

ABSTRACT
We report a Caucasian boy with intractable epilepsy and global developmental delay. Whole-exome sequencing identified the likely genetic etiology as a novel p.K212E mutation in the X-linked gene HSD17B10 for mitochondrial short-chain dehydrogenase/reductase SDR5C1. Mutations in HSD17B10 cause the HSD10 disease, traditionally classified as a metabolic disorder due to the role of SDR5C1 in fatty and amino acid metabolism. However, SDR5C1 is also an essential subunit of human mitochondrial RNase P, the enzyme responsible for 5′-processing and methylation of purine-9 of mitochondrial tRNAs. Here we show that the p.K212E mutation impairs the SDR5C1-dependent mitochondrial RNase P activities, and suggest that the pathogenicity of p.K212E is due to a general mitochondrial dysfunction caused by reduction in SDR5C1-dependent maturation of mitochondrial tRNAs.
KEYWORDS: HSD10 disease, PRORP, SDR5C1, 17β-hydroxysteroid dehydrogenase type 10, TRMT10C
Abbreviations
- AdoMet
S-adenosyl methionine
- CADD
combined annotation dependent depletion
- DTT
dithiothreitol
- ESP
exome sequencing project
- FPLC
fast performance liquid chromatography
- GI
gastrointestinal
- HSD10
17β-hydroxysteroid dehydrogenase type 10
- LOD
logarithm (base 10) of odds
- MRPP
mitochondrial RNase P protein
- mt-tRNA
mitochondrial tRNA
- mtDNA
mitochondrial DNA
- NAD
nicotinamide adenine dinucleotide
- PRORP
protein-only RNase P
- PAGE
polyacrylamide gel electrophoresis
- SDR5C1
short-chain dehydrogenase/reductase 5C1
- SDS
sodium dodecyl sulfate
- TRMT10C
tRNA methyl transferase 10C
Introduction
HSD17B10 (17β-hydroxysteroid dehydrogenase type 10) encodes SDR5C1, a protein of 261 amino acids that is a member of the NAD+/NADP-dependent short-chain dehydrogenase/reductase (SDR) family.1,2 SDR5C1 is the only hydroxysteroid dehydrogenase (HSD) family member localized to mitochondria.3-8 It acts by oxidoreduction of L-2-methyl-3-hydroxy-butyryl-CoA at the penultimate step in the β-oxidation of isoleucine.9 In vitro, SDR5C1 also acts on other substrates, including branched short-chain fatty acids, hydroxysteroids, and sex hormones.3-6,10,11 SDR5C1 deficiency causes a mitochondrial disorder known as ‘HSD10 disease’.7 Biochemical diagnosis of HSD10 disease has traditionally relied upon the detection of elevated metabolites from isoleucine breakdown,12 which are the same metabolites elevated in patients with α-ketothiolase (AKT) deficiency.13 However, the clinical presentations of these two metabolic disorders are distinct. HSD10 disease is characterized by progressive neurodegeneration and cardiomyopathy in early childhood,7 whereas AKT deficiency presents intermittent ketoacidosis with typically normal development and no symptoms between crises.13 Yet, their overlapping biochemical signature has led to clinical misdiagnosis.14 HSD10 disease has been reported only in a small number of families world-wide, mostly occurring in males, consistent with HSD17B10 being located on the X-chromosome.7 DNA sequencing analyses to date have identified 10 missense mutations in HSD17B10.7,14,15, and a splice variant that reduces the protein expression level.16,17 Among the different HSD17B10 missense mutations, the clinical manifestations are diverse,7 including a neonatal-onset form (associated with mutations p.D86G, p.R226Q, and p.N247S), an infantile-onset form (associated with mutations p.L122V, p.R130C, and p.P210S), a milder atypical form (associated with mutation p.Q165H), and a later-onset form (associated with mutation p.E249Q).
Here we report a new case of HSD10 disease in which a novel p.K212E mutation was identified by exome sequencing. The proband was a 9-year-old Caucasian boy with suspected mitochondrial disease based on intractable epilepsy, global developmental delay, static encephalopathy, optic atrophy and blindness, and chronic lactic acidemia. His family history was notable for 2 similarly affected maternal uncles, making particularly likely an X-linked etiology. However, enzymatic analysis of the dehydrogenase activity of SDR5C1 revealed only a modest decrease by the p.K212E mutation. Additional biochemical analyses revealed that this HSD17B10 mutation caused a global deficiency in mitochondrial tRNA processing and maturation.
SDR5C1 (HSD17B10) is naturally present in the mitochondrial RNase P holo-complex required for processing mitochondrial tRNAs (mt-tRNAs).18 The human mitochondrial DNA (mtDNA) genome encodes 22 mt-tRNAs, each of which is used to translate 13 mtDNA-encoded proteins for assembly into the electron transport chain for ATP synthesis. Transcription of the mtDNA genome generates primary polycistronic transcripts, from which individual mt-tRNAs are processed and post-transcriptionally modified. The mitochondrial RNase P complex is composed of 3 subunits.18 SDR5C1 (MRPP2) forms a sub-complex with TRMT10C (MRPP1), a mitochondrial tRNA methyl transferase that synthesizes the methylated m1G9 or m1A9 on mt-tRNAs.19 This methylation is crucial for proper folding of mt-tRNALys 20 and may be important for folding of 19 of the 22 mt-tRNAs that contain purines at position 9. The sub-complex forms a ternary complex with PRORP (MRPP3), which cleaves polycistronic mitochondrial transcripts to generate 5′-processed mt-tRNAs.18 We show here that the p.K212E mutation in HSD17B10 adversely affects the enzymatic activities of both TRMT10C and PRORP. This result demonstrates the secondary propagation of the mutational effect from SDR5C1 to the entire mitochondrial RNase P holo-complex, possibly leading to the global impairment of processing and maturation of mt-tRNAs. Our data suggest that the deficiency in mt-tRNA processing and maturation is the major pathogenetic mechanism underlying HSD10 disease.
Subject description
The proband was a 9-year-old Caucasian boy who presented to the Mitochondrial-Genetics Diagnostic Clinic at The Children's Hospital of Philadelphia for evaluation of a presumed mitochondrial cytopathy. He displayed intractable epilepsy, developmental regression, static encephalopathy, optic nerve atrophy, non-verbal, GI dysmotility, multi-systemic dysfunction, and chronic lactic academia. His family history was suggestive of X-linked disease. His epilepsy onset at age 2½ years coincided with progressive developmental regression that was characterized by the loss of ability to walk and stand by age 4 y as well as increased drooling, constipation, urinary dysfunction, and swallowing dysfunction. His seizures were initially characterized by quick head drop events with syncope, and currently characterized as Lennox Gastaut syndrome occurring in daily clusters under one minute duration involving tonic stiffening with arm extension, typically in the morning and occasionally in the evening. His seizures were treatment refractory to benzodiazepines, clorazepate, topiramate, lamotrigine, rufinamide, levetiracetam, and clobazam. Episodic sustained and uncontrollable laughters with difficulty breathing also occurred, although whether these represent seizures or pseudobulbar effect was unclear. Other neurologic problems include prominent choreoathetotic movements that have worsened overtime with severe spastic tetraparesis. His brain MRIs were normal at 15 months and at 5 y old, but by 9 y old revealed diffuse cerebral atrophy, with minimal worsening by age 12.5 y. His brain MRS revealed a lactate couplet at age 15 months, with decreased n-acetylaspartate seen at 9 y old. His muscle biopsy at age 1 y was suggestive of a mitochondrial myopathy with increased lipid content, abnormal mitochondrial histology, and mitochondrial proliferation. Consistently, succinate dehydrogenase activity was 3-fold increased. This enzyme activity is derived from proteins in complex II, which are nucleus-encoded, whereas all other electron transport chain enzyme complexes contain proteins made within mitochondria. Indeed, although electron transport chain enzyme activities of I, III, and IV were within the normal range, they were below normal when normalized to citrate synthase to compensate for mitochondrial proliferation. No mitochondrial DNA mutation was identified. His additional multi-systemic features, physical examination findings, biochemical and genetic diagnostic investigations, and detailed muscle biopsy testing results are described in the Supplemental File.
Results
Identification of HSD17B10 mutation by research-based whole exome sequencing
The exome in blood from the proband was sequenced using Agilent SureSelect Human All Exon V4 kit capture followed by 2 × 101 basepair paired-end sequencing with an Illumina HiSeq 2000 sequencer. Variants were detected, annotated, and analyzed as described.21-24 A total of 18,466 coding variants were identified in the proband across the genome, including 18,131 single nucleotide variants and 335 indels. Because the proband's 2 maternal half-uncles died of a similar-spectrum disorder, analysis was focused on X-linked genes. Given the apparent rare nature of the maternal half-uncles' disorder, we only examined rare variants in the proband with allele frequencies below 0.5 percent in the general population based on the 1000 Genomes Project and the UK10K Project, and with occurrence less than 10 times in 1,334 internal control exomes that we had analyzed.21-24
After applying these data filters, only one non-synonymous hemizygous variant was found on the X chromosome in the proband. This X-linked variant was located within the gene HSD17B10 (X:g.53458504T>C), resulting in a p.K212E amino acid change. This variant occurs at a highly evolutionally-conserved position (GERP,25 score = 5.88 and a phastCons LOD score,26 of 259). Pathogenicity predictions were discordant: it is predicted to be benign in PolyPhen-2,27 tolerated by SIFT,28 and disease-causing by MutationTaster,29 with the latest and most comprehensive Combined Annotation Dependent Depletion (CADD) algorithm,30 yielding a scaled phred-like score of 21.7. This variant is completely novel, having never been reported by the NHLBI Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/), the 1000 Genomes Project (http://www.1000genomes.org/), the UK10K Project (http://www.uk10k.org/), and the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/) that includes sequence data from 60,706 unrelated individuals. Nor was the variant found in any of the more than 1,400 exomes from different disease cohorts that we have analyzed. No parental or maternal uncles' samples were available for sequence analysis.
Structural mapping of the p.K212E mutation in SDR5C1 (HSD17B10)
Human SDR5C1 is a tetramer of a single-domain structure31 of 261 amino acids (Fig. 1A), which shares 88% sequence identity with its rat counterpart. In crystal structures of the human and rat enzymes in complex with NAD+ and a bound substrate or substrate analog,31,32 this single domain structure adopts a typical dehydrogenase “Rossmann” fold made up of a central β-sheet of 7 parallel strands and 3 connecting α-helices both above and below the β-sheet. NAD+ is bound to a conserved motif GGxxGxG at residues 17-23 near the N-terminus. The catalytic triad for hydride transfer consists of 3 conserved residues S155, Y168, and K172 at the dehydrogenase active site (Fig. 1A). Two insertion loops, between residues 100-110 and 140-150, are unique to SDR5C1 and related enzymes and are absent from other short-chain dehydrogenases. These two insertion loops are involved in subunit-subunit interaction to form a dimer, while the C-terminal residues 256-261 are involved in bridging dimers to form a tetramer (Fig. 1B). Binding of a ketobutyrate substrate is accompanied by closure of the active site using the substrate-specificity loop at residues 203-220, whereas binding of a steroid substrate does not require the closure.32 Indeed, because the enzyme makes no direct interaction with the hydroxyl end of steroids, this explains its capacity for a range of diverse steroid substrates.
Figure 1.
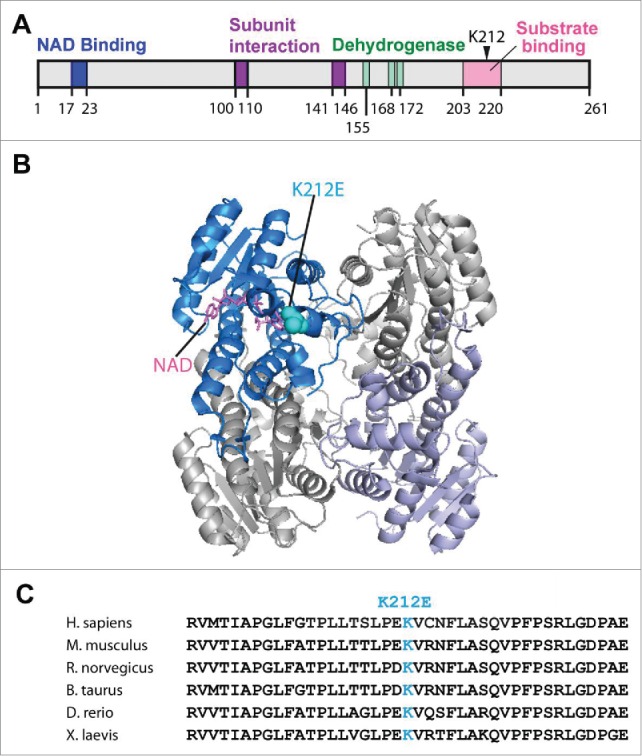
The p.K212E mutation of human SDR5C1. (A), The position of p.K212E in the domain structure of the 261 amino acids of human SDR5C1, showing the NAD+ binding domain (residues 17–23), the insertion segments for subunit-subunit interaction (residues 100–110 and residues 141-146), the catalytic triad for the dehydrogenase (S155, Y168, and K172), and the substrate-specificity loop (residues 203–220)32. (B) The position of p.K212E in the tetramer structure of human SDR5C1. The mutation (in blue) and the bound NAD+ (in red) are only mapped to one subunit for clarity of presentation. (C) Multiple sequence alignment of the region surrounding p.K212E. Abbreviations: H. sapiens (Homo sapiens), M. musculus (Mus musculus), R. norvegicus (Rattus norvegicus), B. taurus (Bos taurus), D. rerio (Danio rerio), X. laevis (Xenopus laevis). The 3D-structure rendering of SDR5C1 was generated with PyMol 1.7.
The p.K212E mutation is in the substrate-specificity loop that moves into the active site upon binding of a ketobutyrate substrate. Among vertebrates, this loop consists of highly conserved residues (Fig. 1C), which form 2 helices (residues 204-208 and residues 211–217) separated by a sharp turn.32 K212 is in the second helix acting as the “lid” to close the active-site cleft and it is strictly conserved among mammalian SDR5C1 enzymes (Fig. 1C). The mutation substitutes a positively charged side-chain with a negatively charged one, thus altering the electrostatic environment of the lid and likely interfering with the active-site closure.
Effects of the p.K212E mutation on SDR5C1 and the mitochondrial RNase P enzyme activities
We determined whether the p.K212E mutation decreases the dehydrogenase activity of SDR5C1. While other HSD10-disease associated mutations had decreased the enzyme's dehydrogenase activity, some of these mutations were analyzed in homogenates of patient fibroblasts,7,33 where SDR5C1 levels among samples were not controlled. We chose to determine the mutational effect by comparing known quantities of purified wild-type and mutant enzyme, each expressed and purified from an E. coli over-expression strain.19,34 The assay used acetoacetyl-CoA (3-ketoacyl-CoA) as the substrate and monitored its reduction to 3-hydroxyacetyl-CoA accompanied by oxidation of NADH to NAD+. While the wild-type enzyme had an activity of 3.18 ± 0.18 units/mg, similar to other reported values,34 the mutant showed an almost 4-fold decrease of the activity to 0.84 ± 0.12 units/mg (Fig. 2A). Notably, while the mutational defect is modest, it is similar to that of the p.P210S mutation,34 which is separated from the p.K212E mutation by 2 residues in the enzyme sequence and is associated with the infantile form of HSD10 disease.7
Figure 2.
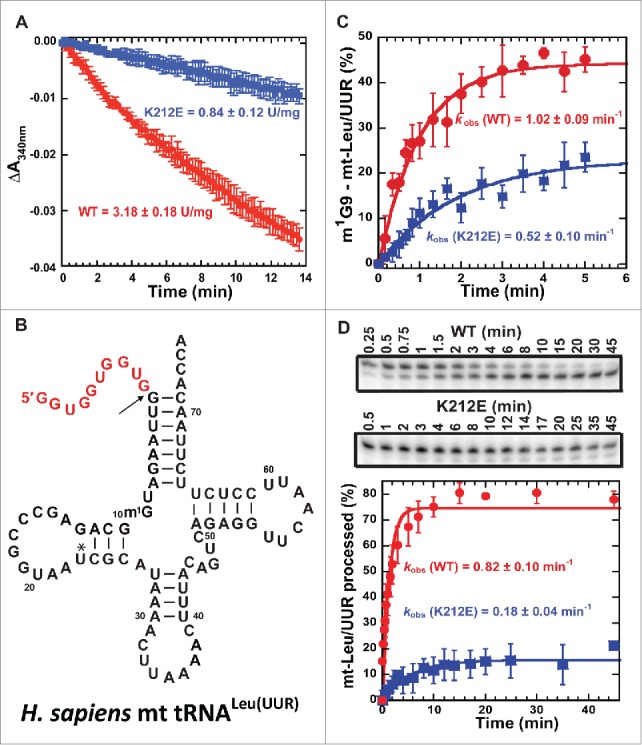
Impairment of SDR5C1-dependent activities by the p.K212E mutation. (A) The mutational effect on the dehydrogenase activity of SDR5C1, showing the rate constant of NADH oxidation to NAD+ upon reduction of acetoacetyl-CoA to 3-hydroxy-acetyl-CoA. (B) The sequence and cloverleaf structure of human mt-tRNALeu(UUR). The mature sequence is in black, whereas the 5′-precursor sequence is shown in red. (C) The mutational effect on the synthesis of m1G9-mt-tRNALeu(UUR), showing the kobs of methyl transfer of the TRMT10C-SDR5C1 sub-complex with wild-type (WT) and p.K212E mutant of SDR5C1. (D) The mutational effect on the 5′-processing activity of mitochondrial RNase P to convert the precursor of mt-tRNALeu(UUR) to the mature form, showing a time-course of the processing activity (top) and the kobs of the processing by the WT and p.K212E mutant forms of SDR5C1 in the holo-complex (bottom).
We next determined whether the p.K212E mutation decreases the methyl transferase activity of TRMT10C, which requires the presence of SDR5C1 to be active.19,34 We assayed a reconstituted and recombinant form of the TRMT10C-SDR5C sub-complex with AdoMet as the methyl donor and the N1 of G9 in the unmodified transcript of mt-tRNALeu(UUR) as the acceptor (Fig. 2B). The binary complex with wild-type SDR5C1 displayed a kobs of methyl transfer of 1.02 ± 0.09 min−1, similar to reported values,34 whereas the mutant complex displayed a 2-fold reduced activity with kobs of 0.52 ± 0.10 min−1 (Fig. 2C). Thus, the p.K212E mutation impairs the methyl transferase activity of the TRMT10C-SDR5C1 complex.
We then determined whether the p.K212E mutation decreases the 5′-processing activity of the mitochondrial RNase P holo-complex. The endonucleolytic subunit of mitochondrial RNase P is PRORP, which requires TRMT10C and SDR5C1 to be active.18 We reconstituted the holo-complex in a molar ratio of 1:2:1 with purified TRMT10C, SDR5C1, and PRORP, respectively. A precursor form of mt-tRNALeu(UUR) carrying a 5′-extension (Fig. 2B) was synthesized by in vitro transcription, 3′-end labeled, and used as a substrate. Quantification of the fractional conversion of substrate to product over time determined the cleavage rate. Under a single turnover assay condition, the mitochondrial RNase P holo-complex containing the wild-type SDR5C1 efficiently catalyzed 5′-processing of the precursor mt-tRNA, with the reaction reaching 80% completion in 45 min and a kobs of 0.8 ± 0.1 min−1. The complex with the p.K212E mutation was much less efficient, with the reaction reaching only 15% completion and a 4.5-fold reduction of kobs to 0.18 ± 0.04 min−1 (Fig. 2D). Thus, the p.K212E mutation of SDR5C1 impairs the processing activity of mitochondrial RNase P. Notably, the rate of 5′-processing of mt-tRNALeu(UUR) by the wild-type complex (kobs = 0.8 ± 0.1 min−1) was slower compared to the reported value for mt-tRNAIle (kobs = 4.63 ± 0.61 min−1),34 possibly reflecting a sequence-dependent tRNA effect and differences in assay conditions. However, the decrease in the 5′-processing activity by the p.K212E mutation is comparable to that seen with the p.P210S mutation in SDR5C1 (4.5-fold vs. 8.0-fold).34
Effects of the p.K212E mutation on the TRMT10C-SDR5C1 sub-complex stability
The ability of the p.K212E mutation to reduce the enzymatic activity of both TRMT10C and PRORP raised the possibility that the mutation decreased the stability of the mitochondrial RNase P holo-complex. In this complex, the sub-complex TRMT10C-SDR5C1 is highly stable and is made up of 2 monomers of the methyl transferase (α2, 86 kDa) and a tetramer of the dehydrogenase/reductase (α4, 116 kDa) in a mass ratio of 3:4. We assembled the sub-complex using purified recombinant proteins in vitro and determined its stability by gel filtration through a Superdex 200 column that had been calibrated with proteins of known molecular weights. The wild-type and the K212E mutant sub-complex eluted at the same position corresponding to a molecular mass of 170 kDa, consistent with its expected molecular weight (˜200 kDa) (Fig. 3A). In both cases, the excess SDR5C1 protein after formation of the sub-complex eluted later, corresponding to the expected molecular mass of 116 kDa. Analysis of the elution peak for the sub-complex on an SDS-PAGE confirmed the co-existence of both proteins, and similar analysis of the later peak confirmed the presence of SDR5C1 (Fig. 3B).
Figure 3.
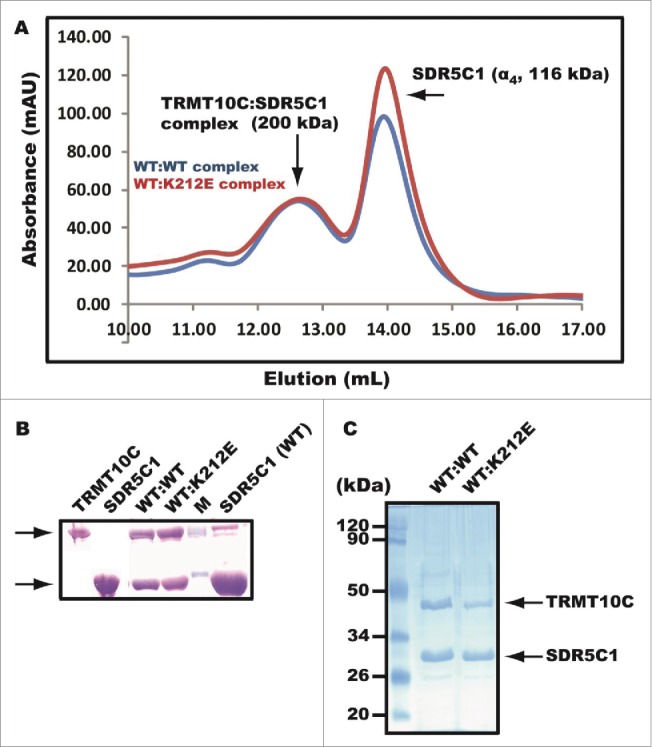
Destabilization of the TRMT10C-SDR5C1 sub-complex by p.K212E. (A) Elution profile of gel filtration of a pre-assembled sub-complex through Superdex 200. The first peak eluted at 12.7 mL corresponding to the molecular mass of 200 kDa of the sub-complex, while the second peak eluted at 14.0 mL corresponding to the molecular mass of 116 kDa of the homotetramer of SDR5C1. The sub-complex WT:WT refers to wild-type TRMT10C and wild-type SDR5C1, while the WT:K212E refers to wild-type TRMT10C and the p.K212E mutant of SDR5C1. (B) SDS-PAGE analysis of gel filtration. From left to right, lane 1 was loaded with a marker of TRMT10C, lane 2 a marker of SDR5C1, lane 3 the first peak of gel filtration of the WT-WT sub-complex, lane 4 the first peak of gel filtration of the WT-K212E sub-complex, lane 5 2 molcular weight markers, and lane 6 the second peak of gel filtration of the WT-WT sub-complex. (C) Pull-down of an untagged TRMT10C from E. coli cell lysates by a His-tagged SDR5C1 bound to a solid support and analyzed by SDS-PAGE for the WT-WT or WT-K212E sub-complex.
Because the gel filtration assay was performed with saturating SDR5C1, there was no apparent effect of the p.K212E mutation on the stability of the sub-complex. We then used a separate pull-down assay to detect the formation of the sub-complex under a non-saturating condition and in the context of potentially competing proteins. As shown previously, when TRMT10C was expressed in E. coli in the native form without any tag, it can be specifically pulled down from cell lysates by a His-tagged SDR5C1 bound to a solid support.34 This pull-down reflects the ability of the 2 proteins to form a stable sub-complex in a cellular environment. Using this assay, we applied the same amount of SDR5C1 (wild-type or the K212E mutant) to an E. coli cell lysate that expressed a tag-free TRMT10C. After extensive washes to remove non-specific binding, we found that the mutant SDR5C1 was less able to pull down TRMT10C than the wild-type. On average, the pull-down ability of the mutant is 2-fold reduced as compared to the wild-type enzyme (Fig. 3C). This suggests that the p.212E mutation has reduced the ability of SDR5C1 to form a stable sub-complex with TRMT10C.
Discussion
The major pathogenic mechanism underlying HSD10 disease has been elusive. The initial mechanism was thought to relate to a primary biochemical defect in isoleucine metabolism. Indeed, both SDR5C1 and AKT are involved in the β-oxidation of isoleucine, where SRD5C1 catalyzes oxidation of 2-methyl-3-hydroxy-butyrate-CoA to 2-methyl-acetoacyl-CoA, and AKT catalyzes breakdown of 2-methyl-acetoacyl-CoA to propionyl-CoA and acetyl-CoA.35 Because these 2 enzymes function in 2 consecutive steps of the same pathway, loss-of-function mutations in either enzyme show nearly identical urine organic acid profiles in patients.36 This has led to misdiagnosis of the more severe clinical disorder caused by SRD5C1 deficiency as AKT deficiency.14 Furthermore, initiating an isoleucine-restricted diet does not alter or improve the neurodegeneration and developmental disabilities of HSD10 disease, despite a biochemical reduction in the excretion of 2-methyl-3-hydroxy-butyrate and tiglyl-glycine.12,37 Thus, the severe clinical course of HSD10 disease cannot be readily explained by a blockade of isoleucine catabolism.
An alternative pathophysiologic mechanism for HSD10 disease postulated that an imbalance occurred in neurosteroid metabolism associated with the dehydrogenase activity of SDR5C1.38 While SDR5C1 is ubiquitously expressed in all tissues, it is predominantly found in liver, heart, and brain. In the brain, it is primarily expressed in neural rather than in glial cells, implying that the loss of the dehydrogenase activity could cause neurodegeneration. However, the residual dehydrogenase activity of the enzyme has little correlation with the clinical manifestations of HSD10 disease. For example, a severe neonatal presentation associated with the p.D86G mutation in SDR5C1 retains 30% of the dehydrogenase activity, whereas an atypical form with normal intellectual and neurological development associated with the p.Q165H mutation has no dehydrogenase activity.39 Also, embryo and animal model studies have demonstrated that the dehydrogenase activity of SDR5C1 is not required for cell survival. In particular, although Xenopus cells with HSD17B10-knockdown and mouse models with conditional HSD17B10-knockout have disintegrated mitochondrial structure and apoptotic cellular features, these defects can be prevented by introducing the “dehydrogenase-dead” p.Q165H variant of SDR5C1, but not by the “partially active” dehydrogenase variants p.R130C or p.D86G.39
A third, and most plausible, pathophysiologic mechanism for the severe phenotype of HSD10 disease relates to the recent realization that SDR5C1 functions as an integral component of human mitochondrial RNase P. This ‘moonlighting’ function raises the possibility that pathogenic mutations in SDR5C1 disrupt mt-tRNA processing and maturation. Rather than relying on metabolites as the primary diagnosis indicators, we performed exome sequencing on the proband and used his family history as a guide to identify the p.K212E mutation in SDR5C1 as the most likely cause of the genetic etiology. The severe multi-system manifestations of the HSD17B10 p.K212E mutation in the proband, including congenital hypotonia, absent speech, static encephalopathy, intractable epilepsy, progressive neurodegeneration, optic atrophy, blindness, gastrointestinal dysfunction, and lactic acidemia, are similar to those reported for infantile-onset HSD10 disease.7 Despite devastating clinical presentations, we show here that the p.K212E mutation causes only a modest defect in the dehydrogenase activity of SDR5C1. However, in additional experiments, we show that the mutation reduces the purine-9 methylation and 5′-processing activities on mt-tRNA and destabilizes the TRMT10C-SDR5C1 sub-complex in the mitochondrial RNase P holo-enzyme. These data suggest that the mutation, by disrupting the methyl transferase sub-complex and in turn the mitochondrial RNase P holo-complex, can compromise the processing and maturation of mt-tRNAs, and thereby impair mitochondrial protein synthesis. Consistently, the patient's muscle biopsy showed clear evidence for reduced electron transport chain activities specifically in complexes that contain proteins synthesized by the mitochondrial translation apparatus.
The p.K212E is a novel mutation. We showed that its effects on the mitochondrial RNase P holo-complex are quantitatively similar to those of other mutations causing HSD10 disease. For example, 2 mutations associated with the infantile form (p.R130C and p.P210S) and 2 mutations associated with the neonatal form (p.R226Q and p.N247S) all impair the processing activity of mitochondrial RNase P.34 In each cases, the degree of damage on the 3 activities of the RNase P holo-complex is similar to the effect of p.K212E reported here. Collectively, these data suggest that the major pathogenic effects of HSD17B10 mutations may be related to their secondary disruption of the mitochondrial RNase P holo-complex, thereby resulting in the impairment of the processing and maturation of mt-tRNAs. We postulate that pathogenic HSD17B10 mutations decrease the processing and maturation of mt-tRNAs, possibly leading to secondary mitochondrial myopathy and dysfunction that culminates in the severe multi-system clinical presentation of HSD10 disease.
Methods
Human subjects research consent
The proband was evaluated in Mitochondrial-Genetics Diagnostic Clinic at The Children's Hospital of Philadelphia (M.J.F.) and his blood DNA was extracted following informed consent per the Children's Hospital of Philadelphia Institutional Review Board approved protocol #08–6177.
Expression and purification of recombinant proteins
Recombinant human proteins TRMT10C (C-terminus myc-His tagged), SDR5C1 (N-terminus His-tagged), and PRORP (C-terminus His-tagged) were each expressed from a pET28-derived plasmid in E. coli BL21 Rosetta 2 cells and purified by binding and elution from Ni-NTA (nickel-nitrilo triacetic acid) agarose resin.19,34 Protein concentration was determined by Bradford assay relative to bovine serum albumin standards and adjusted by image analysis of purity on SDS-PAGE. The mutation p.K212E was introduced into the plasmid-encoded recombinant HSD17B10 gene by site-directed mutagenesis and verified by sequence analysis.
Enzyme activity assays
The dehydrogenase activity of SDR5C1 was measured as described34 in the reverse direction for the reduction of acetoacetyl-CoA to L-3-hydroxyacyl-CoA accompanied by oxidation of NADH (nicotinamide adenine dinucleotide) to NAD+. SDR5C1 (10 nM) was assayed with acetoacetyl-CoA (30 μM) and NADH (100 μM) and the decrease of NADH upon the synthesis of NAD+ was monitored as the decrease of absorbance at 340 nm.
The methyl transferase activity of TRMT10C was measured using [3H-methyl]-S-adenosyl-methionine (AdoMet) as the methyl donor, similar to other AdoMet-dependent methyl transferase assays.40-47 The transcript of human mt-tRNALeu(UUR) was used as the substrate for the synthesis of m1G9. The radioactive methyl donor was purchased from Perkin-Elmer. The tRNA transcript was made by in vitro transcription from an oligonucleotide-derived double-stranded DNA template,48 and was purified by denaturing gel electrophoresis. For each experiment, the tRNA transcript was heat denatured at 85°C for 3 min, and annealed at 37°C for 15 min before use. Recombinant TRMT10C (10 μM) was mixed with SDR5C1 (20 μM) to form the sub-complex, which was used to catalyze [3H-methyl]-AdoMet (15 μM)-dependent methyl transfer to human mt-tRNALeu(UUR) (0.5 μM) in single turnover conditions. The reaction was performed at 30°C in a buffer of 50 mM Tris-HCl, pH 8.0, 20 mM NaCl, 4.5 mM MgCl2, 1 mM DTT, and 0.02 mg/mL BSA. Aliquots were acid precipitated on filter pads. After extensive acid washes, filter pads were measured for radioactivity, corrected for the filter quenching factor, and converted to pmoles of methyl transfer. Data points over a time course were fit to the single-exponential equation:
| (1) |
where yo is the y intercept, A is the scaling constant, kapp (or kobs) is the apparent (or observed) rate constant, and t is the time in seconds to determine kobs.
The processing activity of mitochondrial RNase P was measured on a precursor form of human mt-tRNALeu(UUR) made by in vitro transcription and 3′-end labeled with [α-32P]ATP by Bacillus stearothermophilus CCA enzyme.49-51 The mitochondrial RNase P holo-enzyme complex was formed by mixing recombinant TRMT10C, SDR5C1, and PRORP at a final concentration of 1 μM, 2 μM, and 1 μM, respectively, to catalyze 5′-endonucleolytic cleavage of the precursor tRNA (5 nM) at 22°C in the buffer of 50 mM Tris-HCl, pH 8.3, 50 mM NaCl, 2.5 mM MgCl2, and 1 mM DTT. The cleavage product was separated from the precursor tRNA on a 12% PAGE/7M urea gel in a mini apparatus and the fractional conversion of substrate to product was measured by image analysis of the gel. Data points over a time course were fit to equation 1.
Analysis of the TRMT10C-SDR5C1 sub-complex
Gel filtration analysis of the sub-complex, pre-assembled with purified recombinant proteins, was performed on a Superdex 200 column on Äkta FPLC in the buffer of 50 mM Tris-HCl, pH 7.5, 400 mM NaCl, and 5% glycerol. The column was calibrated with cytochrome c from horse heart (14.4 kDa), carbonic anhydrase from bovine erythrocytes (29 kDa), alcohol dehydrogenase from yeast (150 kDa), β-amylase from sweet potato (200 kDa), and blue dextran (2,000 kDa). Absorbance of protein elution was monitored at A280 and peak fractions were analyzed by a 10% SDS-PAGE. For pull-down analysis of the sub-complex, a native form of the recombinant protein TRMT10C, without any tag, was expressed in E. coli Arctic Express cells at OD600 of 2.0 and at 6°C for 48 hours with IPTG (0.25 mM). Cells (100 mL) were harvested, broken by gentle sonication, and the cleared cell lysates were incubated with His-tagged SDR5C1 (50 μg) in the presence of Ni-NTA resin equilibrated in the buffer of 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 50 mM imidazole, 10% glycerol, 10 mM DTT, and 1 mM NAD. After incubation for 2 hours at 4°C, the resin was collected and washed with 10 volumes of the binding buffer. The proteins stably bound to the resin were resuspended in the SDS loading buffer, heated at 95°C, and analyzed on a 10% SDS-PAGE.
Statistical analysis
Activities and reaction rates were compared between the wild-type and mutant enzymes of SDR5C1. Each data point was the average of at least 3 independent measurements. Error bars represent standard deviation (SD).
Supplementary Material
Contributors
All authors fulfilled the criteria for authorship as defined by the international Committee of Medical Journal editors.
Patient consent
Obtained.
Ethics approval
IRB-approved research protocol #08–6177 (MJF, PI) at the Children's Hospital of Philadelphia.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the patienet's family for their participation and Dr. Isao Masuda for his helpful discussion. This work was funded in part by the National Institutes of Health (U01-GM108972 and R01-GM114343 to Y.M.H., R03-DK082446 to M.J.F., R01-EY012910 to E.A.P., and P30EY014104-MEEI core support), and by the Austrian Science Fund (FWF) [P25983] to W.R. The content is solely the responsibility of the authors, and does not necessarily represent the official views of the sponsors, and is not influenced by the sponsors.
References
- 1.Kallberg Y, Oppermann U, Jornvall H, Persson B. Short-chain dehydrogenases/reductases (SDRs). Eur J Biochem 2002; 269:4409-17; PMID:12230552; http://dx.doi.org,/ 10.1046/j.1432-1033.2002.03130.x [DOI] [PubMed] [Google Scholar]
- 2.He XY, Schulz H, Yang SY. A human brain L-3-hydroxyacyl-coenzyme A dehydrogenase is identical to an amyloid beta-peptide-binding protein involved in Alzheimer's disease. J Biol Chem 1998; 273:10741-6; PMID:9553139; http://dx.doi.org/ 10.1074/jbc.273.17.10741 [DOI] [PubMed] [Google Scholar]
- 3.He XY, Merz G, Mehta P, Schulz H, Yang SY. Human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase is a single-domain multifunctional enzyme. Characterization of a novel 17beta-hydroxysteroid dehydrogenase. J Biol Chem 1999; 274:15014-9; PMID:10329704; http://dx.doi.org/ 10.1074/jbc.274.21.15014 [DOI] [PubMed] [Google Scholar]
- 4.He XY, Wegiel J, Yang YZ, Pullarkat R, Schulz H, Yang SY. Type 10 17beta-hydroxysteroid dehydrogenase catalyzing the oxidation of steroid modulators of gamma-aminobutyric acid type A receptors. Mol Cell Endocrinol 2005; 229:111-7; PMID:15607535; http://dx.doi.org/ 10.1016/j.mce.2004.08.011 [DOI] [PubMed] [Google Scholar]
- 5.He XY, Wegiel J, Yang SY. Intracellular oxidation of allopregnanolone by human brain type 10 17beta-hydroxysteroid dehydrogenase. Brain research 2005; 1040:29-35; PMID:15804423; http://dx.doi.org/ 10.1016/j.brainres.2005.01.022 [DOI] [PubMed] [Google Scholar]
- 6.Adamski J, Jakob FJ. A guide to 17beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol 2001; 171:1-4; PMID:11165003; http://dx.doi.org/ 10.1016/S0303-7207(00)00383-X [DOI] [PubMed] [Google Scholar]
- 7.Zschocke J. HSD10 disease: clinical consequences of mutations in the HSD17B10 gene. J Inherit Metab Dis 2012; 35:81-9; PMID:22127393; http://dx.doi.org/ 10.1007/s10545-011-9415-4 [DOI] [PubMed] [Google Scholar]
- 8.Persson B, Kallberg Y, Oppermann U, Jornvall H. Coenzyme-based functional assignments of short-chain dehydrogenases/reductases (SDRs). Chemico-Biol Interact 2003; 143-144:271-8; PMID: 12604213; http://dx.doi.org/ 10.1016/S0009-2797(02)00223-5 [DOI] [PubMed] [Google Scholar]
- 9.Luo MJ, Mao LF, Schulz H. Short-chain 3-hydroxy-2-methylacyl-CoA dehydrogenase from rat liver: purification and characterization of a novel enzyme of isoleucine metabolism. Arch Biochem Biophys 1995; 321:214-20; PMID:7639524; http://dx.doi.org/ 10.1006/abbi.1995.1388 [DOI] [PubMed] [Google Scholar]
- 10.He XY, Merz G, Yang YZ, Pullakart R, Mehta P, Schulz H, Yang SY.. Function of human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase in androgen metabolism. Biochim Biophys Acta 2000; 1484:267-77; PMID:10760475; http://dx.doi.org/ 10.1016/S1388-1981(00)00014-7 [DOI] [PubMed] [Google Scholar]
- 11.He XY, Yang YZ, Schulz H, Yang SY. Intrinsic alcohol dehydrogenase and hydroxysteroid dehydrogenase activities of human mitochondrial short-chain L-3-hydroxyacyl-CoA dehydrogenase. Biochem J 2000; 345 Pt 1:139-43; PMID:10600649; http://dx.doi.org/ 10.1042/bj3450139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korman SH. Inborn errors of isoleucine degradation: a review. Mol Genet Metab 2006; 89:289-99; PMID:16950638; http://dx.doi.org/ 10.1016/j.ymgme.2006.07.010 [DOI] [PubMed] [Google Scholar]
- 13.Fukao T, Scriver CR, Kondo N, t2 Collaborative Working G . The clinical phenotype and outcome of mitochondrial acetoacetyl-CoA thiolase deficiency (beta-ketothiolase or T2 deficiency) in 26 enzymatically proved and mutation-defined patients. Mol Genet Metab 2001; 72:109-14; PMID:11161836; http://dx.doi.org/ 10.1006/mgme.2000.3113 [DOI] [PubMed] [Google Scholar]
- 14.Fukao T, Akiba K, Goto M, Kuwayama N, Morita M, Hori T, Aoyama Y, Venkatesan R, Wierenga R, Moriyama Y, et al.. The first case in Asia of 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (HSD10 disease) with atypical presentation. J Hum Genet 2014; 59:609-14; PMID:25231369; http://dx.doi.org/ 10.1038/jhg.2014.79 [DOI] [PubMed] [Google Scholar]
- 15.Seaver LH, He XY, Abe K, Cowan T, Enns GM, Sweetman L, Philipp M, Lee S, Malik M, Yang SY. A novel mutation in the HSD17B10 gene of a 10-year-old boy with refractory epilepsy, choreoathetosis and learning disability. PloS One 2011; 6:e27348; PMID:22132097; http://dx.doi.org/ 10.1371/journal.pone.0027348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenski C, Kooy RF, Reyniers E, Loessner D, Wanders RJ, Winnepenninckx B, Hellebrand H, Engert S, Schwartz CE, Meindl A, et al.. The reduced expression of the HADH2 protein causes X-linked mental retardation, choreoathetosis, and abnormal behavior. Am J Hum Genet 2007; 80:372-7; PMID:17236142; http://dx.doi.org/ 10.1086/511527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reyniers E, Van Bogaert P, Peeters N, Vits L, Pauly F, Fransen E, Van Regemorter N, Kooy RF. A new neurological syndrome with mental retardation, choreoathetosis, and abnormal behavior maps to chromosome Xp11. Am J Hum Genet 1999; 65:1406-12; PMID:10521307; http://dx.doi.org/ 10.1086/302638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holzmann J, Frank P, Loffler E, Bennett KL, Gerner C, Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 2008; 135:462-74; PMID:18984158; http://dx.doi.org/ 10.1016/j.cell.2008.09.013 [DOI] [PubMed] [Google Scholar]
- 19.Vilardo E, Nachbagauer C, Buzet A, Taschner A, Holzmann J, Rossmanith W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res 2012; 40:11583-93; PMID:23042678; http://dx.doi.org/ 10.1093/nar/gks910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helm M, Giege R, Florentz C. A Watson-Crick base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry 1999; 38:13338-46; PMID:10529209; http://dx.doi.org/ 10.1021/bi991061g [DOI] [PubMed] [Google Scholar]
- 21.Gai X, Ghezzi D, Johnson MA, Biagosch CA, Shamseldin HE, Haack TB, Reyes A, Tsukikawa M, Sheldon CA, Srinivasan S, et al.. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am J Hum Genet 2013; 93:482-95; PMID:23993194; http://dx.doi.org/ 10.1016/j.ajhg.2013.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falk MJ, Zhang Q, Nakamaru-Ogiso E, Kannabiran C, Fonseca-Kelly Z, Chakarova C, Audo I, Mackay DS, Zeitz C, Borman AD, et al.. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet 2012; 44:1040-5; PMID:22842227; http://dx.doi.org/ 10.1038/ng.2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bujakowska KM, Zhang Q, Siemiatkowska AM, Liu Q, Place E, Falk MJ, Consugar M, Lancelot ME, Antonio A, Lonjou C, et al.. Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum Mol Genet 2015; 24:230-42; PMID:25168386; http://dx.doi.org/ 10.1093/hmg/ddu441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, Wang DY, Au ED, et al.. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med: Off J Am College Med Genet 2015; 17:253-61; PMID:25412400; http://dx.doi.org/ 10.1038/gim.2014.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooper GM, Stone EA, Asimenos G, Program NCS, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005; 15:901-13; PMID:15965027; http://dx.doi.org/ 10.1101/gr.3577405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al.. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005; 15:1034-50; PMID:16024819; http://dx.doi.org/ 10.1101/gr.3715005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7:248-9; PMID:20354512; http://dx.doi.org/ 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31:3812-4; PMID:12824425; http://dx.doi.org/ 10.1093/nar/gkg509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7:575-6; PMID:20676075; http://dx.doi.org/ 10.1038/nmeth0810-575 [DOI] [PubMed] [Google Scholar]
- 30.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46:310-5; PMID:24487276; http://dx.doi.org/ 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kissinger CR, Rejto PA, Pelletier LA, Thomson JA, Showalter RE, Abreo MA, Agree CS, Margosiak S, Meng JJ, Aust RM, et al.. Crystal structure of human ABAD/HSD10 with a bound inhibitor: implications for design of Alzheimer's disease therapeutics. J Mol Biol 2004; 342:943-52; PMID:15342248; http://dx.doi.org/ 10.1016/j.jmb.2004.07.071 [DOI] [PubMed] [Google Scholar]
- 32.Powell AJ, Read JA, Banfield MJ, Gunn-Moore F, Yan SD, Lustbader J, Stern AR, Stern DM, Brady RL. Recognition of structurally diverse substrates by type II 3-hydroxyacyl-CoA dehydrogenase (HADH II)/amyloid-beta binding alcohol dehydrogenase (ABAD). J Mol Biol 2000; 303:311-27; PMID:11023795; http://dx.doi.org/ 10.1006/jmbi.2000.4139 [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Villoria J, Navarro-Sastre A, Fons C, Perez-Cerda C, Baldellou A, Fuentes-Castello MA, González I, Hernández-Gonzalez A, Fernández C, Campistol J, et al.. Study of patients and carriers with 2-methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) deficiency: difficulties in the diagnosis. Clin Biochem 2009; 42:27-33; PMID:18996107; http://dx.doi.org/ 10.1016/j.clinbiochem.2008.10.006 [DOI] [PubMed] [Google Scholar]
- 34.Vilardo E, Rossmanith W. Molecular insights into HSD10 disease: impact of SDR5C1 mutations on the human mitochondrial RNase P complex. Nucleic Acids Res 2015; 43:5112-9; PMID:25925575; http://dx.doi.org/ 10.1093/nar/gkv408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang SY, He XY, Schulz H. 3-Hydroxyacyl-CoA dehydrogenase and short chain 3-hydroxyacyl-CoA dehydrogenase in human health and disease. FEBS J 2005; 272:4874-83; PMID:16176262; http://dx.doi.org/ 10.1111/j.1742-4658.2005.04911.x [DOI] [PubMed] [Google Scholar]
- 36.Pasquali M, Monsen G, Richardson L, Alston M, Longo N. Biochemical findings in common inborn errors of metabolism. Am J Med Genet Part C, Semin Med Genet 2006; 142C:64-76; PMID:16602099; http://dx.doi.org/ 10.1002/ajmg.c.30086 [DOI] [PubMed] [Google Scholar]
- 37.Sutton VR, O'Brien WE, Clark GD, Kim J, Wanders RJ. 3-Hydroxy-2-methylbutyryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 2003; 26:69-71; PMID:12872843; http://dx.doi.org/ 10.1023/A:1024083715568 [DOI] [PubMed] [Google Scholar]
- 38.Yang SY, He XY, Olpin SE, Sutton VR, McMenamin J, Philipp M, Denman RB, Malik M. Mental retardation linked to mutations in the HSD17B10 gene interfering with neurosteroid and isoleucine metabolism. Proc Natl Acad Sci U S A 2009; 106:14820-4; PMID:19706438; http://dx.doi.org/ 10.1073/pnas.0902377106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rauschenberger K, Scholer K, Sass JO, Sauer S, Djuric Z, Rumig C, Wolf NI, Okun JG, Kölker S, Schwarz H, et al.. A non-enzymatic function of 17beta-hydroxysteroid dehydrogenase type 10 is required for mitochondrial integrity and cell survival. EMBO Mol Med 2010; 2:51-62; PMID:20077426; http://dx.doi.org/ 10.1002/emmm.200900055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christian T, Evilia C, Hou YM. Catalysis by the second class of tRNA(m1G37) methyl transferase requires a conserved proline. Biochemistry 2006; 45:7463-73; PMID:16768442; http://dx.doi.org/ 10.1021/bi0602314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christian T, Evilia C, Williams S, Hou YM. Distinct origins of tRNA(m1G37) methyltransferase. J Mol Biol 2004; 339:707-19; PMID:15165845; http://dx.doi.org/ 10.1016/j.jmb.2004.04.025 [DOI] [PubMed] [Google Scholar]
- 42.Christian T, Hou YM. Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J Mol Biol 2007; 373:623-32; PMID:17868690; http://dx.doi.org/ 10.1016/j.jmb.2007.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christian T, Lahoud G, Liu C, Hoffmann K, Perona JJ, Hou YM. Mechanism of N-methylation by the tRNA m1G37 methyltransferase Trm5. RNA 2010; 16:2484-92; PMID:20980671; http://dx.doi.org/ 10.1261/rna.2376210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christian T, Lahoud G, Liu C, Hou YM. Control of catalytic cycle by a pair of analogous tRNA modification enzymes. J Mol Biol 2010; 400:204-17; PMID:20452364; http://dx.doi.org/ 10.1016/j.jmb.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lahoud G, Goto-Ito S, Yoshida K, Ito T, Yokoyama S, Hou YM. Differentiating analogous tRNA methyltransferases by fragments of the methyl donor. RNA 2011; 17:1236-46; PMID:21602303; http://dx.doi.org/ 10.1261/rna.2706011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakaguchi R, Giessing A, Dai Q, Lahoud G, Liutkeviciute Z, Klimasauskas S, Piccirilli J, Kirpekar F, Hou YM. Recognition of guanosine by dissimilar tRNA methyltransferases. RNA 2012; 18:1687-701; PMID:22847817; http://dx.doi.org/ 10.1261/rna.032029.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakaguchi R, Lahoud G, Christian T, Gamper H, Hou YM. A divalent metal ion-dependent N(1)-methyl transfer to G37-tRNA. Chem Biol 2014; 21:1351-60; PMID:25219964; http://dx.doi.org/ 10.1016/j.chembiol.2014.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hou YM. High-purity enzymatic synthesis of site-specifically modified tRNA. Methods Mol Biol 2012; 941:195-212; PMID:23065563; http://dx.doi.org/ 10.1007/978-1-62703-113-4_15 [DOI] [PubMed] [Google Scholar]
- 49.Shitivelband S, Hou YM. Breaking the stereo barrier of amino acid attachment to tRNA by a single nucleotide. J Mol Biol 2005; 348:513-21; PMID:15826650; http://dx.doi.org/ 10.1016/j.jmb.2005.02.023 [DOI] [PubMed] [Google Scholar]
- 50.McLaughlin HM, Sakaguchi R, Liu C, Igarashi T, Pehlivan D, Chu K, Iyer R, Cruz P, Cherukuri PF, Hansen NF, et al.. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet 2010; 87:560-6; PMID:20920668; http://dx.doi.org/ 10.1016/j.ajhg.2010.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLaughlin HM, Sakaguchi R, Giblin W, Program NCS, Wilson TE, Biesecker L, Lupski JR, Talbot K, Vance JM, Züchner S, et al.. A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N). Hum Mutat 2012; 33:244-53; PMID:22009580; http://dx.doi.org/ 10.1002/humu.21635 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.