

Chronic lymphocytic leukemia (CLL) is a B-cell malignancy in which apoptotic-resistant mature lymphocytes relentlessly accumulate in bone-marrow, lymph nodes and peripheral blood. In these body compartments, CLL B cells get survival signals mainly through the B-cell antigen receptor (BCR) pathway. The critical role of BCR signaling in CLL pathogenesis, i.e. proliferation, survival, adhesion, migration, and differentiation, is very well established. Bruton’s tyrosine kinase (BTK) is a key component of the BCR pathway and is required for CLL maintenance and development. BCR pathway inhibition through irreversible inhibition of BTK at Cys481 residue in its ATP-binding domain by ibrutinib has significantly improved the treatment outcome in CLL via disrupting the interactions with microenvironment.
Phase 1 clinical trial of ibrutinib (PCI-32765) in relapsed or refractory CLL malignancy patients resulted in 70% overall response rate [1]. Phase 1b and 2 ibrutinib clinical testing in patients with relapsed and refractory disease also showed similar (70%) response rate which lead to breakthrough status for ibrutinib [2]. Outstanding outcome during clinical trials without untoward toxicity resulted in FDA approval of ibrutinib for patients with CLL who have received at least one prior therapy. Long-term follow-up suggested a revolutionary role of ibrutinib for CLL patients.
Single-agent ibrutinib induced durable responses in the majority of CLL patients; however, these were mostly partial remissions, less than 10% achieved complete remission and, importantly, patients are not cured. A major part of the reason for partial remission is lymphocytosis after ibrutinib treatment which was due to the egress of CLL lymphocytes from lymph node niches. A key feature of ibrutinib therapy was early and dramatic reduction in lymph nodes with a parallel increase in CLL cells in blood circulation [2]. Ibrutinib-mediated BTK inhibition does not induce cell death directly but impairs BCR-associated integrin-mediated adhesion and migration, which leads to CLL cells efflux from lymphoid tissues (lymph node microenvironment) to peripheral blood. In peripheral blood due to lack of sustained survival factors which are in lymphoid tissues, these lymphocytosed CLL cells slowly undergo cell death. However, this slow process might allow the opportunity of selection or outgrowth of resistant clones of CLL cells. In fact, during therapy, in many individuals, peripheral blood lymphocytosis was prolonged. Furthermore, some of the patients on continuous therapy with ibrutinib developed resistance to ibrutinib therapy [3]. Among these patients, mutation at the ibrutinib-binding site on BTK, mutation in additional molecules in the BTK axis such as PLCγ2, and non BCR pathway modulations were identified [3]. Moreover driver mutation SF3B1 which is associated with poor prognosis also contributed to ibrutinib-acquired resistance [4]. Finally, increased Bcl-2 protein with a decline in Mcl-1 and Bcl-XL has also been observed and suggested as a survival mechanism for ibrutinib-treated CLL cells [5]. These data underscore a need to identify pharmacological agents that can synergistically partner with ibrutinib. Continuous and prolonged use of ibrutinib incurs a high cost of therapy. Finding optimal drug combinations would not only help achieve deeper responses but may also taper the high cost of chronic ibrutinib intake.
TP-0903 is a selective inhibitor of Axl receptor tyrosine kinase. Axl receptor, a member of TAM (Tyro3, Axl and Mertk) family of receptor tyrosine kinases, is overexpressed in CLL and plays an oncogenic survival role [6]. Previously Sinha and colleagues [7] demonstrated Axl receptor inhibition by TP-0903 as an effective cytotoxic approach for inducing cell death in CLL. They showed that application of TP-0903 during in vitro investigations of primary CLL cells disrupted constitutively phosphorylated status of Axl and induced marked Bcl-2-family anti-apoptotic members’ depletion-dependent apoptosis. This was independent of CLL stage, prognostic and cytogenetic markers, or microenvironmental protection. B-PAC-1 is a second generation procaspase-activating compound (PAC) [8], which can convert inactive dimers of executioner procaspases to their active cleaved forms by removing labile Zinc ions from inactive dimer states. These agents overcome resistance inflicted by survival factors such as Bcl-2 family protein overexpression, a phenomenon observed in primary CLL cells. B-PAC-1, as a single agent, induced dose- and time-dependent selective cytotoxicity to malignant CLL and not normal cells and also overcame microenvironment-mediated apoptosis resistance. The cell death was bax/bak-independent and required only the presence of executioner procaspases (−3, −6, and −7). These terminal procaspases are present in abundance in primary CLL cells [9].
Based on this background information about these two drugs and the over-abundance of Bcl-2 in lymphocytosed cells after ibrutinib therapy, we hypothesized that both TP-0903 and B-PAC-1 would result in the cell death of CLL lymphocytes that are spared after ibrutinib intake. Furthermore, we postulated that lymphocytosed cells obtained from patients undergoing ibrutinib therapy would be ideal to test these two novel agents.
TP-0903 and B-PAC-1 were obtained from Tolero Pharmaceuticals and Vanquish Oncology, respectively. All patients were on ibrutinib single agent protocol [see Supplementary Table 1 – online only] and signed informed consent; these were approved by our Institutional Review Board.
To test our hypothesis, we used CLL cells from patients prior to or 1, 2, 4, and 12 weeks after ibrutinib therapy. Using 50–175 nM levels, we demonstrated that TP-0903 treatment induced significant dose-dependent CLL cell death after a 24-hr treatment; LD50 was similar during our base-line ex vivo and prior in vitro treatments [7] [Fig. 1A]. TP-0903 LD50 values, for week 0 samples (white color) were 120, 130, and 160 nM for top, middle and bottom panels, respectively; for week 1 (gray color) samples, the values were 115 and 55 nM for top and middle panels; for week 4 (lined bar), the values were 160 nM (top panel) while for the rest of the samples, the values were not reached at the concentration used. Data were similar in an additional two patient samples at weeks 2, 4, and 12 [Supplementary Fig. 1 – online only]. For these two patients (only post ibrutinib therapy), at week 2 (WBC:327; ALC:307) and week 4 (WBC:358; ALC:347), LD50 were 160 nM and 140 nM, respectively, while for the other patient sample at week 12, LD50 was not reached. Furthermore, cell death was highest at peak lymphocytosis; suggesting weeks 1–2 post-ibrutinib as the optimal time to add TP-0903 for maximal apoptosis. In fact, at weeks 4 and 12, the extent of apoptosis declined due to a percent decrease of CLL cells in the population [Fig. 1A; Supplementary Fig. 1]. The dose-dependent cell death induced by TP-0903 and cell death response dependent on peak lymphocytosis using patient samples on ibrutinib therapy suggested the potential utility of this combination. In this regard, it is important to mention that AXL inhibitors are entering Phase I clinical trials [10].
Figure 1.
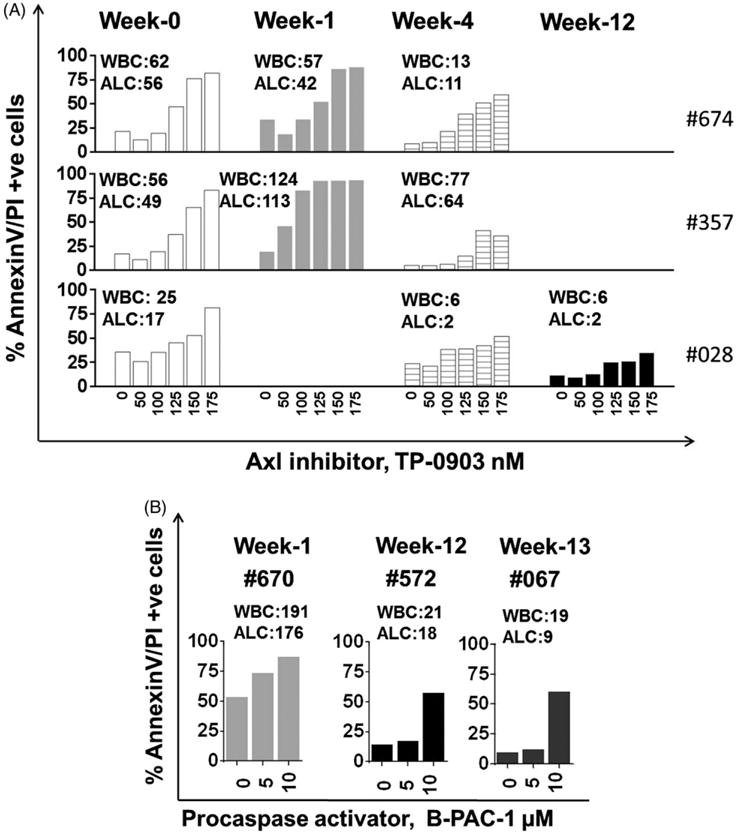
Induction of apoptosis by TP-0903 and B-PAC-1 treatment in lymphocytes obtained from CLL patients before and after oral ibrutinib therapy. All experiments were conducted in freshly isolated CLL lymphocytes. For this, peripheral blood samples were obtained from patients with CLL undergoing oral bid ibrutinib at 420 mg dose. CLL cells were isolated from blood and suspended in RPMI medium with 10% human serum. (A) Peripheral blood of CLL patients (n = 3) was collected at week 0 (pre-ibrutinib treatment), and at weeks 1, 4 and 12 (post-ibrutinib treatment). Cells were treated ex-vivo with DMSO, or increasing concentrations of Axl inhibitor, TP-0903 (50, 100, 125, 150, 175 nM) for 24 hours. Each panel (Top [patient sample #674], middle [patient sample #357], and bottom [patient sample #028]) contain samples taken from the same patient during pre- and post- ibrutinib therapy. (B) Lymphocytes were isolated from the peripheral blood of three CLL patients. Patient sample #670, #572 and #067 were collected at week 1, 12 and 13 post-ibrutinib. For these patients, pre-ibrutinib therapy samples were not obtained. Cells were treated ex-vivo with DMSO, or 5 and 10 μM B-PAC-1 (executioner caspases activator) for 18 hours. AnnexinV/PI staining was performed by flow cytometry and annexin/PI positive cells were taken as cells undergoing apoptosis. The white blood cell count (WBC; # × 103/mL) and absolute lymphocyte count (ALC; # × 103/μL) for each sample were determined from clinical blood count and are mentioned in the figure. Protocol was approved by the institutional review board at The University of Texas MD Anderson Cancer Center and patients signed informed consent.
For the second agent, B-PAC-1, we have used B cells isolated from three CLL patients on ibrutinib therapy at different durations, i.e. week 1, 12, and 13 [Fig. 1B]. Using these leukemic B cells, we demonstrate that B-PAC-1 induced significant dose-dependent cell death (5 and 10 μM B-PAC-1, for 18 hours). Regardless of their duration of treatment and lymphocyte counts, B-PAC-1 was able to induce cytotoxicity in leukemic cells. Previously, we have established that in primary CLL cells during in vitro incubation, IC50 concentration of B-PAC-1 at 24 hours was 10 μM. In the current work, a similar concentration for 18 hours induced 53% cell death in CLL cells from patients on ibrutinib therapy indicating similar or higher sensitivity of this agent on post ibrutinib samples [Fig. 1B]. Our results suggested the potential of combining this novel compound with ibrutinib. Currently, PAC-1 is in clinical trials (NCT02355535- clinicaltrials.gov) and B-PAC-1 is being developed to move to clinic.
Prior reports have identified pharmacologic agents such as ABT-199 (Bcl-2 antagonist) [5], carfilzomib (proteasome inhibitor) [11], dinaciclib (cyclin-dependent kinase inhibitor) [12] and selinexor (exportin inhibitor) [13] as agents that induced significant apoptosis in lymphocytosed cells post-ibrutinib therapy during ex-vivo investigations. Besides these agents, during in vitro settings pharmacological agents such as TP-0903 [7] and sudemycin (spliceosome modulator) [14] resulted in synergistic or additive cytotoxicity in CLL B cells when applied in combination with ibrutinib [14] or with a reversible BTK-inhibitor [7]. In contrast, BCR pathway inhibitors such as ibrutinib, idelalisib, duvelisib, MK2206, AEB-071, dasatinib or alkylating agent such as bend-amustine did not result in synergy [5,12]. These results suggest that compounds that induce apoptosis were generally beneficial with ibrutinib. Consistent with these drugs, TP-0903 and B-PAC-1 are also highly proapoptotic in CLL cells [9,15]. In addition, TP-0903 was also found to be active in CLL with 17p del; a poor-prognosis with single agent ibrutinib.
Clinical trials are either being designed or underway with ibrutinib and either ABT-199 or carfilzomib for treatment of CLL. The two new agents introduced for combination with ibrutinib suggest a good potential for their clinical progress. Ibrutinib couplet with TP-0903 or B-PAC-1 can be easily translated to the clinic to test safety and utility of these combinations. Concentrations of TP-0903 are physiological [7]. Moreover, B-PAC-1’s parent compound PAC-1 is currently in clinical trial. These combinations may avoid development of potential therapy-induced resistance to ibrutinib through mitigation of prolonged lymphocytosis and through acceleration of faster and deeper responses leading to potentially complete remissions for patients with CLL.
Supplementary Material
Acknowledgments
This work was supported in part by grant P01CA81534 from the NCI, DHHS, sponsored research agreements from Pharmacyclics and Vanquish, and funds from the MD Anderson Moon Shot Program.
Footnotes
Potential conflict of interest: Disclosure forms provided by the authors are available with the full text of this article at http://dx.doi.org/10.3109/10428194.2015.1102243.
Supplemental data for this article can be accessed http://dx.doi.org/10.3109/10428194.2015.1102243
References
- 1.Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byrd JC, O’Brien S, James DF. Ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:1278–1279. doi: 10.1056/NEJMc1309710. [DOI] [PubMed] [Google Scholar]
- 3.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burger JA, Landau D, Hoellenriegel J, et al. Clonal evolution in patients with chronic lymphocytic leukemia (CLL) developing resistance to BTK inhibition. Blood. 2013;122:866. doi: 10.1038/ncomms11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cervantes-Gomez F, Lamothe B, Woyach JA, et al. Pharmacological and protein profiling suggests venetoclax (ABT-199) as optimal partner with ibrutinib in chronic lymphocytic leukemia. Clin Cancer Res. 2015;21:3705–3715. doi: 10.1158/1078-0432.CCR-14-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boysen J, Sinha S, Price-Troska T, et al. The tumor suppressor axis p53/miR-34a regulates Axl expression in B-cell chronic lymphocytic leukemia: implications for therapy in p53-defective CLL patients. Leukemia. 2014;28:451–455. doi: 10.1038/leu.2013.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha S, Boysen J, Nelson M, et al. Targeted axl inhibition primes chronic lymphocytic leukemia B Cells to apoptosis and shows synergistic/additive effects in combination with BTK inhibitors. Clin Cancer Res. 2015;21:2115–2126. doi: 10.1158/1078-0432.CCR-14-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Putt KS, Chen GW, Pearson JM, et al. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat Chem Biol. 2006;2:543–550. doi: 10.1038/nchembio814. [DOI] [PubMed] [Google Scholar]
- 9.Patel V, Balakrishnan K, Keating MJ, et al. Expression of executioner procaspases and their activation by a procaspase-activating compound in chronic lymphocytic leukemia cells. Blood. 2015;125:1126–1136. doi: 10.1182/blood-2014-01-546796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheridan C. First Axl inhibitor enters clinical trials. Nat Biotechnol. 2013;31:775–776. doi: 10.1038/nbt0913-775a. [DOI] [PubMed] [Google Scholar]
- 11.Lamothe B, Cervantes-Gomez F, Sivina M, et al. Proteasome inhibitor carfilzomib complements ibrutinib’s action in chronic lymphocytic leukemia. Blood. 2015;125:407–410. doi: 10.1182/blood-2014-07-585364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2014;123:1810–1817. doi: 10.1182/blood-2013-09-527853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hing ZA, Mantel R, Beckwith KA, et al. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood. 2015;125:3128–3132. doi: 10.1182/blood-2015-01-621391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xargay-Torrent S, Lopez-Guerra M, Rosich L, et al. The splicing modulator sudemycin induces a specific anti-tumor response and cooperates with ibrutinib in chronic lymphocytic leukemia. Oncotarget. 2015;6:22734–22749. doi: 10.18632/oncotarget.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh AK, Secreto C, Boysen J, et al. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: implications for therapy. Blood. 2011;117:1928–1937. doi: 10.1182/blood-2010-09-305649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.