.
Glossary
Abbreviations
- CASP
Critical Assessment of methods for protein Structure Prediction
- CHARMM
Chemistry at Harvard Molecular Mechanics
- GDT
Global Distance Test
- GDT-TS
GDT total score
- GDT-HA
GDT high accuracy
- iRMSD
initial RMSD
- locPREFMD
local Protein structure REFinement via Molecular Dynamics
- locPREFMin
local Protein Structure REFinement via Minimization;MD molecular dynamics
- MMTSB
Multiscale Modeling Tools in Structural Biology
- MP
MolProbity
- NMR
nuclear magnetic resonance
- PC
Procheck
- PDB
Protein Data Bank
- RMSD
root-mean-square deviation
- SCWRL
side-chain reconstruction with rotamer library
- SICHO
SIde-CHain-Only model
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.6b00222.
Information as mentioned in the text. (PDF)
Author Contributions
M.F. designed and carried out the research and wrote the manuscript.
The author declares no competing financial interest.
Supplementary Material
References
- Drenth J.Principles of Protein X-ray Crystallography; Springer: New York, 2007. [Google Scholar]
- Wuthrich K. Protein Structure Determination in Solution by NMR-Spectroscopy. J. Biol. Chem. 1990, 265, 22059–22062. [PubMed] [Google Scholar]
- Kühlbrandt W. Cryo-EM Enters a New Era. eLife 2014, 3, e03678. 10.7554/eLife.03678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook J.; Feng Z. K.; Chen L.; Yang H. W.; Berman H. M. The Protein Data Bank and Structural Genomics. Nucleic Acids Res. 2003, 31, 489–491. 10.1093/nar/gkg068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D.; Sali A. Protein Structure Prediction and Structural Genomics. Science 2001, 294, 93–96. 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Dror R. O.; Shaw D. E. How Fast-Folding Proteins Fold. Science 2011, 334, 517–520. 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- Osguthorpe D. J. Ab initio Protein Folding. Curr. Opin. Struct. Biol. 2000, 10, 146–152. 10.1016/S0959-440X(00)00067-1. [DOI] [PubMed] [Google Scholar]
- Kryshtafovych A.; Fidelis K.; Moult J. CASP10 Results Compared to Those of Previous CASP Experiments. Proteins: Struct., Funct., Genet. 2014, 82, 164–174. 10.1002/prot.24448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D.; Luthy R.; Bowie J. U. VERIFY3D: Assessment of Protein Models with Three-Dimensional Profiles. Methods Enzymol. 1997, 277, 396–404. 10.1016/S0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- Wiederstein M.; Sippl M. J. ProSA-web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASP11 Web Site. http://www.predictioncenter.org/casp11/index.cgi (accessed May 27, 2016),.
- Zemla A.; Venclovas C.; Moult J.; Fidelis K. Processing and Analysis of CASP3 Protein Structure Predictions. Proteins: Struct., Funct., Genet. 1999, 37, 22–29. . [DOI] [PubMed] [Google Scholar]
- Mirjalili V.; Feig M. Protein Structure Refinement through Structure Selection and Averaging from Molecular Dynamics Ensembles. J. Chem. Theory Comput. 2013, 9, 1294–1303. 10.1021/ct300962x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirjalili V.; Noyes K.; Feig M. Physics-Based Protein Structure Refinement through Multiple Molecular Dynamics Trajectories and Structure Averaging. Proteins: Struct., Funct., Genet. 2014, 82, 196–207. 10.1002/prot.24336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headd J. J.; Immormino R. M.; Keedy D. A.; Emsley P.; Richardson D. C.; Richardson J. S. Autofix for Backward-Fit Sidechains: Using MolProbity and Real-Space Refinement to Put Misfits in Their Place. J. Struct. Funct. Genomics 2009, 10, 83–93. 10.1007/s10969-008-9045-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo L.; Park H.; Seok C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41, W384–W388. 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H.; Kim T.-R.; Ahn S.; Ji S.; Lee J. Protein NMR Structures Refined wtihout NOE Data. PLoS One 2014, 9, e108888. 10.1371/journal.pone.0108888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig M.; Mirjalili V. Protein Structure Refinement via Molecular-Dynamics Simulations: What Works and What Does Not?. Proteins: Struct., Funct., Genet. 2015, 10.1002/prot.24871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig M.; Karanicolas J.; Brooks C. L. III MMTSB Tool Set: Enhanced Sampling and Multiscale Modeling Methods for Applications in Structural Biology. J. Mol. Graphics Modell. 2004, 22, 377–395. 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Feig M.; Rotkiewicz P.; Kolinski A.; Skolnick J.; Brooks C. L. I. Accurate Reconstruction of All-Atom Protein Representations From Side-Chain-Based Low-Resolution Models. Proteins: Struct., Funct., Genet. 2000, 41, 86–97. . [DOI] [PubMed] [Google Scholar]
- Bower M. J.; Cohen F. E.; Dunbrack R. L. Prediction of Protein Side-chain Rotamers from a Backbone-dependent Rotamer Library: A New Homology Modeling Tool. J. Mol. Biol. 1997, 267, 1268–1282. 10.1006/jmbi.1997.0926. [DOI] [PubMed] [Google Scholar]
- Krivov G. G.; Shapovalov M. V.; Dunbrack R. L. Improved Prediction of Protein Side-Chain Conformations with SCWRL4. Proteins: Struct., Funct., Genet. 2009, 77, 778–795. 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L.; Mackerell A. D.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabs A.; Weiss M. S.; Hilgenfeld R. Non-Proline cis Peptide Bonds in Proteins. J. Mol. Biol. 1999, 286, 291–304. 10.1006/jmbi.1998.2459. [DOI] [PubMed] [Google Scholar]
- Feig M.; Harada R.; Mori T.; Yu I.; Takahashi K.; Sugita Y. Complete Atomistic Model of a Bacterial Cytoplasm Integrates Physics, Biochemistry, and Systems Biology. J. Mol. Graphics Modell. 2015, 58, 1–9. 10.1016/j.jmgm.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P.; Mittal J.; Feig M.; MacKerell A. D. Jr Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- locPREFMD Web Service. http://feig.bch.msu.edu/web/services/locprefmd/ (accessed May 27, 2016).
- Zhang Y.; Skolnick J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins: Struct., Funct., Genet. 2007, 68, 1020–1020. 10.1002/prot.21643. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Skolnick J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins: Struct., Funct., Genet. 2004, 57, 702–710. 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- Engh R. A.; Huber R. Accurate Bond and Angle Parameters for X-Ray Protein-Structure Refinement. Acta Crystallogr., Sect. A: Found. Crystallogr. 1991, 47, 392–400. 10.1107/S0108767391001071. [DOI] [Google Scholar]
- Feig M.; MacKerell A. D. Jr.; Brooks C. L. III Force Field Influence on the Observation of π-Helical Protein Structures in Molecular Dynamics Simulations. J. Phys. Chem. B 2003, 107, 2831–2836. 10.1021/jp027293y. [DOI] [Google Scholar]
- MacKerell A. D. Jr.; Feig M.; Brooks C. L. III Improved Treatment of the Protein Backbone in Empirical Force Fields. J. Am. Chem. Soc. 2004, 126, 698–699. 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D. Jr.; Feig M.; Brooks C. L. III Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25, 1400–1415. 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.