

Abstract
Background
Increased reports of human infections have led fasciolosis, a widespread disease of cattle and sheep caused by the liver flukes Fasciola hepatica and Fasciola gigantica, to be considered an emerging zoonotic disease. Chemotherapy is the main control measure available, and triclabendazole is the preferred drug since is effective against both juvenile and mature parasites. However, resistance to triclabendazole has been reported in several countries urging the search of new chemical entities and target molecules to control fluke infections.
Methodology/Principle Findings
We searched a library of forty flavonoid derivatives for inhibitors of key stage specific Fasciola hepatica cysteine proteases (FhCL3 and FhCL1). Chalcones substituted with phenyl and naphtyl groups emerged as good cathepsin L inhibitors, interacting more frequently with two putative binding sites within the active site cleft of the enzymes. One of the compounds, C34, tightly bounds to juvenile specific FhCL3 with an IC50 of 5.6 μM. We demonstrated that C34 is a slow-reversible inhibitor that interacts with the Cys-His catalytic dyad and key S2 and S3 pocket residues, determinants of the substrate specificity of this family of cysteine proteases. Interestingly, C34 induces a reduction in NEJ ability to migrate through the gut wall and a loss of motility phenotype that leads to NEJ death within a week in vitro, while it is not cytotoxic to bovine cells.
Conclusions/Significance
Up to date there are no reports of in vitro screening for non-peptidic inhibitors of Fasciola hepatica cathepsins, while in general these are considered as the best strategy for in vivo inhibition. We have identified chalcones as novel inhibitors of the two main Cathepsins secreted by juvenile and adult liver flukes. Interestingly, one compound (C34) is highly active towards the juvenile enzyme reducing larval ability to penetrate the gut wall and decreasing NEJ´s viability in vitro. These findings open new avenues for the development of novel agents to control fluke infection and possibly other helminthic diseases.
Author Summary
Despite the widespread prevalence of liver fluke infections and many efforts aimed at developing a preventive vaccine just one drug, triclabendazole, is effective in killing all parasite stages. This is aggravated by the increasing number of countries reporting parasite isolates resistant to triclabendazole treatment, highlighting the urgent need to develop new agents to control fluke infections. Here, we report the discovery of chalcones that effectively inhibits key cysteine proteases essential for parasite development and survival within the host. We further characterized the mode of inhibition of the most active compound against cathepsins secreted by both adult and larval stages of the parasite. This class of compounds is generally considered safe for clinical use and we showed that the most effective derivative is not cytotoxic to bovine sperm cells. Importantly, in vitro approaches showed that it reduces host penetration and larvae viability, finally leading to parasite death. These findings point at this derivative as a good starting point for the development of novel agents to control fluke infection and possibly other helminthic diseases.
Introduction
Parasitic flatworms are the causative agents of serious human and livestock infections many of which have been considered neglected tropical diseases in urgent need for research efforts. Liver flukes (Fasciola spp.) cause fasciolosis, traditionally a relevant disease of cattle, sheep and goats [1], currently considered an emerging zoonotic disease by the World Health Organization due to an increased incidence of human infections [2]. Despite many efforts to develop a vaccine to prevent mammalian infection [3], chemotherapy is the only Fasciola control mechanism currently available. Triclabendazole is the first choice drug since it is effective in killing juvenile and mature parasites, but resistance is emerging in several countries [4, 5]. This highlights the urgency of finding novel strategies and target molecules for developing innovative drugs to treat fluke infections.
Many virulence factors have been identified as primary targets for parasite control, since they can be used for developing therapies based on drugs or immunogens. Cysteine proteases play essential roles in numerous protozoan (like Trypanosoma cruzi and Plasmodium falciparum) and other helminth parasites [6], were they have been explored as appropriate targets for antiparasitic chemotherapy [7, 8, 9, 10, 11]. Liver flukes secrete high amounts of cysteine proteases being key players in host-parasite interaction at all stages of their life cycle [12, 13, 14, 15]. Cathepsin L3 is expressed by the newly excysted juveniles (NEJ) and consequently take part in the first steps of mammalian host infection [12, 13, 15, 16]. A different set of cathepsins L are produced by adult flukes residing in the bile ducts [15,16] and are involved mainly in digesting nutrients and developing immune responses mechanisms [17, 18]. Therefore, F. hepatica cathepsins are interesting targets for drug development in an effort to avoid parasite infection or reduce parasite burden and the pathogenic effects of the infection.
Due to their role in human disease and tumour progression, inhibitors targeting cysteine proteases have been extensively studied. Most efforts were focused on peptidic inhibitors with different substituents such as aminoacetonitriles, heterocyclic ketones, nitriles, epoxides and vinyl sulfones [19, 20, 21]. Many of these small molecules contain electrophilic groups that bind in the active-site through covalent interactions with the catalytic cysteine either in a reversible or irreversible way. Non-peptidic compounds have also been reported as cathepsin inhibitors, which are considered a better strategy for in vivo inhibition in order to avoid degradation by proteases. Among these, chalcones and other flavonoids can be found [22, 23, 24]. Flavonoids are biologically active compounds that possess remarkable properties, being presented as antioxidant, anticancer, antidiabetes, anti-inflammatory, antiprotozoal, antiHIV, antituberculosis, among many other interesting activities [25, 26, 27]. What is more, several flavonoids, particularly chalcones, have shown good pharmacological potential and have been approved for clinical use or tested in humans [27]. There have also been described flavonoid derivatives with cathepsin L-like cysteine protease inhibitory activity [23, 28, 29, 30, 31] as well as some natural flavonoids with fasciolicide activity [32, 33]. However, up to date there are no reports of in vitro screening for non-peptidic inhibitors of Fasciola cathepsins (FhCLs) or in vitro screening of synthetic chalcones with fasciolicide activity.
Taking this into account, we performed a search for small molecular weight compounds from our own library of synthetic flavonoids that may inhibit key Fasciola cysteine proteases as FhCL3 and FhCL1. FhCL3 is the only cathepsin L found in the early NEJ excretion/secretion products [12, 13, 14, 16], and FhCL1 is the main cathepsin expressed by adult flukes [14, 15]. This kind of compounds was also previously assayed as anti-tumoral and cancer chemopreventive agents showing non-mutagenic effects and being well tolerated in vivo [34, 35, 36]. Here, we identified novel inhibitors of F. hepatica cathepsins with in vitro fasciolicide activity which shall contribute in the design of novel drugs to control fluke infection.
Methods
Selection of compounds
Since flavonoids have been reported as able to inhibit cysteine protease family enzymes, we evaluated 39 synthetic flavonoids (S1–S3 Tables) from our chemical library. In order to test a variety of chemical entities, we included chalcones without (C1-C8, C20 and C21) or with (C9-C19 and C22-C26) a 2'-substituent in the A ring, chalcones with extended aromaticity (C27-C35) and flavones (C36-C39) along with the natural flavonol quercetin (C40).
Production of recombinant FhCL1 and FhCL3
FhCL1 and FhCL3 recombinant enzymes were expressed in the yeast Hansenula polymorpha as previously described [17, 39]. Briefly, yeast transformants were cultured in 500 mL YEPD broth (1% glucose, 1% tryptone, 1% yeast extract) at 37°C to an OD600 of 2–6, harvested by centrifugation at 3000xg for 10 min and induced by resuspending in 50 mL of buffered minimal media (0.67% yeast nitrogen base; 0.1M phosphate buffer pH 6.0; 1% methanol) for 36 h at 30°C. Recombinant propeptidases were secreted to the culture media, and recovered by 20–30 fold concentration of culture supernatants by ultrafiltration with a 10 kDa cut-off membrane. The proenzymes were autocatalytically activated to the mature form by incubation for 2 h at 37°C in 0.1 M sodium citrate buffer (pH 5.0) with 2 mM DTT and 2.5 mM EDTA, dialyzed against PBS pH 7.3 and stored in aliquots at -20°C until used. Functionally active recombinant enzyme was determined by titration against the cysteine protease inhibitor E-64c.
FhCL1 and FhCL3 inhibition screening of the selected compounds
A 10 mM stock of each evaluated compound was prepared in dimethylsulfoxide (DMSO). Enzyme activity assays were conducted to evaluate the inhibitory capacity of the different compounds. FhCL1 and FhCL3 were used at nanomolar concentrations to measure initial rates during the first 10 min of the assay and compounds were tested at a moderate fixed dose of 10 μM. Briefly, each enzyme and compound were preincubated 5 minutes in a 96-well plate in 0.1M sodium phosphate buffer pH 6, 1 mM DTT and 1 mM EDTA at room temperature. The reaction was initiated by the addition of 20 μM of substrate, a synthetic peptide conjugated to the fluorophore 7-amino-4-methylcoumarin (AMC). Enzyme activity was measured by the increase in AMC fluorescence as peptide substrates were hydrolyzed (Z-VLK-AMC for FhCL1 and Tos-GPR-AMC for FhCL3) at an excitation wavelength of 340 nm and emission wavelength of 440 nm using a spectrofluorometer (Varioskan Thermo). Enzyme activity was expressed as RFU/s (relative fluorescence units of AMC released per unit of time). Each compound was tested in duplicate. A progress curve without enzyme was performed to control for non-catalyzed reactions between substrates and inhibitors and a spectrum from 300 to 450 nm wavelength was done for each inhibitor to verify that none of the compounds has optical activity in the measurement range. The percentage of enzyme inhibition was calculated as: 100 - (vi/vo) x 100, where vi and vo correspond to the initial rate of AMC fluorescence increase (RFU/s) with and without inhibitor, respectively.
Further C34 inhibition studies
IC50 was calculated at 12 different concentrations of the inhibitor compound, 0, 0.625, 0.937, 1.25, 1.875, 2.5, 3.75, 5, 7.5, 10, 15 and 20 μM. The measurement of enzyme activity was performed in triplicate in a 96-well plate as previously described. We plotted initial rates (RFU/s) versus log10 of inhibitor concentration and obtained the IC50 value from a linear regression of the data. We also carried out slow-binding assays to evaluate if inhibition was time-dependent. C34 at 10 μM was incubated with each enzyme for increasing lengths of time from 3 to 120 min and the percentage of inhibition was determined as described for the screening assay. To test for reversibility of the enzyme-compound interaction, we performed rapid dilution assays [37]. Briefly, samples containing each enzyme at a 100-fold concentration (compared with standard assays) were preincubated with 10-fold the IC50-equivalent concentration of the inhibitor for 20 min at room temperature. Control reactions for each enzyme without inhibitor were carried out in parallel. Samples were then diluted 100-fold with assay buffer containing the appropriate substrate to initiate reactions, and the time course of AMC release was measured as previously describe.
Preparation of protein structures for molecular docking
FhCL1 and FhCL3 structures previously obtained by homology modelling were used [38, 39] (Robinson 2011, Corvo 2013). In order to improve structures accuracy for molecular docking, MD simulations were performed using the pmemd module implemented in the AMBER12 package [40], with the ff03.r1 force field [41]. Hydrogen atoms and sodium ions (to neutralize charge) were added to each protein with the leap utility. Each system was placed in a truncated octahedral box of TIP3P explicit water [42], extended 10 Å outside the protein on all sides. The structures of FhCL1 and FhCL3 were treated as follows: a) water and counterions were relaxed to minimize energy during 2,500 steps (500 steepest descent steps, SD, and 2,000 conjugate-gradient steps, CG) with the protein restrained with a force constant of 500 kcal/molÅ2; b) the system was minimized without restraints during 20,000 steps (5,000 SD and 15,000 CG). Long range electrostatic interactions were considered using the particle-mesh Ewald (PME) method [43] and a non-bonded interactions cutoff of 10 Å was used. After minimization, each system was gradually heated in a NVT ensemble from 0 to 300 K over 100 ps using the Berendsen coupling algorithm [44]. This procedure was followed by 20 ns of NPT simulations at 300 K and 1 atm pressure using the Langevin dynamics algorithm [45]. All bonds involving hydrogen atoms were constrained using the SHAKE algorithm [46]. The equations of motion were integrated with a time step of 2.0 fs and coordinates of the systems were saved every 2 ps. Representative structures of FhCL1 and FhCL3 from the last 30 ns of the trajectories were obtained through cluster analysis using the average-linkage algorithm [47] and used for subsequent docking calculations. Clustering, RMSD, RMSF and hydrogen bond analysis were performed using the cpptraj module in AmberTools14. For trajectories visualization the VMD program was used [48].
Preparation of ligand structures
Compounds 1–39 (S1–S3 Tables) were fully optimized at the ωB97XD/6-31+G(d,p) level [49, 50] in water using the IEF-PCM continuum model [51] with Bondi atomic radii. The nature of the optimized structures as stable species was inspected checking the eigenvalues of the analytic Hessian matrix, calculated at the same level of theory, to be positive in all the cases. All these calculations were performed using the Gaussian09 software [52].
Ligand-protein molecular docking
To predict the binding site of flavonoids C1-C39 into FhCL1 and FhCL3 flexible-ligand docking was performed using a grid box of 126×94×116 points with a grid spacing of 0.60 Å in order to cover the entire protein surface (blind docking). The grid box was centered on the macromolecule. Results differing by less than 2.0 Å in root-square deviation were grouped in the same cluster. The conformation with the lowest binding energy was chosen from the most populated cluster and the corresponding ligand-protein complex was used for further MD studies. All docking calculations were done with the AutoDock 4.2 [53] software package using the Lamarckian genetic algorithm. A population size of 150 individuals and 2.5×106 energy evaluations were used for 50 search runs.
Ligand-protein molecular dynamics
MD simulations of C34 with FhCL1 and FhCL3 were performed as described above using the GAFF [54] force field for the ligand. RESP partial charges [55] for C34 were derived using Gaussian09 at the HF/6-31G* level and the antechamber module in AMBER12 was employed to obtain the force field parameters. 40 ns of productive MD were simulated and coordinates of the systems were saved every 10 ps.
In vitro NEJ treatment with C34 and gut invasion assay
F. hepatica metacercariae were acquired from Baldwin Aquatics Inc. (Monmouth, Oregon) for in vitro NEJ treatment and from Instituto Miguel C. Rubino (DILAVE, MGAP, Uruguay) for the gut invasion assay. NEJ were obtained by in vitro excystement as previously described with minor modifications [56], with no differences observed between metacercariae from different origin. Briefly, 100 metacercariae were incubated with 1% sodium hypochlorite for 5 min at room temperature to remove the outer cyst wall and then washed exhaustively with PBS. Metacercarie were activated by incubation at 39°C in a medium prepared by mixing equal volumes of solution A (0.4% sodium taurocholate, 120 mM NaHCO3, 140 mM NaCl pH 8.0) and solution B (50 mM HCl and 33 mM L-cysteine). A 100 μm filters were used to retain the cyst wall as NEJ began to emerge. The excystment process was monitored for m90-180 min under the microscope. Collected NEJ were washed three times with RPMI-1640 supplemented with 200 U/mL Penicillin G sulfate, 200 mg/mL streptomycin sulfate, 500 ng/mL amphotericin B, 10 mM HEPES, counted and divided in groups of around 20 parasites that were transferred to 12 wells tissue culture plates. Parasites were maintained at 37°C, 5% CO2 in modified Basch’s medium [57]. At day 1, C34 (50 μM) was added to treated groups and 0.5% DMSO to control groups, each condition tested by duplicate. Medium was changed twice a week and fresh compound was added, control NEJ were cultured for 20 days. NEJ behavior was monitored under a light microscope (Olympus BX41), every day each well was recorded for a minute in order to assess parasite motility and registered using the following score: 3-normally active; 2- reduced activity (sporadic movement); 1- immotile (adapted from [58]). For the gut invasion assay we performed an in vitro excystement of metacercarie as previously described and incubated the NEJ for 4 h either in the presence of 50 μM C34 or 0.5% DMSO (control group) and immediately transferred them to gut sacs. For gut preparation we used the protocol previously described by Burden et al. with some modifications [59]. Brifely, 6 weeks old male Wistar rats were euthanized by cervical dislocation and approximately 25 cm of small intestine was excised using the caecum as a reference. The intestine was washed several times with RPMI-1640 media warmed at 37°C and cut in 5 cm sacs. One end of each sac was ligated, 20–30 NEJ from treated or control groups were pipetted inside the sac and then the other end was ligated. Gut sacs were maintained in RPMI-1640 media at 37°C, 5% CO2 in 12 wells plates and parasites that migrated through the mucosal wall within 3 h were recovered at the plate bottom and counted. Each condition was assayed in triplicate.
Cytotoxicity assay on bovine spermatozoa
Semen samples were obtained from a healthy fertile Hereford bull and kept frozen in 0.5 mL straws (extended in Andromed, Minitube, Germany) under liquid nitrogen until use. The semen used belonged to a single freezing batch that was obtained during a regular collection schedule with an artificial vagina. Samples from three straws were thawed and a sperm pool was prepared in PBS at a concentration of 40 million spermatozoa per mL, then 50 μL of this sperm suspension was carefully mixed with 50 μL of C34 diluted to 100, 50, 25, 12.5 and 6.25 μM or with 1% DMSO in control experiments. Each condition was assayed by duplicate in 96-well plates and controls were assayed by triplicate. Plates were incubated at 37°C for 1 h with moderate shaking. The motility analysis was carried out using a CASA (Computer Assisted Semen Analyzer) system Androvision (Minitube, Tiefenbach, Germany) with an Olympus BX 41 microscope (Olympus, Japan) equipped with a warm-stage at 37°C. Each sample (10 μL) was placed onto a Makler Counting Chamber (deph 10 μm, Sefi-Medical Instruments, Israel) and the following parameters were evaluated: percentage of total motile spermatozoa (motility >5 μm/s) and velocity curved line (VCL, >24 μm/s). At least 400 spermatozoa were analysed from each sample from at least four microscope fields.
Results and Discussion
Aromatic chalcones are good cathepsin inhibitors
We evaluated forty flavonoids from three structural clusters of our own chemistry library as inhibitors of FhCL1 and FhCL3. Cluster I contained twenty-six chalcones modified in ring B (C1-C26, Fig 1A), cluster II contained naphtolchalcone derivatives (C27-C35, Fig 1B) and cluster III was composed of flavones modified in ring B (C36-C40, Fig 1B). Initially, we screened the compounds at a low concentration of 10 μM measuring initial rates during the first 10 min, finding activities ranging from 0% to 75% enzyme inhibition (Fig 1). Roughly, a larger amount of compounds decreased FhCL1 activity considerably, while FhCL3 proved more difficult to inhibit. Thus, eleven compounds showed more than 50% inhibition of FhCL1 (C3, C10, C22-C24, C27, C30, C31, C33, C34 and C35) (Fig 1), while two of them, belonging to cluster II, were above this cut-off for FhCL3, C34 and C35 (Fig 1B), being C34 the best global inhibitor. None of the assayed flavones (cluster III) displayed relevant activities against both FhCL1 and FhCL3 (% inhibition at 10 μM lower than 20%, Fig 1B) for this reason the studied population of flavones was lower than the chalcones one.
Fig 1. Screening of chalcones as FhCL1 and FhCL3 inhibitors.

The % of inhibition of FhCL1 (black bars) and FhCL3 (gray bars) activity is shown for each compound of: A. cluster I (chalcones modified on ring B, represented as R), B. clusters II and III (naphtochalcones and flavone derivatives, respectively). All compounds were screened at 10 μM. A general structure of each cluster of compounds is shown above the graphic bars: A. X can be -H or -OH and R is phenyl with different substituents or a heterocycle. B. X can be -H or -OH at position 1 or 2, dashed lines represent phenyl or naphtyl at position 1 or 2 and R is -H, -Cl, -Br or -OCH3. C40 is the natural flavone quercetine (the structure of each compound can be found in S1–S3 Tables).
In general, chalcones containing heterocycles as B-ring in cluster I (C22-C26) and those with extended aromaticity belonging to cluster II showed the highest inhibition percentages. Clearly, this kind of compounds inhibited more readily FhCL1 than FhCL3 (fourteen showing at least 50% inhibition of FhCL1 vs two with FhCL3 at 10 μM). These might be explained considering the active site pocket structure and substrate preferences of the enzymes. While FhCL1 has a wide S2 pocket which easily accommodates bulky and aromatic residues, FhCL3 has a narrow and restricted site that preferentially interacts with small moieties like proline and glycine [39]. Strikingly, C34 substituted by a hydroxyl at position 2 of the A-ring and with a naphthyl on both rings, was the best inhibitor of FhCL3, showing 75% inhibition for FhCL1 and 65% for FhCL3 in the screening. Likewise, the highest percentages of FhCL3 inhibition correspond to the same type of compounds (C28, C30, C31, C35) (Fig 1B). We hypothesized that the narrower conformation of FhCL3 active site compared to FhCL1 diminishes the number of compounds that manage to accommodate in such a position as to allow enzyme inhibition.
There are some interesting compounds showing selectivity towards FhCL1, for example derivatives C4, C13 and C22-C25 exhibiting around 50% inhibition of the former enzyme and poor or no inhibition of FhCL3. These compounds belong to the group containing heterocycles as B-ring (C22-C25) or 4-methoxy moiety on the B-phenyl ring (C4, C13), that might establish hydrogen bonds with specific residues of this enzyme (S4 Table). Particularly, C20, the non-hydroxylated analogue of C22, does not inhibit FhCL1, suggesting the 2´-OH might also be playing an important role in this inhibition. A chalcone with extended aromaticity (C27) also exhibits 65% inhibition of FhCL1 and only 13% of FhCL3, being the only one with a phenyl substituent as B-ring. Notably, among these bulky compounds, the less active towards FhCL1 was C29, which lacks Michael-acceptor motive susceptible of nucleophilic attack by enzyme residues (Fig 1B, S2 Table).
The p-Cl substituted chalcone C10 also showed good inhibition for FhCL1 (63%) (Fig 1A, S1 Table). Likewise, this compound offers some interesting structure-activity relationships. Here, a positive influence of the 2´-OH in the inhibition might be addressed again when comparing C10 with its de-2´OH analogue C2 that exhibits only 25% of inhibition. Besides, its bioisostere p-Br substituted on the B-ring (C11) depicts poor inhibition of FhCL1 (19%), suggesting that it is the combination of both 2´-OH and p-Cl substituents that exerts optimum inhibition of this enzyme (Fig 1A, S1 Table).
Different mode of inhibition of C34 with FhCL1 and FhCL3
We selected C34 to perform additional characterization as it was the best inhibitor of both target enzymes. Consequently, a dose-response study was performed. The IC50 was similar with both enzymes, being 7.7 μM and 5.6 μM for FhCL1 and FhCL3 respectively (Table 1). Interestingly, the interaction mode with each enzyme seems to differ. Longer incubation times at 10 μM resulted in higher inhibition of FhCL3 but not FhCL1, thus C34 interacts in a slow-binding time-dependent fashion only with the former enzyme. Furthermore, when increasing the preincubation time with the enzyme, FhCL3 is almost completely inhibited (96%) (Table 1).
Table 1. IC50 and effect of preincubation time, at 10 μM, on enzyme inhibition by C34.
| % inhibition over time (min) | ||||||
|---|---|---|---|---|---|---|
| Enzyme | IC50 (μM) | 3 | 30 | 60 | 90 | 120 |
| FhCL1 | 7.7 ± 0.4 | 73 | 81 | 75 | 67 | 79 |
| FhCL3 | 5.6 ± 0.1 | 53 | 87 | 96 | 96 | 93 |
We then evaluated the reversibility of the inhibition by measuring the recovery of enzymatic activity after a rapid and large dilution of the enzyme–inhibitor complex. When the initial rate of the reaction is similar in the presence and absence of the inhibitor, it indicates a rapid recovery of activity after compound dilution meaning the reaction is reversible. This was the case for the interaction of C34 with FhCL1 (Fig 2, left panel), where the progress curve after inhibitor dilution has a slope similar to that of the control sample (enzyme incubated and diluted in the absence of inhibitor); however, we found a different behavior with FhCL3. Here, the progress curves have a lag phase followed by a linear phase (Fig 2, right panel), reflecting the slow recovery of activity as inhibitor dissociates from the enzyme and suggesting that the interaction of C34 with FhCL3 is slowly reversible being the compound tightly bound to the enzyme.
Fig 2. Reversibility of C34 inhibition.
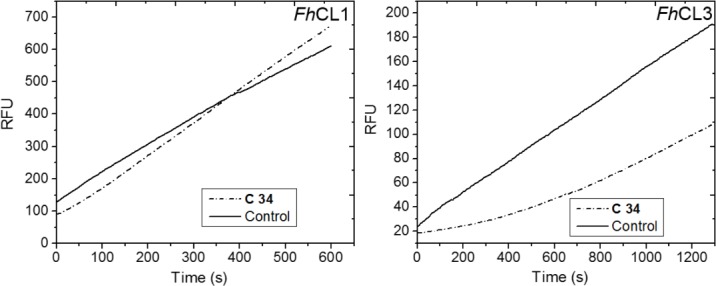
Rapid dilution assay to test reversibility of C34 interaction with cathepsins. Recovery of enzymatic activity was measured after incubation of each enzyme in the absence (Control) or presence of inhibitor (C34). Initial rates were measured for 10–20 min after dilution of the enzyme-inhibitor complex with substrate to initiate reaction, and plotted as Relative Fluorescence Units (RFU) of AMC released from substrate per unit of time (s).
Flavonoids can bind along the active site
According to docking results the studied compounds most frequently interact with a putative binding site, represented in cyan for FhCL1 and orange for FhCL3, and found at a similar position within the active site cleft of each enzyme (Fig 3). A second putative binding site (represented in blue) was observed for FhCL3, which has a different orientation and is not as deep located inside the binding site (Fig 3).
Fig 3. Flavonoids binding sites predicted by molecular docking.

Flavonoid interaction is predicted to occur along the active site cleft of the target enzymes. Above: Site represented with cyan compounds superimposed has 100% occupation with flavonoids evaluated in FhCL1. Below: Sites represented in orange and blue are the more populated clusters found in FhCL3. Around 40% of compounds have only one cluster (represented in orange) while about 33% of flavonoids have also a second cluster with similar probability (colored in blue). C34 is included in the second group. Enzyme structures are represented by new cartoon and compounds structures in licorice. The catalytic dyad Cys25 and His162 is represented in balls and sticks. Subsites residues are depicted in beads colored in red (S2) and yellow (S3).
To obtain a deeper knowledge of C34 interaction with target enzymes, 40 ns molecular dynamics simulations were performed allowing us to confirm the binding sites predicted by docking, as we obtained stable complexes in the last 35 ns of the simulation (S1 Fig). Thus, the high percentage of inhibition experimentally observed can be attributed to the compound locating next to residues directly implicated in substrate positioning and catalysis (Figs 4 and 5). From a population cluster analysis, we determined the most representative structure for the enzyme-inhibitor complex for FhCL1, with an occurrence of 51% (Fig 4 upper panel). We found two more clusters with occurrences of 24% and 22% that, while changing the relative position and interaction with certain residues, still occupy the active site (Fig 4 middle and lower panels, respectively), and two last clusters with lower occurrences of 2 and 1% (not shown). For FhCL3, C34 occupies the orange site (Fig 3) along the entire simulation time. A major cluster with an occurrence of 59% is observed (Fig 5 upper panel), followed by another two clusters with lower occurrences of 23% and 11% (Fig 5 middle and lower panels, respectively), and two with less occurrences of 6 and 1% (not shown), similar in location and relative position to the major cluster.
Fig 4. FhCL1-C34 interactions from MD simulations.

Structure of the three most populated clusters for FhCL1. Left panels: C34 structure is represented in balls and sticks. Residues belonging to sub-sites involved on complex formation are represented in beads and colored in red (S2) and yellow (S3). Residues outside the active site pockets involved on complex formation are translucent (light blue). Catalytic dyad is depicted by VDW. Right panels: 2D diagram of interactions between C34 and FhCL1 for each cluster generated using LigPlot+ [60]. C34 is shown in violet. Protein residues involved in non-polar contacts are represented as red semicircles with radiating spokes and their names colored according to left panels. A hydrogen bond (green dashed line) is shown together with its distance value (green).
Fig 5. FhCL3-C34 interactions from MD simulations.

Structure of the three most populated clusters for FhCL3. Left panels: C34 structure is represented in balls and sticks. Residues belonging to sub-sites involved on complex formation are represented in beads and colored in red (S2) and yellow (S3). Residues outside the active site pockets involved on complex formation are translucent (light blue). Catalytic dyad is depicted by VDW. Right panels: 2D diagram of interactions between C34 and FhCL3 for each cluster generated using LigPlot+. C34 is shown in violet. Protein residues involved in non-polar contacts are represented as red semicircles with radiating spokes and their names colored according to left panels. A hydrogen bond (green dashed line) is shown together with its distance value (green).
The experimentally observed variation in inhibition percentages might be explained by the high number of interactions established by the ligand with each target. It is worth noticing that the experimental results were in good correlation with computational observations. C34 is positioned along the active site cleft in both enzymes and interacts with Cys-His catalytic dyad and residues from the S2 and S3 pockets (Figs 4 and 5), determinants of the substrate specificity of this family of cysteine proteases [39, 61]. FhCL1 establishes several hydrophobic interactions with the inhibitor, particularly through residues Gly66, Gly67, Val160 and Asn161 from S2 and S3 subsites, and Cys25 from the catalytic dyad (Fig 4). Likewise, we observed many hydrophobic interactions of C34 with FhCL3, with a larger number of residues belonging to the enzyme sub-sites: Trp69, Met70, Ala135, Val160, Thr161, Val163 and Ala209, which form the entire S2 subsite, plus Gly68 belonging to S3 subsite, both decisive in FhCL3 substrate positioning for catalysis (Fig 5). In this sense, the high inhibition experimentally observed for C34 with both enzymes is confirmed.
Interestingly, Trp69 can contribute to inhibitor binding by establishing Pi-Pi stacking interactions. This residue is specific of FhCL3 and when mutated for Leu as in FhCL1 it renders the enzyme almost inactive highlighting its importance for substrate positioning for catalysis [39]. A recent in silico search for FhCL3 inhibitors provides additional evidence that the non-polar side chain of Trp69 establishes critical interaction with ligands and adopts variable conformations to accommodate different groups in the enzyme binding site [62]. Furthermore, it showed that aromatic moieties with high hydrophobicity can establish favorable interactions with non-polar residues from FhCL3 binding cleft [62]. The greater number of interactions between inhibitor and residues from FhCL3 sub-sites compared with FhCL1, might explain its lower IC50 (Table 1), its behavior as a slowly reversible inhibitor (Fig 2), and its highest percentage of inhibition reached after 120 min (Table 1).
No hydrogen bonds with a significant occupancy (greater than 50) along the simulation time were found in any of the enzyme-inhibitor complexes, except for the bond between carbonyl oxygen in C34 and Gly68 with an occupancy of 22 in FhCL1 (Fig 4), analogous to the interaction with Glu72 in FhCL3, with only 11 of occupancy (Fig 5). However, there are significant differences in the hydrogen bonds at the protein structural level, where some intra-molecular protein hydrogen bonds have an occupancy greater than 90 in the absence of inhibitor but disappear in its presence, mainly in the FhCL3-inhibitor complex (Fig 6).
Fig 6. Hydrogen bonds varying in the presence or absence of C34 bound to FhCL1 (blue, top panel) and FhCL3 (orange, low panel).

Residues involved in hydrogen bonds (HB) are depicted in ball and sticks and colored by ΔOcc (delta of occupancy). ΔOcc is calculated with the formula ΔOcc = Occ FhCL—Occ FhCL_34 from the data in S5 and S6 Tables. HB are represented by black dashed lines. The catalytic Cys-His dyad is colored by atom.
If we analyze the hydrogen bonds involving residues from sub-sites S2 and S3 in FhCL1 (Fig 6 and S5 Table, residues in bold), we see a tendency to diminish its occupancy in the presence of C34, supporting its interaction along the active site alters local protein conformation. A similar behavior is observed in FhCL3, with a more remarkable loss in occupancy seen for hydrogen bonds involving Val136-Thr161, Asp137- Thr161 and His63-Arg58 (Fig 6 and S6 Table, residues in bold). Moreover, upon ligand binding new hydrogen bonds are established between backbone and side chains of sub-site residues (with occupancy greater than 50) in the proximity of C34 binding site (Fig 6, HB with ΔOcc.˂0), named His63-Gly66, Trp69-Asn62 and Ala135-Ala163 (S6 Table, residues in bold), again suggesting the interaction with C34 dramatically alters protein conformation and thus its ability for catalysis.
Fasciolicide activity of C34 over in vitro cultured NEJ
We evaluated the effect of C34 on NEJ parasites cultured in vitro. NEJ were incubated in the presence of 50 μM C34 and the assessment of parasite movement is shown in Fig 7.
Fig 7. Viability assessment of NEJ cultured with C34.

Top: Microscopic appearance of control (left) and treated parasites (right) after 7 days of culture. * NEJ dead with protrusions. Bottom: pie charts showing the percentage of NEJ in each score category when incubated with 50 μM C34 (duplicates taken together for percentage calculation): 3-normally active; 2- reduced activity (sporadic movement); 1- immobile. Control NEJ were incubated in 0.5% DMSO.
The addition of C34 resulted in a decrease in parasite motility leading to parasite death. At day 4 the percentage of immobile flukes was considerably higher in treated vs control groups, 67% vs 0% respectively. The percentage of sporadic movement and immobile parasites increased in treated parasites and evident signs of internal and tegument damage appeared. Culture media was dirty as compared to control flukes due to the release of body components and some NEJ died protruding their anterior region (Fig 7, S1 and S2 Videos). After day 10 no movement was detected in treated NEJ while control parasites continue moving until the experiment was finished on day 20, thus, motility seems to adequately estimate worm viability.
There have been controversial findings regarding the effect of cathepsin L knock down by RNAi. While a recent work described no phenotypic changes in NEJ even after 21 days of exposure to long dsRNA or siRNA [63], an earlier report had documented immobile NEJ after a 4h interference with Cat-L and B siRNA [64]. Here, we observed a reduction in fluke motility consistent with this first report. Additional evidence supporting this result comes from an in vitro treatment of NEJ with the cathepsin L and B inhibitors E64-d and CA-074Me, which resulted in the loss of worm motility accompanied by structural damage of parasites [65]. In a similar fashion, they observed a progressive loss of motility starting 5 to 12 days after treatment initiation. As a consequence of decreased motility, a diminished ability of parasites to traverse the duodenum wall was reported [64]. In order to check if C34 treatment can also impair invasion, we performed a gut penetration assay. NEJ incubated 4h with C34 showed the expected phenotype of reduced migration through the gut wall, suggesting the impairment of cathepsin L function (Fig 8). While the in vitro treatment effect of movement reduction is seen after longer incubation times with C34, a reduced gut penetration is readily detected in the invasion assay. This is indicative that the main role of juvenile cathepsin L3 in parasite host invasion is promptly affected, and a secondary effect on parasite movement might emerge later. In the host environment, this might translate into a considerable reduction in the number of NEJ able to reach the bile ducts and establish a successful infection.
Fig 8. NEJ gut penetration assay.
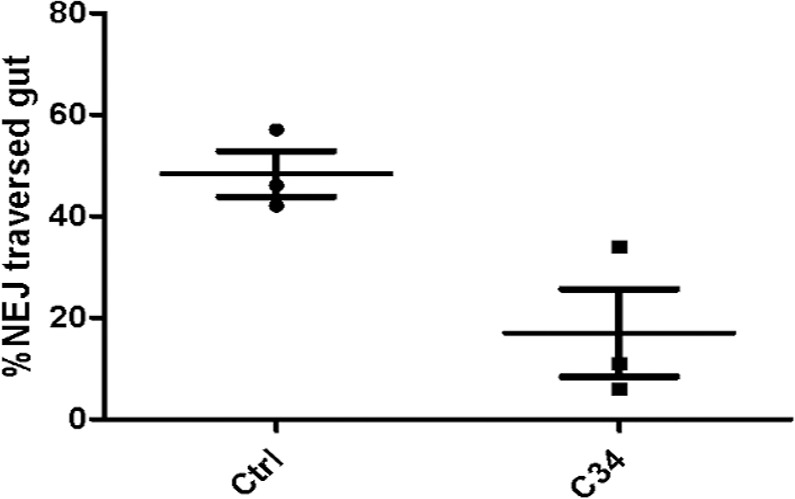
Scattered plot showing the percentage of NEJ that traverse the gut wall within 3 h in control (circles) and C34 treated groups (squares). Horizontal lines and bars represent means ± SD percent gut penetration, respectively.
C34 is not toxic to bovine spermatozoa
Citotoxicity assays using spermatozoa are considered a suitable approach for preclinical toxicology screening during drug development processes [66]. The cytotoxicity assay on bovine spermatozoa showed C34 is not toxic to reproductive cells even at the highest tested dose of 100 μM. We observed no significant differences on percentage of motile spermatozoa and velocity curved line between treated and control samples (S7 Table). Also, our previous studies showed C34 is not mutagenic in Ames test using Salmonella typhimurium TA98 strain with and without metabolic activation and no genotoxic effect was observed measuring DNA damage in the alkaline comet assay [36], supporting the fact that this class of compounds exhibits low toxicity and are promising agents for clinical usage.
Conclusions
In this work we have identified novel chalcones that inhibit Fasciola hepatica major cathepsins L from both adult and juvenile parasites. Chalcones with heterocycles as B-ring (C22-C26) and chalcones containing phenyl and naphtyl moieties (C27-C35) showed the highest inhibition percentages. Among these, C34 resulted a slow-reversible tight binding inhibitor of FhCL3, exerting an almost complete inactivation of the enzyme. In addition, we performed an extensively computational analysis characterizing C34 interaction with cathepsins providing evidence of the different inhibition seen with each target enzyme. We demonstrated C34 is not toxic to bovine sperm, exhibits in vitro fasciolicide activity over cultured NEJ and reduces larval penetration of the gut wall, suggesting is a suitable candidate for further drug development against worm infection.
Supporting Information
(TIF)
% inh.: percentage of inhibition at 10 μM dose. Values represent means ± SE. n = 2. R: represent B ring.
(DOCX)
% inh.: percentage of inhibition at 10 μM dose. Values represent means ± SE. n = 2. *C29 is a derivative of C28, which lacks the α,β-unsaturated system: 3-(naphthalen-1-yl)-1,5-diphenylpentane-1,5-dione.
(DOCX)
% inh.: percentage of inhibition at 10 μM dose. Values represent means ± SE. n = 2.
(DOCX)
(DOCX)
Residues of catalytic sub-sites are highlighted in bold. ΔOcc is calculated with the formula ΔOcc = Occ FhCL1—Occ FhCL1_34
(DOCX)
Residues of catalytic sub-sites are highlighted in bold. ΔOcc is calculated with the formula ΔOcc = Occ FhCL3—Occ FhCL3_34.
(DOCX)
% MOT: percentage of motile spermatozoa (motility > 5 μm/s) normalized to untreated control. VCL: Velocity curved line (> 24 μm/s). Spermatozoa were treated 1h with C34 at 37°C.
(DOCX)
(WMV)
(WMV)
Acknowledgments
We would like to thank Dr. R.J.S. Baerends and Dr. J.A.K.W. Kiel, Molecular Cell Biology Lab, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, The Netherlands, for kindly providing the yeast expression system. We would also like to thank the help of Dra. Beatriz Álvarez and Martina Steglich from Laboratorio de Enzimología, Facultad de Ciencias and Dra. Verónica Fernández from Cátedra de Inmunología, Dpto. de Biociencias, Facultad de Química, UdelaR.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was partially funded by Programa de Desarrollo de las Ciencias Básicas (www.pedeciba.edu.uy) and Agencia Nacional de Investigación e Innovación (www.anii.org.uy) granted an scolarship INI_X_2013_1_100964 to FF. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Robinson MW, Dalton JP. Zoonotic helminth infections with particular emphasis on fasciolosis and other trematodiases. Philos Trans R Soc Lond B Biol Sci. 2009; 364(1530):2763–76. 10.1098/rstb.2009.0089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fürst T, Keiser J, Utzinger J. Global burden of human food-borne trematodiasis: a systematic review and meta-analysis. Lancet Infect Dis. 2012; 12(3):210–221. 10.1016/S1473-3099(11)70294-8 [DOI] [PubMed] [Google Scholar]
- 3.Molina-Hernández V, Mulcahy G, Pérez J, Martínez-Moreno Á, Donnelly S, O'Neill SM, et al. Fasciola hepatica vaccine: we may not be there yet but we're on the right road. Vet Parasitol. 2015; 208 (1–2):101–111. 10.1016/j.vetpar.2015.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brockwell YM, Elliott TP, Anderson GR, Stanton R, Spithill TW, Sangster NC. Confirmation of Fasciola hepatica resistant to triclabendazole in naturally infected Australian beef and dairy cattle. Int. J. Parasitol. 2014; 4(1): 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moll L, Gaasenbeek CPH, Vellema P, Borgsteede FHM. Resistance of Fasciola hepatica against triclabendazole in cattle and sheep in the Netherlands. Vet. Parasitol. 2000; 91: 153–158. [DOI] [PubMed] [Google Scholar]
- 6.Sajid M, McKerrow JH. Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 2002; 120: 1–21. [DOI] [PubMed] [Google Scholar]
- 7.Engel JC, Doyle PS, Hsieh I, McKerrow JH. Cysteine Protease Inhibitors Cure an Experimental Trypanosoma cruzi Infection. J. Exp. Med. 1998; 188: 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sijwali PS, Rosenthal PJ. Gene disruption confirms a critical role for the cysteine protease falcipain-2 in hemoglobin hydrolysis by Plasmodium falciparum. Proc. Natl. Acad. Sci. 2004; 101: 4384–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Istvan ES, Gluzman IY, Gross J, Goldberg DE. Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc. Natl. Acad. Sci. 2006; 103: 8840–8845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdulla MH, Lim KC, Sajid M, McKerrow JH, Caffrey CR. Schistosomiasis Mansoni: Novel Chemotherapy Using a Cysteine Protease Inhibitor. PLoS Medicine. 2007; 4(1):e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vermeire JJ, Lantz LD, Caffrey CR. Cure of Hookworm Infection with a Cysteine Protease Inhibitor. PLoS NTD. 2012; 6(7): e1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancela M, Acosta D, Rinaldi G, Silva E, Durán R, Roche L, et al. A distinctive repertoire of cathepsins is expressed by juvenile invasive Fasciola hepatica. Biochimie. 2008; 10: 1461–1475. [DOI] [PubMed] [Google Scholar]
- 13.Cancela M, Ruétalo N, Dell'Oca N, da Silva E, Smircich P, Rinaldi G, et al. Survey of transcripts expressed by the invasive juvenile stage of the liver fluke Fasciola hepatica. BMC Genomics. 2010; 11:227 10.1186/1471-2164-11-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson MW, Tort JF, Lowther J, Donnelly SM, Wong E, Xu W, et al. Proteomics and phylogenetic analisys of the cathespin L protease family of the helmith pathogen Fasciola hepatica: expansion of a repertoire of virulence associated factors. Mol Cell Prot. 2008; 6: 1111–1123. [DOI] [PubMed] [Google Scholar]
- 15.Robinson MW, Menon R, Donnelly SM, Dalton JP, Ranganathan S. An integrated transcriptomics and proteomics analysis of the secretome of the helminth pathogen Fasciola hepatica proteins associated with invasion and infection of the mammalian host Mol Cell Proteomics. 2009; 8(8):1891–907. 10.1074/mcp.M900045-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corvo I, Cancela M, Cappetta M, Pi-Denis N, Tort JF, Roche L. The major cathepsin L secreted by the invasive juvenile Fasciola hepatica prefers proline in the S2 subsite and can cleave collagen. Mol Biochem Parasitol. 2009; 167(1): 41–47. 10.1016/j.molbiopara.2009.04.005 [DOI] [PubMed] [Google Scholar]
- 17.Tort J, Brindley PJ, Knox D, Wolfe KH, Dalton JP. Proteinases and associated genes of parasitic helminths. Adv Parasitol. 1999; 43:161–266. [DOI] [PubMed] [Google Scholar]
- 18.Stack C, Dalton JP, Robinson MW. The phylogeny, structure and function of trematode cysteine proteases, with particular emphasis on the Fasciola hepatica cathepsin L family. Adv Exp Med Biol. 2011; 712:116–135. 10.1007/978-1-4419-8414-2_8 [DOI] [PubMed] [Google Scholar]
- 19.Moran BW, Anderson FP, Ruth DM, Fágáin CO, Dalton JP, Kenny PT. Fluorobenzoyl dipeptidyl derivatives as inhibitors of the Fasciola hepatica cysteine protease cathepsin L1. J Enzyme Inhib Med Chem. 2010; 25(1):1–12. 10.3109/14756360902888184 [DOI] [PubMed] [Google Scholar]
- 20.Ang KH, Ratnam J, Gut J, Legac J, Hansell E, Mackey ZB, et al. Mining a Cathepsin Inhibitor Library for New Antiparasitic Drug Leads. PLoS NTD. 2011; 5(5): e1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Löser R, Pietzsch J. Cysteine cathepsins: their role in tumor progression and recent trends in the development of imaging probes. Front. Chem. 2015; 3, Article 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raghav N, Kaur R. Chalcones, semicarbazones and pyrazolines as inhibitors of cathepsins B, H and L. Int. J. Biol. Macromol. 2015; 80: 710–724. 10.1016/j.ijbiomac.2015.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baek KH, Karki R, Lee ES, Na Y, Kwon Y. Synthesis and investigation of dihydroxychalcones as calpain and cathepsin inhibitors. Bioorg. Chem. 2013; 51: 24–30. 10.1016/j.bioorg.2013.09.002 [DOI] [PubMed] [Google Scholar]
- 24.Lee E, Jang I, Shin MJ, Cho HJ, Kim J, Eom J, et al. Chalcones as Novel Non-peptidic μ-Calpain Inhibitors. Bull. Korean Chem. Soc. 2011, 32 (9): 3459–3464. [Google Scholar]
- 25.Sahu NK, Balbhadra SS, Choudhary J, Kohli DV. Exploring pharmacological significance of chalcone scaffold: a review. Curr Med Chem. 2012: 19 (2): 209–225. [DOI] [PubMed] [Google Scholar]
- 26.Singh P, Anand A, Kumar V. Recent developments in biological activities of chalcones: a mini review. Eur J Med Chem. 2014; 85: 758–777. 10.1016/j.ejmech.2014.08.033 [DOI] [PubMed] [Google Scholar]
- 27.Batovska DI, Todorova IT. Trends in utilization of the pharmacological potential of chalcones. Curr Clin Pharmacol. 2010: 5 (1): 1–29. [DOI] [PubMed] [Google Scholar]
- 28.Troeberg L, Chen X, Flaherty TM, Morty RE, Cheng M, Hua H, et al. Chalcone, acyl hydrazide, and related amides kill cultured Trypanosoma brucei brucei. Mol Med. 2000: 6 (8): 660–669. [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SH, Lee E, Baek KH, Kwon HB, Woo H, Lee ES, et al. Chalcones, inhibitors for topoisomerase I and cathepsin B and L, as potential anti-cancer agents. Bioorg Med Chem Lett. 2013: 23 (11): 3320–3324. 10.1016/j.bmcl.2013.03.106 [DOI] [PubMed] [Google Scholar]
- 30.Burger MC, Fernandes JB, da Silva MF, Escalanate A, Prudhomme J, Le Roche KG, et al. Structure and bioactivities of dihydrochalcones from Metrodorea stipularis. J Nat Prod. 2014: 77 (11): 2418–2422. 10.1021/np500453x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Sousa LR, Wu H, Nebo L, Fernandes JB, da Silva MF, Kiefer W, et al. Natural products as inhibitors of recombinant cathepsin L of Leishmania mexicana. Exp Parasitol. 2015: 156: 42–48. 10.1016/j.exppara.2015.05.016 [DOI] [PubMed] [Google Scholar]
- 32.del Rayo-Camacho M, Sanchez B, Quiroz H, Contreras JL, Mata R. Pinocembrine: a bioactive flavanone from Teloxys graveolens. J Ethnopharmacol. 1991: 31 (3): 383–389. [DOI] [PubMed] [Google Scholar]
- 33.Toner E, Brennan GP, Wells K, McGeown JG, Fairweather I. Physiological and morphological effects of genistein against the liver fluke Fasciola hepatica. Parasitology, 2008, 135 (10): 1189–1203. 10.1017/S0031182008004630 [DOI] [PubMed] [Google Scholar]
- 34.Cabrera M, Simoens M, Falchi G, Lavaggi ML, Piro OE, Castellano EE, et al. Synthetic chalcones, flavanones, and flavones as antitumoral agents: biological evaluation and structure-activity relationships. Bioorg Med Chem. 2007: 15 (10): 3356–3367. [DOI] [PubMed] [Google Scholar]
- 35.Cabrera M, Lavaggi ML, Croce F, Celano L, Thomson L, Fernández M, et al. Identification of chalcones as in vivo liver monofunctional phase II enzymes inducers. Bioorg Med Chem. 2010: 18 (14): 5391–5399. 10.1016/j.bmc.2010.05.033 [DOI] [PubMed] [Google Scholar]
- 36.Cabrera M. Flavonoides y análogos como moduladores de enzimas detoxificantes de xenobióticos. Potenciales agentes quimiopreventivos para el cáncer. PhD. Thesis, Universidad de la República, Uruguay. 2012. http://riquim.fq.edu.uy/archive/files/44c15f32f388abc51410721ffbfaf760.pdf
- 37.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists. 2 nd. Ed. USA, John Wiley & Sons, Inc; 2013. [PubMed] [Google Scholar]
- 38.Robinson MW, Corvo I, Jones PM, George AM, Padula MP, Tort JF, et al. Collagenolytic activities of the major secreted cathepsin L peptidases involved in the virulence of the helminth pathogen, Fasciola hepatica. PLoS NTD. 2011; 5(4): e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corvo I, O´Donoghue A, Pastro L, Pi-Denis N, Eroy-Reveles A, Roche L. Dissecting the active site of the collagenolytic cathepsin L3 protease of the invasive stage of Fasciola hepatica. PLoS NTD 2013; 7(7): e2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Case DA, Darden TA, Cheatham TE 3rd, Simmerling CL, Wang J, Duke RE, et al. AMBER 12, University of California, San Francisco: 2012. [Google Scholar]
- 41.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003; 24 (16): 1999–2012. [DOI] [PubMed] [Google Scholar]
- 42.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW & Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys.1983; 79: 926–935. [Google Scholar]
- 43.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H & Pedersen L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995; 103: 8577–8593. [Google Scholar]
- 44.Berendsen HJC, Postma JPM, Van Gunsteren WF, Haak ARHJ & Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984; 81: 3584–3690. [Google Scholar]
- 45.Pastor RW. Brooks BR, Szabo A. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 1988; 65: 1409–1419. [Google Scholar]
- 46.Ryekaert JP, Ciecotti G, Berendsen HJC. Numerical-integration of cartesian equations of motion of a system with constraints -molecular-dynamics of N-alkanes. J. Comput. Chem. 1997; 23: 327–341. [Google Scholar]
- 47.Shao J, Tanner SW, Thomson N, Cheatham TE. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J Chem Theory Comput. 2007; 3(6): 2312–2334. 10.1021/ct700119m [DOI] [PubMed] [Google Scholar]
- 48.Humphrey W, Dalke A, Schulten K. VMD ‐ Visual Molecular Dynamics. J. Mol. Graph. 1996; 14: 33‐38. [DOI] [PubMed] [Google Scholar]
- 49.Chai JD, Head-Gordon M. Long-range corrected hybrid density fuctionals with dumped atom-atom dispersion corrections. J. Chem. Phys. 2008; 10: 6615–6620. [DOI] [PubMed] [Google Scholar]
- 50.Krishnan R, Binkley JS, Seeger R, Pople JA. Self-Consistent Molecular Orbital Methods. XX. A Basis set for correlated wave-functions. J. Chem. Phys. 1980; 72: 650–654. [Google Scholar]
- 51.Cances MT, Mennucci B, Tomasi J. A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropics dielectrics. J. Chem. Phys. 1997; 107: 3032–3041. [Google Scholar]
- 52.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 09, Revision A.02, Gaussian Inc., Wallingford CT, 2009. [Google Scholar]
- 53.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009; 30(16): 2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J. Comput. Chem. 2004; 25: 1157–1174. [DOI] [PubMed] [Google Scholar]
- 55.Breneman CM, Wiberg KB. Determining atom-centered monopoles from molecular electrostatics potentials–the need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990; 11: 361–373. [Google Scholar]
- 56.Carmona C, Dowd AJ, Smith AM, Dalton JP. Cathepsin L proteinase secreted by Fasciola hepatica in vitro prevents antibody-mediated eosinophil attachment to newly excysted juveniles. Mol. Biochem. Parasitol. 1993; 62: 9–17. [DOI] [PubMed] [Google Scholar]
- 57.Basch P. Cultivation of Schistosoma in vitro. I. Establishment of culture from cercariae and development until pairing. J. Parasitol. 1981; 67: 179–185. [PubMed] [Google Scholar]
- 58.Paveley RA, Bickle QD. Automated imaging and other developments in whole-organism anthelmintic screening. Parasit. Immunol. 2013; 35: 302–313. [DOI] [PubMed] [Google Scholar]
- 59.Burden DJ, Bland AP, Hughes DL, Hammet NC. Fasciola hepatica: a technique for the study of gut penetration by juvenile flukes. Parasitology. 1981; 83(2):249–52. [DOI] [PubMed] [Google Scholar]
- 60.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT. A program to generate schematic diagrams of protein–ligand interactions, Protein Eng. 1995; 8: 127–134. [DOI] [PubMed] [Google Scholar]
- 61.Lecaille F, Choe Y, Brandt W, Li Z, Craik CS, Bromme, D. Selective inhibition of the collagenolytic activity of human cathepsin K by altering its S2 subsite specificity. Biochemistry 2002; 41: 8447–8454. [DOI] [PubMed] [Google Scholar]
- 62.Hernández-Álvarez L, Naranjo Feliciano D, Hernández González JE, Soares RO, Barreto Gomes DE, Pascutti PG. Insights into the interactions of Fasciola hepatica cathepsin L3 with a substrate and potential novel inhibitors through in silico approaches. PLoS NTD 2015; 9(5): e0003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McVeigh P, McCammick EM, McCusker P, Morphew RM, Mousley A, et al. RNAi dynamics in Juvenile Fasciola spp. Liver flukes reveals the persistence of gene silencing in vitro. PLoS NTD. 2014; 8(9): e3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McGonigle L, Mousley A, Marks NJ, Brennan GP, Dalton JP, Spithill TW, et al. The silencing of cysteine proteases in Fasciola hepatica newly excysted juveniles using RNA interference reduces gut penetration Int J Parasitol. 2008; 38(2):149–155. [DOI] [PubMed] [Google Scholar]
- 65.Beckham SA, Piedrafita D, Phillips CI, Samarawickrema N, Law RH, Smooker PM, et al. A major cathepsin B protease from the liver fluke Fasciola hepatica has atypical active site features and a potential role in the digestive tract of newly excysted juvenile parasites. Int J Biochem Cell Biol. 2009; 41(7):1601–1612. 10.1016/j.biocel.2009.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vicente-Carrillo A, Edebert I, Garside H, Cotgreave I, Rigler R, Loitto V, Magnusson KE, Rodríguez-Martínez H. Boar spermatozoa successfully predict mitochondrial modes of toxicity: Implications for drug toxicity testing and 3R principles. Toxicol. in Vitro. 2015; 29: 582–591. 10.1016/j.tiv.2015.01.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
% inh.: percentage of inhibition at 10 μM dose. Values represent means ± SE. n = 2. R: represent B ring.
(DOCX)
% inh.: percentage of inhibition at 10 μM dose. Values represent means ± SE. n = 2. *C29 is a derivative of C28, which lacks the α,β-unsaturated system: 3-(naphthalen-1-yl)-1,5-diphenylpentane-1,5-dione.
(DOCX)
% inh.: percentage of inhibition at 10 μM dose. Values represent means ± SE. n = 2.
(DOCX)
(DOCX)
Residues of catalytic sub-sites are highlighted in bold. ΔOcc is calculated with the formula ΔOcc = Occ FhCL1—Occ FhCL1_34
(DOCX)
Residues of catalytic sub-sites are highlighted in bold. ΔOcc is calculated with the formula ΔOcc = Occ FhCL3—Occ FhCL3_34.
(DOCX)
% MOT: percentage of motile spermatozoa (motility > 5 μm/s) normalized to untreated control. VCL: Velocity curved line (> 24 μm/s). Spermatozoa were treated 1h with C34 at 37°C.
(DOCX)
(WMV)
(WMV)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.