

Abstract
Development of Heat shock protein 90 (Hsp90) C-terminal inhibitors has emerged as an exciting strategy for the treatment of cancer. Previous efforts have focused on modifications to the natural products novobiocin and coumermycin. Moreover, variations in both the sugar and amide moieties have been extensively studied, whereas replacements for the coumarin core have received less attention. Herein, 24 cores were synthesized with varying distances and angles between the sugar and amide moieties. Compounds that exhibited good anti-proliferative activity against multiple cancer cell lines and Hsp90 inhibitory activity, were those that placed the sugar and amide moieties between 7.7 to 12.1 Å apart along with angles of 180°.
Keywords: Hsp90, C-terminal inhibitors, Luciferase refolding, Anti-cancer agents, Novobiocin
Graphical abstract

Introduction
Heat shock protein 90 (Hsp90) is a molecular chaperone that is responsible for folding nascent polypeptides into their biologically active conformation. More than 200 client proteins depend upon Hsp90 for their maturation/activation, and many are essential to cancer cell survival and proliferation.[1–4] Unlike drugs that target these individual client proteins, inhibition of Hsp90 results in the degradation of more than 30 anti-cancer targets simultaneously. Furthermore, Hsp90 is overexpressed and can be selectively targeted in cancer, thus its inhibition can diminish potential side effects.[5–7]
17 Hsp90 inhibitors have entered clinical trials for the treatment of cancer. All of these inhibitors bind the N-terminal ATP binding site.[8–10] The major problem associated with these inhibitors is induction of the heat shock response (pro survival) at the same concentration that leads to protein degradation. Due to increased Hsp90 levels observed upon administration of N-terminal inhibitors, the scheduling and dosing of patients is often difficult.[11], Presently, no Hsp90 inhibitor is FDA-approved for the treatment for cancer.
In contrast to N-terminal inhibitors, C-terminal inhibitors can be prepared that do not induce the pro-survival heat shock response that is responsible for these dosing difficulties. [12] In fact, Hsp90 C-terminal inhibitors have proven to exhibit distinct properties when compared to N-terminal inhibitors.[13,14] Therefore, the development of Hsp90 C-terminal inhibitors could provide a clinically useful alternative for the treatment of cancer.[15–19]
Studies have shown that natural product C-terminal inhibitors,[20–24] such as novobiocin and coumermycin A1, do not induce the heat shock response. Unfortunately, these compounds manifest IC50 values of 700 µM and 70 µM, respectively.[25,26] Although no crystal structure of the Hsp90 C-terminus bound to inhibitors exists, homology models have been developed to aid the design of new inhibitors.[27–29] Using these models, structure-activity-relationship studies on novobiocin and coumermycin have led to compounds that manifest activity in the mid to low nanomolar range against a variety of cancers,[30] which supports the potential use of these compounds as anti-cancer therapeutics.
To date, SAR studies on novobiocin have primarily focused on finding replacements for the noviose sugar and the prenylated aryl side chain. (Figure 1) In fact, more potent Hsp90 inhibitors can be accessed by replacement of the noviose sugar with N-methylpiperidine (2) or by substitution of the prenylated aryl side chain with a biaryl side chain (3).[31] Both of these modifications not only lead to more efficacious analogs, but also a simplified synthesis, as the noviose sugar requires more than 10 synthetic transformations. In addition to the coumarin core, biaryl cores (4 and 5) have been discovered that manifest superior anti-cancer activity.[32] Since compounds containing a biaryl core exhibit improved inhibitory activity over the coumarin-based compounds, it suggests that some flexibility within the C-terminal binding pocket may exist. Additionally, orientation of the N-methylpiperidine and the biaryl side chain may also be important for the increased efficacy. In fact, compounds prepared to date project the sugar and amide side chains linearly at ~180°. Therefore, the focus of this study was to determine the optimal distance and angle between the N-methylpiperidine and the biaryl side chain.
Figure 1.

Novobiocin and analogs previously evaluated
Results and Discussion
Design and synthesis of novel scaffolds
In order to determine the optimal distance and angle between the N-methylpiperidine and the biaryl side chain, cores were designed to place these groups between 6.2 and 14.3 Å apart as presented in Table 1. From a design perspective, initial efforts were focused on modification of the bicyclic core represented by the coumarin ring in novobiocin. First, the biaryl side chain was placed at the 1-position of both the tetrahydronaphthalene and naphthalene cores. (6 and 7). Second, the biaryl side chain was placed at the 2-position of the tetrahydronaphthalene core. Compounds 6–8 would provide insight into both the position and planarity of the biaryl side chain on the bicyclic ring system.
Table 1.
List of cores with varying distance and angles
 | |||
|---|---|---|---|
| Compound | Structure | Distance[a,b] | Angle(θ)[b] |
| 6 | 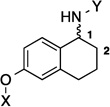 |
6.2 Å | 73.4° |
| 7 | 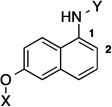 |
6.3 Å | 120° |
| 8 | 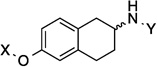 |
7.8 Å | 160.9° |
| 9 | 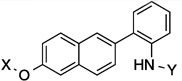 |
7.7 Å | 115° |
| 10 | 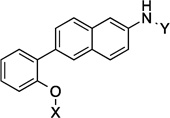 |
7.9 Å | 115° |
| 11 | 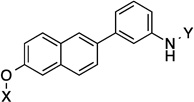 |
10.4 Å | 139.3° |
| 12 | 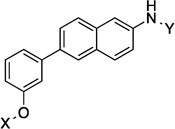 |
10.5 Å | 141.6° |
| 13 | 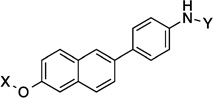 |
11.8 Å | 168.9° |
| 14 | 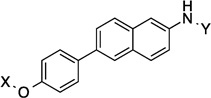 |
11.8 Å | 172.1° |
| 15 | 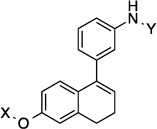 |
7.8 Å | 79.4° |
| 16 | 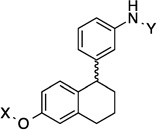 |
9.1 Å | 118.8° |
| 17 | 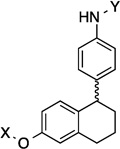 |
9.2 Å | 108.3° |
| 18 | 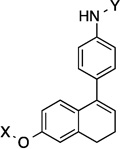 |
9.8 Å | 104.3° |
| 19 | 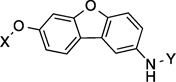 |
9.1 Å | 124.6° |
| 20 | 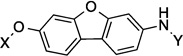 |
9.4 Å | 154.5° |
| 21 | 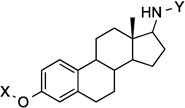 |
9.2 Å | 168.3° |
| 22 | 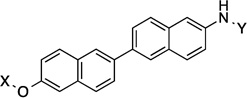 |
14.1 Å | 180° |
| 23 | 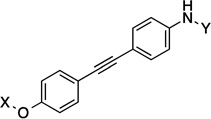 |
12.1 Å | 180° |
| 24 | 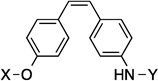 |
9.1 Å | 120° |
| 25 | 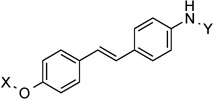 |
11.8 Å | 170.7° |
| 26 | 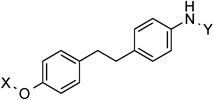 |
11.9 Å | 166.6° |
| 27 | 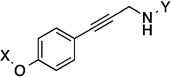 |
8.7 Å | 163° |
| 28 | 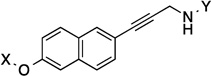 |
10.9 Å | 172.2° |
| 29 | 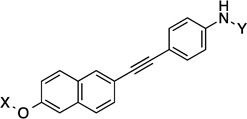 |
14.3 Å | 160.5° |
Distance measured between the oxygen bound to N-methylpiperidine and amide nitrogen of the biaryl side chain
Both distance and angles were measured in PyMOL after the 3D structures had been minimized through molecular dynamic simulations.
Next, a series of compounds with a three-ring core was designed. The first set of compounds contained a phenyl-naphthalene core, wherein the biaryl side chain or the N-methylpiperidine was placed at all three positions on the benzene ring. (Figure 2) A variety of distances and angles could be quickly accessed with this scaffold, as advanced intermediates could be used for the preparation of additional compounds.
Figure 2.

Analogs with phenyl-naphthalene core
The second set of compounds investigated were the 1-phenyl-di(tetra)hydronaphthalene analogs. (15–18) Initially, the 1-phenyl-dihydronaphthalene scaffold was designed to compare placement of the phenyl ring on the 1- versus 2-position of the naphthalene ring. Later, 1-phenyl-tetrahydronaphthalene analogs could also be accessed by use of an intermediate. The 1-phenyl-tetrahydronaphthalene scaffold would provide data on planarity of the three-ring system and its effect on anti-proliferative activity. The third set of compounds contained two fused three-ring cores. (19 and 20). Unlike the previous sets of compounds, where one of the rings was able to rotate freely, the fused ring system provides ridged counterparts for investigation. A comparison of anti-proliferative activities between these compounds would help establish the role played by the core.
Compounds 21 and 22 contain 4-ring analogs that were designed for this investigation. Additionally, compound 23 represents a derivative of 5, where an alkyne was placed between the phenyl rings to increase distance between the N-methylpiperidine and the biaryl side chain by two carbons, while maintaining linearity. The alkyne in 23 could be reduced to both the alkene (24 and 25) and alkane (26) to provide insights into the flexibility and geometry of the two-carbon linker. Finally, an additional series of alkyne-containing compounds was envisioned (27–29) that extend from an aromatic core.
Various angles between the N-methylpiperidine and the biaryl side chain were accessed by placement of these groups at different positions within the same core structure. Compounds 9, 11 and 13 are representative examples wherein this strategy was utilized. Alternatively, different angles were observed when the biaryl side chain was placed out of plane with regards to the core (Figure 3). Scheme 1 illustrates the synthetic protocol used to prepare 1-amido and 2-amido analogs. Amines 30a/b, which were synthesized from the corresponding 6-methoxy-tetralones (See supporting information), were subjected to an EDCI coupling with carboxylic acid A. After removal of the benzyl group via hydrogenolysis, phenol 32a/b was coupled with N-methyl-4-piperidinol (B) through Mitsunobu etherification.[33]
Figure 3.

Graphic representation of the biaryl side chain that rests either in or out-of-plane
Scheme 1.

Synthesis of 1 and 2-amido novobiocin analogs
Synthesis of select analogs
A representative synthesis of the phenyl-naphthalene analogs is shown in scheme 2. Synthesis began by the coupling of commercially available 2-amino-6-bromonaphthalene (33) with C to form amide 34 in good yield. Amide 34 was the common intermediate used to prepare analogs wherein the N-methylpiperidine was placed at all three positions on the phenyl substituent. A Suzuki coupling between amide 34 and boronic acids 35a–c produced the phenyl-naphthalenes, 36a–c, in moderate yield. Finally phenols 36a–c were coupled with B via a Mitsunobu reaction to generate the final products (10, 12, 14) in 53–60% yield.
Scheme 2.

Synthesis of 2-Phenyl-Naphthalene Analog
Synthesis of 1-phenyl-di(tetra)hydronaphthalene analogs commenced via a Suzuki coupling between commercially available boronic acids, 38a/b, and vinyl triflate 37[34] to yield the corresponding nitro aromatics, 39a/b (Scheme 3). Following demethylation of 39a/b, N-methyl-4-piperidinol was coupled with phenols 40a/b under Mitsunobu conditions. At this point, the synthesis diverged and the nitro group in 41a/b selectively reduced to the corresponding aniline using iron-catalyzed conditions (42a/b); or both the nitro group and the alkene were reduced via a palladium-catalyzed hydrogenation (43a/b). The resulting anilines were then coupled with acid chloride C to form amide 15/18 or 16/17, respectively.
Scheme 3.

Synthesis of 1-phenyl-di(tetra)hydronaphthalene analogs
Preparation of the dibenzo[b,d]furan cores is shown in scheme 4.[35] Synthesis of the dibenzofuran-containing analogs began by a Suzuki coupling between boronic acid 44 and aryl bromides 45a/b to yield 46a/b in moderate yield. Pyridine-HCl was used to cleave the methyl ethers of 46a/b and provide resorcinols 47a/b. An intramolecular nucleophilic substitution of the resorcinol moiety (47a/b) generated dibenzofuranols 48a/b in good yield. N-methyl-4-piperidinol was added to 48a/b through a Mitsunobu reaction, which produced 49a/b in good yield. Following reduction of the nitro group present in 49a/b, the resulting aniline was coupled with acid chloride C to furnish 19 and 20. The synthesis of an alkyne-containing compound is outlined in Scheme 5. Commercially available 4-ethynylanisole (51) was coupled with 1-bromo-4-nitrobezene (52) under Sonogashira conditions to yield alkyne 53 in 82% yield. Following cleavage of the methyl ether present in 53, phenol 54 was treated with N-methyl-4-piperidinol under Mitsunobu conditions to furnish 55 in moderate yield. The nitro group on 55 was subsequently reduced,[36] and the resulting aniline was coupled with C to produce the final product, 23.
Scheme 4.

Synthesis of dibenzo[b,d]furan analogs
Scheme 5.

Synthesis of 1,2-Diphenyl-Ethyne Analog
Evaluation of anti-proliferation activity of analogs
Upon preparation of the compounds listed table 1, the anti-proliferation activity manifested by these compounds was evaluated against MCF-7 (estrogen receptor positive breast cancer cells), SKBr3 (estrogen receptor negative, HER2 overexpressing breast cancer cells), PC3-MM2 (androgen-independent prostate cancer cells) and MDA-MB-468Ln (human breast cancer triple negative) cell lines (Table 2). In the bicyclic core series, the position of the amide was important for anti-proliferation activity. When the amide was attached at the 1-position, no anti-proliferative activity observed for the aromatic core (in-plane) (7), whereas moderate activity was observed when the amide was placed out-of plane (6). In contrast, when the amide was placed at the 2-position, an increase in anti-proliferation activity was observed when the amide was in-plane (57) vs. out-of-plane (8).
Table 2.
Anti-proliferative activity manifested by novobiocin analogs
| Compd | Structure | Dist. | Angle | MCF-7[a] | SKBr3[a] | PC3-MM2 [a] |
MDA-MB- 468Ln[a,d] |
|---|---|---|---|---|---|---|---|
| 6 | 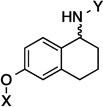 |
6.2 Å | 73.4° | 5.4±0.80 | 5.4±0.00 | 8.7±0.37 | 7.0 |
| 7 | 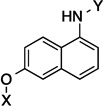 |
6.3 Å | 120° | >50 | >50 | >50 | 22.1 |
| 8 | 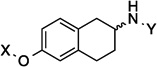 |
7.8 Å | 160.9° | 6.7± 0.00 | 7.6± 0.00 | 10.7±0.10 | 12.83 |
| 57[b] | 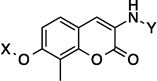 |
7.8 Å | 180° | 1.5 ± 0.30 | 1.3 ± 0.22 | 3.1±0.67 | NT |
| 9 | 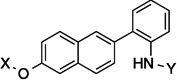 |
7.7 Å | 115° | 3.9±0.016 | 2.0±0.016 | 2.2±0.03 | 3.0 |
| 10 | 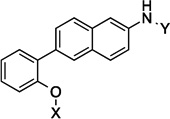 |
7.9 Å | 115° | 2.6±0.738 | 3.3±0.05 | 7.2±1.1 | 4.3 |
| 11 | 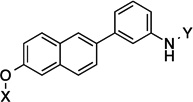 |
10.4 Å |
139.3° | 2.9±0.639 | 2.3±0.06 | 7.5±0.26 | 3.0 |
| 12 | 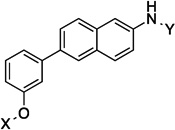 |
10.5 Å |
141.6° | 2.3±0.13 | 3.5±0.02 | 5.7±0.12 | 3.3 |
| 13 | 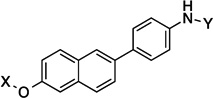 |
11.8 Å |
168.9° | 1.5±0.139 | 1.4 ± 0.09 | 7.9±1.2 | 2.2 |
| 14 | 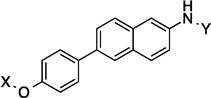 |
11.8 Å |
172.1° | 3.4±0.09 | 2.4±0.17 | 7.49±0.17 | 4.2 |
| 15 | 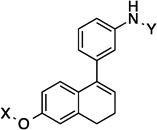 |
7.8 Å | 79.4° | 2.1± 0.03 | 1.8± 0.64 | 5.2±1.0 | 3.8 |
| 16 | 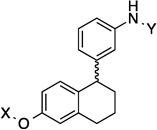 |
9.1 Å | 118.8° | 1.8±0.43 | 1.7±0.70 | 3.3±0.25 | 2.6 |
| 17 | 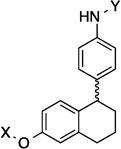 |
9.2 Å | 108.3° | 2.6±0.63 | 3.2±0.90 | 5.4±1.1 | 1.9 |
| 18 | 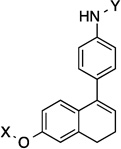 |
9.8 Å | 104.3° | 2.5±0.40 | 2.2±0.06 | 5.6±0.27 | 3.5 |
| 19 |  |
9.1 Å | 124.6° | 2.9±0.58 | 3.3±0.35 | 10.3±0.35 | 5.5 |
| 20 | 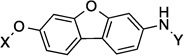 |
9.4 Å | 154.5° | 4.2±0.80 | 6.4±0.21 | 12.0±0.21 | >10 |
| 21 | 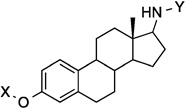 |
9.2 Å | 168.3° | 3.3±0.37 | 4.9±0.43 | 7.9±0.76 | 5.2 |
| 22 | 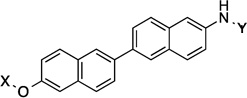 |
14.1 Å |
180° | 20.9±2.54 | 11.8±0.94 | >50 | >10 |
| 23 | 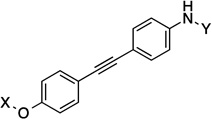 |
12.1 Å |
180° | 2.7±0.79 | 4.3±0.01 | 5.8±0.32 | 4.9 |
| 24 | 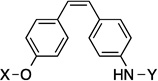 |
9.1 Å | 120° | 5.9±0.00 | 7.5±0.00 | 7.8±1.1 | 6.0 |
| 25 | 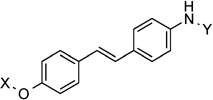 |
11.8 Å |
170.7° | 1.3±0.59 | 0.68±0.10 | 1.3±0.26 | 0.50 |
| 26 | 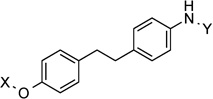 |
11.9 Å |
166.6° | 9.8±3.4 | 2.9±1.108 | 5.3 | 4.6 |
| 5[c] | 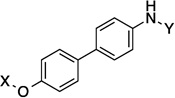 |
9.6 Å | 180° | 0.71±0.02 | 0.47±0.06 | 0.98 | NT |
| 27 | 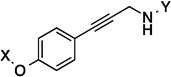 |
8.7 Å | 163° | 8.7±1.9 | 14.3±1.20 | >50 | 26.8 |
| 28 | 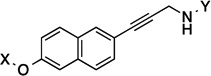 |
10.9 Å |
172.2° | 8.8 | 2.2±0.14 | 2.1±0.41 | 1.9 |
| 29 |  |
14.3 Å |
160.5° | 4.3±0.42 | 3.5±0.63 | 5.4±0.07 | 2.2 |
The anti-proliferative activity manifested by the 2-phenyl-napthalene cores were slightly worse than the 1-phenyl-di(tetra)hydronaphthalene cores and better than the dibenzo[b,d]furan cores in the three-ring series. Compounds containing the amide side chain on the phenyl ring (9, 11 and 13) were more active than compounds containing the N-methylpiperidine on the phenyl ring (Entries 10, 12 and 14), with the para substituted amide analog being most efficacious for the 2-phenyl-naphthalene series (13). When the amide was attached to the para-position of the 1-phenyl-di(tetra)hydronaphthalene core, a marginal difference in activity for the di- versus tetrahydronaphthalene core was observed (17 and 18). On the other hand, when the amide was placed at the ortho-position, a slight increase in activity against SKBr3 cells and a 2-fold increase in activity against PC3-MM2 cells was observed (15 and 16). Finally, there was a 2-fold increase in activity when the N-methylpiperidine was located at the 6-position (19) versus the 7-position (20) of the dibenzo[b,d]furan core. The trends observed for these three-analogs indicate that compounds are less potent when the rings are fused, versus compounds that can rotate freely.
The incorporation of alkynes into the structure did not produce more potent compounds than their counterparts. For example, 5 exhibits mid-nanomolar activity against all of the cell lines, whereas 23 (which contains an alkyne between the two phenyl rings) manifested a 6-fold decrease in activity. A similar trend was also observed between 13 and 29. Interestingly, when the alkyne of 23 was reduced to the E-alkene (25), an increase in activity was observed. In contrast, the Z-alkene (24) exhibited a slight decrease in activity when compared to 23. However, when 23 was fully saturated, a 3-fold decrease in activity was observed against the MCF-7 cell line, whereas a 2-fold increase in activity was observed against SKBr3 cells. These observations highlight the importance of identifying the optimal distance and angle between the N-methylpiperidine and the biaryl side chain.
Based on the data outlined in table 2, three-dimensional graphs[37] were generated to compare the anti-proliferative activity versus the angle and distance between the N-methylpiperidine and the biaryl side chain for MCF-7. SKBr3 and PC3-MM2 cell lines. (Figure 4a) As the angle approaches 180°, the anti-proliferation activity increased in all cell lines. The optimal distance for good anti-proliferation activity (<2µM) was variable in these cell lines. In the MCF-7 cell line, the optimal distance for activity was 7.5 to 10.5Å, although a small pocket was located from 11.5 to 12 Å. In the SKBr3 cell line, the optimal distance was 7.5 to 14Å. In the PC3-MM2 cell line, the optimal distance for good activity was 7.5 to 10Å. In general, the most potent compounds (<1µM) were those that placed the N-methylpiperidine and the biaryl side chain at a 180° angle and between 7.7 to 9.6Å apart from one another.
Figure 4.

A. 3D graphs for the anti-proliferation vs. distance vs. angles for MCF-7, SKBr3 and PC3-MM2 cell lines. B. 3D graph for the Hsp90 inhibitory activity vs. distance vs. angles
Evaluation of Hsp90 inhibitory activity of analogs
After measuring the anti-proliferation activity manifested by these compounds, the molecules were further investigated in the Hsp90-dependent luciferase-refolding assay to correlate Hsp90 inhibitory activity with cell viability.[38] Table 3 lists the compounds that exhibited Hsp90 inhibitory activity. The data in table 3 was then displayed in a three-dimensional graph, which compares Hsp90 inhibitory activity versus the distance and angle between the N-methylpiperidine and the biaryl side chain. (Figure 4b)
Table 3.
Hsp90 inhibitory activity of analogs
| Entry | Distance | Angle | Hsp90 IC50 (µM) [a] |
|---|---|---|---|
| 6 | 6.2 Å | 73.4° | 54.7 |
| 8 | 7.8 Å | 160.9° | 118 |
| 9 | 7.7 Å | 115° | 102 |
| 14 | 11.8 Å | 172.1° | 391 |
| 63 | 7.8 Å | 180° | 61.7 |
| 15 | 7.8 Å | 79.4° | 967 |
| 16 | 9.1 Å | 118.8° | 491 |
| 18 | 9.8 Å | 104.3° | 292 |
| 19 | 9.1 Å | 124.6° | 61.2 |
| 20 | 9.4 Å | 154.5° | 149 |
| 21 | 9.2 Å | 168.3° | 115 |
| 5 | 9.6 Å | 180° | 15.8 |
| 23 | 12.1 Å | 180° | 40.9 |
| 24 | 9.1 Å | 120° | 179 |
| 25 | 11.8 Å | 170.7° | 64.9 |
| 26 | 11.9 Å | 166.6° | 85.1 |
| 27 | 8.7 Å | 163° | 266 |
| 28 | 10.9 Å | 172.2° | 821 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
A distance of 6.2 and 12.1Å between the N-methylpiperidine and the biaryl side chain was optimal for Hsp90 inhibitory activity. In general, the distance observed for the compounds in table 3 fit within the optimal distance identified for the manifestation of good anti-proliferative activity, though there were some exceptions (6, 23). Similar to the anti-proliferation data, the Hsp90 inhibitory activity increased as the angle approached 180°, although exceptions were noted for compounds with a distance of less than 8 Å. There were several compounds that fit within the parameters for distance and angle but did not exhibit Hsp90 inhibitory activity. This information suggests that identification of Hsp90 inhibitors cannot be limited simply to distance and angle. Interestingly, the fused 3- and 4-ring systems (19–21) were generally more potent Hsp90 inhibitors as compared to the 3-ring systems (14, 15, 16, 18) that contained a freely rotatable ring, which was opposite to the observations in the anti-proliferation studies. Similarly, 23 was the most potent Hsp90 inhibitor, even though its counterpart, 5, was the most potent anti-proliferative agent. This data suggests that the Hsp90 inhibitory activity is best for compounds that are flat and exhibit a distance greater than 8 Å.
In order to provide further evidence that the anti-proliferation activity was due to Hsp90 inhibition, Western blot analysis was performed on 25, which exhibited the most potent anti-proliferation activity and good Hsp90 inhibitory activity. As shown in figure 5, 25 induced degradation of Hsp90-dependant clients Her2, Raf-1 and CDK6. Actin was used as a control, since this protein is not an Hsp90-dependent substrate.
Figure 5.

Western blot analyses of Hsp90 client protein degradation in PC3-MM2 cells treated with 25. L represents a concentration 1/2 × IC50 value while H represents a concentration of 5 × IC50 value. GDA (500 nM) represents a positive control, while DMSO, represents the negative control.
Conclusions
In this study, the distance and angle between the N-methylpiperidine and the biaryl side chain was analyzed in an effort to develop more potent Hsp90 C-terminal inhibitors. The best anti-proliferative and Hsp90 inhibitory activities were obtained when the distance was between 7.7 to 12.1 Å and the angle was close to 180°. (Figure 6) As a result of this investigation, a compound (25) was developed, which exhibited mid-nanomolar activity against SKBr3 cells. This study also revealed other structural aspects such as planarity and flexibility are important to achieve good anti-proliferative and Hsp90 inhibitory activities. Based on the information gathered in this study, the compounds that exhibited good anti-proliferative and Hsp90 inhibitory activities can be used as a template to develop more novel and useful Hsp90 inhibitors.
Figure 6.

Summary of optimal distance and angle between the N-methylpiperidine and the biaryl side chain
Experimental Section
1H NMR were recorded at 400 or 500 MHz (Bruker DRX- 400 Bruker with a H/C/P/F QNP gradient probe) spectrometer and 13C NMR spectra were recorded at 125 MHz (Bruker DRX 500 with broadband, inverse triple resonance, and high resolution magic angle spinning HR-MA probe spectrometer); chemical shifts are reported in δ (ppm) relative to the internal reference CDCl3-d (CDCl3, 7.27 ppm). FAB (HRMS) spectra were recorded with a LCT Premier (Waters Corp., Milford, MA) spectrometer. The purity of all compounds was determined to be >95% as determined by 1H NMR and 13C NMR spectra, unless otherwise noted. TLC was performed on glass- backed silica gel plates (Uniplate) with spots visualized by UV light. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions and extractions involved the use of a rotary evaporator operating at reduced pressure.
Supplementary Material
Acknowledgments
We would like to thank Yusuf Oluwasegun Adeshina and Arnab Bandyopadhyay for producing the 3D graphs for this study. Also, we gratefully acknowledge support of this project by the National Institute of Health (CA120458).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Trepel J, Mollapour M, Giaccone G, Neckers L. Nature Rev. Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Travers J, Sharp S, Workman P. Drug Disc. Today. 2012;17:242–252. doi: 10.1016/j.drudis.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 3.Seo YH. Arch. Pharm. Res. 2015;38:1582–1590. doi: 10.1007/s12272-015-0636-1. [DOI] [PubMed] [Google Scholar]
- 4.Neckers L. J. Biosci. 2007;32:517–530. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 5.a) Picard D. Cell. Mol. Life Sci. 2002;59:1640–1648. doi: 10.1007/PL00012491. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Issacs JS, Xu W, Neckers L. Cancer Cell. 2003;3:213–217. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]; c) Pratt WB, Toft DO. Exp. Biol. Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]; d) Whitesell L, Lindquist SL. Nature Rev. Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 6.Moser C, Lang SA, Stoeltzing O. Anticancer Res. 2009;29:2031–42. [PubMed] [Google Scholar]
- 7.Koga F, Kihara K, Nechers L. Anticancer Res. 2009;29:797–807. [PubMed] [Google Scholar]
- 8.Franke J, Eichner S, Zeilinger C, Kirschning A. Nat. Prod. Rep. 2013;30:1299–1323. doi: 10.1039/c3np70012g. [DOI] [PubMed] [Google Scholar]
- 9.Khandelwal A, Crowley V, Blagg SJB. Med. Res. Rev. 2015;36:92–118. doi: 10.1002/med.21351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jhaveri K, Modi S. Onco Targets Ther. 2015;8:1849–1858. doi: 10.2147/OTT.S65804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong Banerji U, Tavana B, George GC, Aaron J, Kurzock R. Cancer Treatment Reviews. 2013;39:375–387. doi: 10.1016/j.ctrv.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 12.a) Shelton SN, Shawgo ME, Comer SB, Lu Y, Donnelly AC, Szabla K, Tanol M, Vielhauer GA, Rajewski RA, Matts RL, Blagg BS, Robertson JD. Mol. Pharmacol. 2009;76:1314–1322. doi: 10.1124/mol.109.058545. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Conde R, Belak ZR, Nair M, O’Carroll RF, Ovsenek N. Biochem. Cell Biol. 2009;87:845–851. doi: 10.1139/o09-049. [DOI] [PubMed] [Google Scholar]
- 13.Donnelly A, Blagg BSJ. Curr. Med. Chem. 2008;15:2702–2717. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomez-Monterrey I, Sala M, Musella S, Campiglia P. Recent Pat. Anticancer Drug Discov. 2012;7:313–336. doi: 10.2174/157489212801820066. [DOI] [PubMed] [Google Scholar]
- 15.Song X, Zhao Z, Qi X, Tang S, Wang Q, Zhu T, Gu Q, Liu M, Li J. Oncotarget. 2015;6:5263–5274. doi: 10.18632/oncotarget.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, McAlpine SR. Org. Biomol. Chem. 2015;13:4627–4631. doi: 10.1039/c5ob00044k. [DOI] [PubMed] [Google Scholar]
- 17.Strocchia M, Terracciano S, Chini MG, Vassallo A, Vaccaro MC, Dal Piaz F, Leone A, Riccio R, Bruno I, Bifulco G. Chem Commun. 2015;51:3850–3853. doi: 10.1039/c4cc10074c. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, McAlpine SR. Chem. Commun. 2015;51:1410–1403. doi: 10.1039/c4cc07284g. [DOI] [PubMed] [Google Scholar]
- 19.Gavenonis J, Jonas NE, Kritzer JA. Bioorg. Med. Chem. 2014;22:3989–3993. doi: 10.1016/j.bmc.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davenport J, Manjarrez JR, Peterson L, Krumm B, Blagg BSJ, Matts RL. J. Nat. Prod. 2011;74:1085–1092. doi: 10.1021/np200029q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khandelwal A, Hall JA, Blagg BSJ. J. Org. Chem. 2013;78:7859–7884. doi: 10.1021/jo401027r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin Z, Henry EC, Gasiewicz TA. Biochemistry. 2009;48:336–345. doi: 10.1021/bi801637q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hadden MK, Galam L, Gestwicki JE, Matts RL, Blagg BSJ. J. Nat. Prod. 2007;70:2014–2018. doi: 10.1021/np070190s. [DOI] [PubMed] [Google Scholar]
- 24.Zhao H, Brandt GE, Galam L, Matts RL, Blagg BSJ. Bioorg. Med. Chem. Lett. 2011;21:2659–2664. doi: 10.1016/j.bmcl.2010.12.088. [DOI] [PubMed] [Google Scholar]
- 25.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. J. Biol. Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 26.Marcu MG, Schulte TW, Neckers L. J. Natl. Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 27.Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, et al. ACS Chem. Biol. 2011;6:800–807. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moroni E, Zhao H, Blagg BSJ, Colombo G. J. Chem. Inf. Model. 2014;54:195–208. doi: 10.1021/ci4005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sattin S, Tao J, Vettoretti G, Moroni E, Pennati M, Lopergolo A, et al. Chem. Eur. J. 2015;21:13598–15608. doi: 10.1002/chem.201502211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Garg G, Zhao H, Blagg BSJ. ACS Med. Chem. Lett. 2015;6:204–209. doi: 10.1021/ml5004475. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hall JA, Forsberg LK, Blagg BSJ. Future Med. Chem. 2014;6:1587–1605. doi: 10.4155/fmc.14.89. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhao J, Zhao H, Hall JA, Brown D, Brandes E, Bazzill J, et al. MedChemComm. 2014;5:1317–1323. doi: 10.1039/C4MD00102H. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhao H, Moroni E, Colombo G, Blagg BSJ. ACS Med. Chem. Lett. 2014;5:84–88. doi: 10.1021/ml400404s. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhao H, Yan B, Peterson LB, Blagg BSJ. ACS Med. Chem. Lett. 2012;3:327–331. doi: 10.1021/ml300018e. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kusuma BR, Peterson LB, Zhao H, Vielhauer G, Holzbeierlein Blagg BSJ. J. Med. Chem. 2011;54:6234–6253. doi: 10.1021/jm200553w. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. J. Am. Chem. Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]; h) Burlison JA, Blagg BSJ. Org. Lett. 2006;8:4855–4858. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]; i) Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. J. Am. Chem. Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 31.a) Zhao H, Kusuma BR, Blagg BSJ. ACS Med. Chem. Lett. 2010;1:311–315. doi: 10.1021/ml100070r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao H, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. J. Med. Chem. 2011;54:3839–3853. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.a) Zhao H, Garg G, Zhao J, Moroni E, Girgis A, Franco LS, et al. Eur. J. Med. Chem. 2015;89:442–466. doi: 10.1016/j.ejmech.2014.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao H, Anyika M, Girgis A, Blagg BSJ. Bioorg. Med. Chem. Lett. 2014;24:3633–3637. doi: 10.1016/j.bmcl.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsunoda T, Yamamiya Y, Kawamura Y, Itô S. Tet. Lett. 1995;36:2529–2530. [Google Scholar]
- 34.Richmond E, Duguet N, Slawin AMZ, Lébl T, Smith AD. Org. Lett. 2012;14:2762–2765. doi: 10.1021/ol300982f. [DOI] [PubMed] [Google Scholar]
- 35.Jepsen TH, Larsen M, Jørgensen M, Nielsen MB. Synth. 2013;45:1115–1120. [Google Scholar]
- 36.Sharma U, Verma PK, Kumar N, Kumar V, Bala M, Singh B. Chem. Eur. J. 2011;17:5903–5907. doi: 10.1002/chem.201003621. [DOI] [PubMed] [Google Scholar]
- 37.Wolfram Research, Inc. Mathematica, Version 10.3. Champaign, IL: 2015. [Google Scholar]
- 38.a) Galam L, Hadden MK, Ma Z, Ye Q, Yun B, Blagg BSJ, et al. Bioorg. Med. Chem. 2007;15:1939–1946. doi: 10.1016/j.bmc.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sadikot T, Swink M, Eskew JD, Brown D, Zhao H, Kusuma BR, et al. Assay Drug Dev. Tech. 2013;11:478–488. doi: 10.1089/adt.2012.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.