

Abstract
Malignant cells from breast cancer, as well as other common cancers such as prostate and melanoma, may persist in bone marrow as quiescent, nondividing cells that remain viable for years or even decades before resuming proliferation to cause recurrent disease. This phenomenon, referred to clinically as tumor dormancy, poses tremendous challenges to curing patients with breast cancer. Quiescent tumor cells resist chemotherapy drugs that predominantly target proliferating cells, limiting success of neoadjuvant and adjuvant therapies. We recently developed a 3-dimensional spheroid model of quiescent breast cancer cells in bone marrow for mechanistic and drug testing studies. We combined this model with optical imaging methods for label-free detection of cells, preferentially using glycolysis versus oxidative metabolism to investigate the metabolic state of co-culture spheroids with different bone marrow stromal and breast cancer cells. Through imaging and biochemical assays, we identified different metabolic states of bone marrow stromal cells that control metabolic status and flexibilities of co-cultured breast cancer cells. We tested metabolic stresses and targeted inhibition of specific metabolic pathways to identify approaches to preferentially eliminate quiescent breast cancer cells from bone marrow environments. These studies establish an integrated imaging approach to analyze metabolism in complex tissue environments to identify new metabolically targeted cancer therapies.
Keywords: optical metabolic imaging, quiescence, metastasis, bone marrow, fluorescence lifetime imaging
Introduction
Quiescent cancer cells are relatively insensitive to cytotoxic therapies and may contribute to late relapse even decades after initial diagnosis of disease (1–3). Disseminated cancer cells reside in microenvironments that support cancer cell quiescence and confer resistance to chemotherapy, allowing these cells to persist in a viable state after adjuvant treatment (4, 5). In particular, bone marrow is regarded as a major reservoir for quiescent cancer cells. Bone marrow is considered to maintain cellular quiescence of cancer cells in a similar fashion as hematopoietic stem cells, using metabolism, cell contacts, and secreted factors to control distribution (endosteal versus vascular niches) and proliferation of these cells (6–9). One strategy toward eliminating quiescent cancer cells is to selectively target unique metabolic demands of these cells, a hallmark feature of cancer that may be exacerbated by hypoxia in bone marrow niches (10). Understanding differences in metabolism between disseminated cancer cells and stromal cells in bone marrow and developing therapies by selectively targeting cancer metabolism offer the potential to eliminate quiescent cancer cells and prevent late recurrence of disease.
Growing cancer cells often have modified energetic profiles compared with normal cells (8); yet, because of few representative experimental models, relatively little is known about metabolism of quiescent cancer cells in bone marrow. Cancer cells, hematopoietic stem cells, and other mesenchymal stem cells (MSCs) have commonalities and differences in energetic and biosynthetic demands during growth, quiescence, and in response to hypoxia that may inform potential therapies (11). Similar to hematopoietic stem cells in a hypoxic bone marrow, cancer cells become increasingly dependent on glycolysis via activation of the hypoxia inducible factor pathway, which inhibits glucose-derived pyruvate from entering the mitochondria (12). MSCs exhibit metabolic flexibility between oxidative versus glycolytic states; differentiate toward chondrogenic and not osteogenic lineages under hypoxia; and are locked in glycolytic or oxidative metabolisms upon differentiation into chondrogenic or osteogenic/adipogenic lineages, respectively (13). Under hypoxic conditions in bone marrow, hematopoietic stem cells, cancer cells, and mesenchymal stromal cells produce higher levels of molecules that scavenge reactive oxygen species, increase pentose phosphate pathway metabolism, and replace mitochondrial flux with reductive glutamine pathways (13, 14).
Despite commonalities in hypoxia responsiveness among resident bone marrow cells, hypoxia induces specific cell-type differences that may be leveraged to selectively eliminate cancer cells. For example, hematopoietic stem cells decrease mitochondrial mass under hypoxia, decreasing production of reactive oxygen species (15, 16). In contrast, hypoxic cancer cells enlarge their mitochondria, inhibiting apoptosis with a concomitant elevation in reactive oxygen species (17). In response to reactive oxygen species, cancer cells may regain proliferative capacity and/or acquire additional mutagenic stress, but hematopoietic stem cells terminally differentiate and/or undergo apoptosis (18, 19). Leveraging a fine balance between responsiveness to reactive oxygen species, energetic demands, and biosynthetic needs may reveal ways to selectively eliminate quiescent cancer cells in heterogeneous bone marrow microenvironments (20).
Recent studies show heterogeneous metabolic microenvironments in bone marrow, which may differently regulate cancer cell quiescence. For example, high metabolic demands of bone marrow create intravascular hypoxia, with additional oxygen depletion proceeding away from the vascular compartment toward endosteal niches (10). Sharply graded hypoxic microenvironments lead to different locations; levels of differentiation; and abundance of hematopoietic, mesenchymal, immune, vascular, and cancer cells (8). In vivo models integrated with imaging provide approaches to analyze heterogeneous microenvironments in intact tissues; but, these methods suffer from limitations of cost, low throughput, and limited opportunities for biochemical manipulations. Therefore, simple, but representative, experimental models are needed to recreate such heterogeneous microenvironments that harbor quiescent cancer cells.
Metabolic status in vivo is the summation of all of these interacting components; but, as a model system, we begin by considering the metabolically diverse status of MSC lineages represented by the MSC-derived cell lines, HS-5 and HS-27A. HS-5 and HS-27A cells support either expansion or maintenance of hematopoietic stem cells, respectively (21). Multiple studies of these cells describe dichotomous regulation of gene expression, secretion of growth factors, matrix deposition, and support of bone marrow transplant efficiency among other effects on cancer and hematopoietic stem cells (22, 23). In addition, recent data suggest that unlike HS-5 cells, HS-27A cells are more pluripotent and express markers of hematopoietic niche formation. HS-27A cells also have greater metabolic flexibility between glycolytic and oxidative metabolic states (13). How the metabolic microenvironment supported by these stromal cells and their metabolic coupling with cancer cells supports cancer growth or quiescence remains largely unknown.
While prior studies have emphasized interconnected signaling networks in tumor environments, recent research reveals that metabolic links among malignant and stromal cells control growth, quiescence, and drug sensitivity of cancer cells. Malignant cells promote metabolic reprogramming of stromal cells to supply metabolites, such as lactate and glutamine, necessary to fuel growth of neighboring epithelial cancer cells (24). Cancer and stromal cells may exhibit different capacities to metabolize glutamine to produce energy and metabolic intermediates (glutaminolysis), suggesting the potential to uncouple tumor–stromal metabolism without toxicity to normal cells (25). To capitalize on cancer metabolism as a target to eliminate disseminated tumor cells from bone marrow, there is an unmet need for cell-based assays that reproduce intercellular interactions in vivo while enabling facile analyses of metabolic states and drug efficacy.
Here we investigate how the metabolic status of 3-dimensional (3D) cancer–stromal spheroids determines growth and drug sensitivity of breast cancer cells. We use optical imaging of endogenous nicotinamide adenine dinucleotide (NADH), a metabolic coenzyme that shifts between protein-bound and -free states in oxidative metabolism and glycolysis, respectively (26). We combined optical imaging of NADH with extracellular metabolic flux measurements as a label-free method for determining relative metabolic status of cancer and stromal cells and their responses to metabolic perturbation and drug treatments within the 3D culture environments. We also independently assess viability of cancer and stromal cells by dual-color bioluminescence imaging. We found that metabolic differences in bone marrow stromal cells differentially support quiescence of multiple cancer cells, interdependence on glucose and glutamine, and sensitivity to molecular inhibition of metabolic pathways. In particular, we found HS-5 stromal cells to rely on glycolysis, induce quiescence of multiple breast cancer subtypes (including aggressive, triple-negative MDA-MB-231 cells), and provide limited support to cancer cells upon metabolic perturbations. We also found HS-27A stromal cells to be more metabolically flexible, supporting cancer cell resistance to nutrient withdrawal. These data highlight the ability for label-free optical imaging approaches to analyze metabolic states of cancer and stromal cells in complex 3D environments, enabling mechanistic studies of quiescent cancer cells in bone marrow and testing of therapies for effective targeting of these cells.
Results
Bone Marrow Stromal Spheroids Support Growth or Quiescence of Breast Cancer Cells
We used long-term co-cultures of bone marrow stromal cell lines (HS-5 and HS-27A) to recreate growth or quiescence of disseminated cancer cells in bone marrow (27). To investigate relative effects of HS-5 and HS-27A stromal cells on quiescence or growth of triple-negative (ER−/PR−/Her2−) MDA-MB-231 cells, we varied the composition of the stromal environment and independently monitored the growth of cancer and stromal cells with stably expressed click beetle (CB)Green and CBRed luciferases, respectively. We determined that a relatively low fraction of HS-5 cells (effective concentration for 50% maximal biologic effect [EC50], ∼27%) in the stromal environment limited the growth of MDA-MB-231 cells (Figure 1A). Compared with the quiescence-supporting HS-5 cells, HS-27A cells supported rapid growth of cancer cells, similar to the results of our previous report (27). MDA-MB-231 co-cultures with untransformed primary human MSC grew rapidly compared with HS-5 cells, suggesting that HS-27A cells represent the subtype of mesenchymal stromal cells typically enriched through in vitro isolation (data not shown).
Figure 1.

HS-5 and HS-27A bone marrow spheroids differentially consume glucose and support cancer cell growth. Spheroids composed of varying ratios of HS-5 and HS-27A stromal cells show different fractional glucose consumption and preference for cancer cell proliferation. Of the 2 stromal cell types, HS-5 cells are greater consumers of glucose, while HS-27A cells better support cancer cell proliferation. Graph shows mean values ± standard error of the mean (SEM) (N = 28 per condition across 3 experiments) of the fold-change in cancer cell bioluminescence and fractional glucose consumption from day 2 to day 10 in spheroids composed of varying ratios of HS-5 and HS-27A stromal cells. *P < .05, **P < .01 (A). Metabolic status of stromal cells in co-culture spheroids queried by fluorescence lifetime imaging (N = 4–5 per condition). Glycolytic cells appear near the bottom right of the graph, whereas oxidative cells appear near the upper left (B). The graph shows mean values ± SEM (N = 6 per condition) of extracellular acidification and oxygen consumption rates of HS-5 and HS-27A stromal cells in co-culture spheroids; higher extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) indicate greater metabolic activity. i, baseline; ii, after glucose injection; and iii, after oligomycin injection. Mean normalized frequency ± SEM (N = 4 per condition) of fluorescent intensity corresponding to nicotinamide adenine dinucleotide (NADH) (D), flavin adenine dinucleotide (FAD) (E), and redox ratio for 3 stromal cell lines (F). Inset shows box and whiskers plot of mean values of redox ratios (N ≥ 20 cells each). *P < .05.
Despite quiescence of cancer cells and stromal cells in the HS-5 spheroid environment, we observed substantial acidification of their extracellular environments compared with HS-27A cells based on phenol red in culture medium (data not shown). To measure whether lactate-based acidification corresponded to decreased glucose levels in HS-5 spheroids, we measured glucose levels in the medium after 2 days in culture. Corresponding with decreased pH in the HS-5 cultures, these cells reduced glucose in the bulk media by ∼20% relative to ∼10% for HS-27A cells (Figure 1A). Although this is a relatively small decrement considering the typical nonphysiologically high glucose culture medium (25 mM) compared with that in vivo (5 mM), we expect that local, cell-scale depletion of glucose for cancer cells within HS-5 spheroids may surpass 20% depletion.
To further confirm differences in glucose consumption between HS-5 and HS-27A stromal cell types in 3D co-culture spheroids, we used optical metabolic imaging based on fluorescence lifetime imaging (FLIM) of NADH. This metabolic coenzyme functions in both glycolysis and oxidative metabolism, serving as the principal electron acceptor and donor in these pathways, respectively. In cells relying predominantly on glycolytic metabolism, relatively more NADH exists in the free state, resulting in a shorter fluorescence lifetime of this autofluorescent molecule (26). By comparison, NADH predominantly is bound to metabolic enzymes in the mitochondria during oxidative phosphorylation, lengthening the fluorescence lifetime. We used frequency-domain FLIM with phasor transformation of NADH autofluorescence lifetime to determine metabolic states of stromal cells in spheroids (28). Optical imaging showed greater glycolytic metabolism in HS-5 than in HS-27A cells in spheroids with either MDA-MB-231 or T47D breast cancer cells (Figure 1B).
To further investigate differences between glucose consumption between these cell types in 2 dimensions, we used the Seahorse extracellular flux analyzer (XFe96) to compare the levels of oxygen consumption and extracellular acidification. Under baseline conditions of glutamine only, or sequential glucose and oligomycin (inhibition of oxidative metabolism) injections, we found the HS-5 cells to have both higher oxygen consumption and lactate production (Figure 1C; i baseline; ii after glucose injection; and iii after oligomycin injection). HS-5 and HS-27A cells were both relatively unaffected by inhibition of oxidative metabolism by oligomycin, as shown by the relatively small shift between points ii and iii of Figure 1C.
On a per cell basis, these results suggest that glucose consumption is different between stromal cells and between 2-dimensional (2D) and 3D cultures. From Seahorse data (Figure 1C, ii), assuming that 2 lactate protons are derived from a single glucose molecule (an imperfect but reasonable estimate), we calculate glucose consumption rates of about 3.1 and 2.5 fmol/min/cell for HS-5 and HS-27A cells in the 2D culture, respectively, after normalizing to 1.5 × 104 cells per well. Assuming a constant per-cell glucose consumption, glucose consumption rates are 14.4 and 7.1 fmol/min/cell for HS-5 and HS-27A cells in 3D spheroids, respectively, after normalizing to 3 × 103 cells per spheroid. These results suggest that on a per cell basis, cells are more glycolytic in 3D cultures, which is consistent with other reports comparing 2D and 3D cultures (29).
Optical Metabolic Imaging to Study Metabolic Microenvironment
To confirm the glucose consumption of our bone marrow stromal cells, we used complementary optical metabolic imaging parameters to capture intensities and redox ratio of endogenous NADH and flavin adenine dinucleotide (FAD), another metabolic coenzyme. While NADH exhibits autofluorescence, the oxidized counterpart of this molecule, NAD+, has no intrinsic fluorescence. Oxidative metabolism in the mitochondria consumes NADH and produces NAD+, while cells using glycolysis have higher amounts of NADH. Therefore, higher fluorescence intensity from NADH is another label-free optical metric of a shift to glycolytic metabolism (30). In contrast, fluorescence intensity of FAD increases in cells with greater utilization of oxidative phosphorylation. The optical redox ratio normalizes fluorescence from these molecules as the ratio of NADH to FAD fluorescence intensities; the redox ratio increases as cells preferentially use glycolysis (31).
We measured distributions of intensities of autofluorescence between NADH and FAD in HS-5 and HS-27A cells. Populations of HS5 cells had relatively more cells with higher NADH intensities and lower FAD. Likewise, the background-adjusted pixel-by-pixel ratio of NADH to FAD fluorescence intensities showed a similar rank order, with HS5 cells having a higher redox ratio. Overall, these data for autofluorescence of NADH and FAD reinforce the conclusion that HS-5 cells are more glycolytic than HS-27A cells.
To assess the impact of stromal cell type on the metabolic microenvironment of 3D spheroids, we measured the intensity-independent fluorescence lifetime profile of NADH across the diameter of spheroids with different cell types and sizes. The following 2 factors determine the metabolic status of these spheroids: the exterior NADH lifetime and the slope of the NADH lifetime over the first 60 μm of the spheroid. The former reflects the non-nutrient limited condition and the latter measures the responsiveness of stromal cell types to radial nutrient density and distance to the center of the spheroid. To measure radial lifetime profiles, we used 4 radial sections of multiple spheroids and a photon count-weighted average of above-threshold pixels to calculate average lifetime for radial positions (Supplemental Figure 1). As proof of concept for this approach, we show that the size of MSC spheroids dictated the slope of the NADH lifetime gradient but not the exterior starting value (Figure 2). Small spheroids showed the limited slope of NADH lifetime from exterior to interior, and we observed a rank-order increase in the slope with increasing size of the spheroid (Figure 2, B and C). Increasing the slope reflects a shift from bound to free NADH as cells in the interior of larger spheroids use glycolysis to a greater extent. Lifetime did not change through depth of small nonnutrient limited spheroids (Supplemental Figure 2). Previous reports describe hypoxia and/or anoxia at approximately 100 μm from blood vessels in rat mesentery in vivo, 100-150 μm in tumor sections, and beyond several cell-diameters in silico bone marrow models (32, 33). Although it is difficult to decouple oxygen and other metabolic gradients from NADH lifetime profiles, our data suggest sensing of metabolic changes at distances smaller than described for hypoxia.
Figure 2.

Mesenchymal stem cell (MSC) spheroids show size-dependent radial NADH lifetime gradients. Pseudocolored fluorescence lifetime imaging (FLIM) images of cross sections of 120-, 160-, or 200-μm diameter MSC spheroids (A). Graph shows mean values ± SEM (N = 4 per condition) of NADH phase lifetimes for different-sized spheroids at given distances from spheroid centers (B). The graph shows the mean values ± SEM (N = 4 per condition) of NADH phase lifetime slope in spheroids of various diameters, where a smaller value indicates less NADH lifetime change throughout a spheroid (C).
Comparing among large spheroids of different stromal cell types, we found that the nonnutrient limited exterior condition reflects the NADH phase lifetimes measured in 2D cultures, with MSCs having the lowest exterior lifetime, followed by HS-5 and HS-27A cells (Figure 3, A and B). This result suggests that the exterior of these spheroids has similar metabolic status as 2D cultures with no metabolic limitations. HS-5 spheroids produced the lowest slope of NADH lifetime at 0.00283 ns/μm (95% CI: 0.0025–0.0032), followed by MSC at 0.0039 ns/μm (95% CI: 0.0034–0.0044), and HS-27A at 0.0041 ns/μm (95% CI: 0.0038–0.0046). High slopes reflect flexibility between oxidative and glycolytic metabolism. These data reflect that HS-5 spheroids are the least responsive to radial nutrient gradients, followed by MSCs and HS-27A cells, with both having similarly high levels of responsiveness.
Figure 3.

Bone marrow stromal cell types produce different 3-dimensional (3D) metabolic gradients in spheroids. The graph shows mean values ± SEM (N = 4 per condition) of NADH phase lifetimes in MSC, HS-5, or HS-27A spheroids at given distances from spheroid centers (A). The graph shows the mean values ± SEM (N = 4 per condition) of NADH phase lifetime slope in spheroids of various stromal cell compositions (B). NADH phase lifetime of HS-5 cells in spheroids shows less depth-dependence, whereas NADH phase lifetime of HS-27A stromal cells shows greater fluctuation as we query the interior of the spheroid (B). Fluorescence image of spheroids composed of MDA-MB-231 cells (indicated by fluorescence) and HS-5 stromal cells (autofluorescent cells) (C). Fluorescence image of spheroids composed of MDA-MB-231 cells (indicated by fluorescence) and HS-27A stromal cells (autofluorescent cells) (D).
To establish how MDA-MB-231 cells are distributed in spheroids with different metabolic profiles, we imaged locations of these breast cancer cells 24 hours after spheroids were formed. MDA-MB-231 cells were distributed solely on the exterior of the more glycolytic, less metabolically responsive, HS-5 spheroids. By comparison, we identified MDA-MB-231 cells both in the interior and exterior of spheroids with the more metabolically responsive HS-27A cells (Figure 3, C and D). Because spheroids are seeded with a homogenous mix of stromal and cancer cells, the interior–exterior distribution must be an active migratory process by the cancer and/or stromal cells. These localization differences persist at longer time points. Over time, the exterior cancer cells in HS-27A spheroids grow substantially, whereas the interior MDA-MB-231 cells do not (Figure 3, C and D; data not shown).
Metabolic Balance Between Glycolysis and Glutaminolysis for Cancer–Stromal Co-culture
Increased glycolysis by HS-5 cells depletes bulk glucose levels by about 20%, which, we expect, limits the local concentration available to the cancer cells. With depleted glucose, we anticipate that cancer cells will become increasingly dependent on other sources of carbon, ie, lactate, glutamine, amino acids, or fatty acids. We tested the interdependence of 2 breast cancer subtypes (MDA-MB-231 and T47D) on combinations of glucose and glutamine when cultured with more metabolically responsive HS-27A or unresponsive HS-5 stromal cells. To selectively quantify relative proliferation or toxicity of different metabolic conditions on cancer versus stromal cells, we used dual-color bioluminescence imaging with CBGreen and CBRed luciferases stably expressed in cancer and stromal cells, respectively (27). After allowing 2 days for the formation of spheroids in complete media and 8 days of culture in various glucose and glutamine concentrations, we quantified differences in the viability of cancer versus stromal cells in different co-culture spheroids by dual-color bioluminescence imaging. We plotted the fold change for cancer cell bioluminescence compared with the high glucose and high glutamine condition, color-coded for the log-2-based ratio of cancer to stromal signals (Figure 4). Positive values (warmer colors with red) reflect preferential survival of cancer cells relative to stromal cells with negative values (cooler blue colors), denoting greater survival of stromal cells. Corresponding surface plots for stromal components with the same color coding are found in Supplemental Figure 3.
Figure 4.
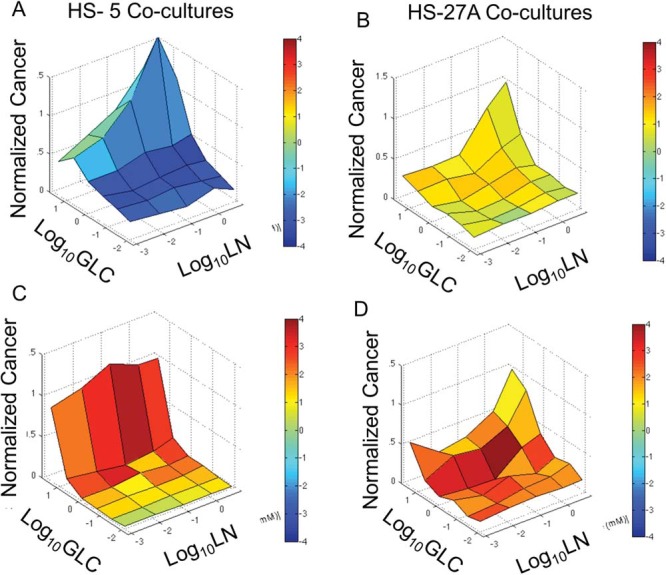
Spheroid stromal cells differentially regulate cancer cell sensitivity to glucose and/or glutamine deprivation. Graphs show mean (N = 4 per condition) fold-change in bioluminescent signal from cancer cells in spheroids composed of MDA-MB-231 cancer cells and HS-5 (A) or HS-27A stromal cells (B) or T47D cancer cells and HS-5 (C) or HS-27A stromal cells (D). GLC, glucose; LN, glutamine.
The 2 breast cancer subtypes show unique responses to combinations of glucose and glutamine in the 2 stromal environments. MDA-MB-231 cells were very responsive to both glucose and glutamine in both HS-5 and HS-27A co-cultures (Figure 4, A and B). In contrast, counterpart HS-5 stromal cells were insensitive to glutamine but highly sensitive to glucose, and counterpart HS-27A cells were largely insensitive to both glucose and glutamine. These data correspond with our NADH lifetime profiles, showing HS-5 stromal cells to be highly glycolytic, while HS-27A cells show more metabolic flexibly between glycolysis and oxidative metabolism. Warmer color coding for low glutamine and high glucose conditions highlights the selective response of MDA-MB-231 cells toward glutamine deprivation in HS-5 co-cultures. The stromal cells' glucose–glutamine response surface profile was independent of counterpart cancer cells (Supplemental Figure 3), suggesting that the larger fraction of stromal cells in each spheroid dominates their response. T47D cells showed minimal sensitivity to changes in extracellular glutamine concentrations but extremely high sensitivity to glucose withdrawal in HS-5 co-cultures (Figure 4C), which may reflect dependence on glycolysis that is associated with ER+-dependent signaling (34). In HS-27A co-cultures, T47D cells were moderately responsive to glucose and glutamine withdrawal (Figure 4D). Recreating pharmacological sensing of glutamine and/or glucose deprivation may provide an avenue for metabolically targeted therapy to selectively eliminate some subsets of breast cancer metastases.
Metabolic Inhibition Selectively Eliminates Cancer Cells
Using known inhibitors of glycolysis, we tested whether we could selectively eliminate growing and quiescent cancer cells by inhibiting differential metabolic profiles. Using 2-deoxy-glucose, which inhibits the first enzyme in glycolysis, we confirmed that HS-5 cells are more dependent on glycolysis than HS-27A and MDA-MB-231 cells (Figure 5A). Compared with HS-5 cells, HS-27A and MDA-MB-231 cells grown with either stromal environment are similarly less sensitive to both 2-deoxy-glucose and glucose deprivation. Using dichloroacetate, which inhibits pyruvate dehydrogenase kinase and forces pyruvate away from lactate production into mitochondria, we selectively eliminated cancer cells in both their quiescent and growing states in both HS-5 and HS-27A spheroids (Figure 5B).
Figure 5.

Metabolic inhibition selectively eliminates MDA-MB-231 cancer cells. The graphs show mean values ± SEM (N = 7 per condition) of fold-change in cancer cell bioluminescence and drug selectivity in spheroids composed of MDA-MB-231 cancer cells and HS-5 or HS-27A stromal cells treated with 2-deoxy-glucose (A), dichloroacetate (B), oligomycin (C), rotenone (D), DHEA (E), or PD0325901 (F).
In addition, we tested known inhibitors of oxidative metabolism via electron transport decoupling and inhibition of oxidative metabolism. As predicted by minimal responsiveness of HS-5 and HS-27A to oligomycin in Seahorse assays, inhibition of complex I and V with rotenone and oligomycin, respectively, had minimal effect on stromal cells (Figure 5, C and D). MDA-MB-231 cells were selectively eliminated in both HS-5 and HS-27A spheroids, suggesting a required function of the role of oxidative metabolism in both quiescent and growing situations in these cells. Inhibition of oxidative metabolism was most effective in the growth-supporting HS-27A microenvironment and less positively selective in HS-5 spheroids. Inhibition of oxidative metabolism was positively selective in both stromal environments. However, elimination of cancer cells was incomplete with 10%–20% and 30%–50% CBGreen signal from cancer cells remaining at maximal levels of rotenone and oligomycin for HS-27A and HS-5 co-cultures, respectively. The incomplete response to inhibition of oxidative metabolism and the cancer cell location within HS-27A spheroids (Figure 3D) suggests that there is a reservoir of glycolytic cancer cells housed in the center of the HS-27A spheroid. Therefore, molecular interventions that could suppress both oxidative and glycolytic metabolism may better eliminate cancer cells in both metabolic states. Analogous to a different disease model, coinhibition of glycolysis and oxidative metabolism was a successful strategy in reversing systemic lupus erythematosus by deactivating CD4+ T cells (35).
Contact-inhibition-induced quiescence of stromal fibroblasts previously has been coupled with maintenance of glycolytic throughput, shuttling glucose into the pentose phosphate shunt, and backward flux of glutamine into the tricarboxylic acid cycle (36). To test whether flexible metabolism of HS-27A and their support of MDA-MB-231 cell growth are dependent on the pentose phosphate pathway, we measured their dose response to the steroid hormone inhibitor of this pathway, dehydroepiandrosterone (DHEA) (Figure 5E) (37). Interestingly, cancer cells were eliminated with positive selectivity in both HS-5 and HS-27A spheroids, suggesting the involvement of the pentose phosphate pathway for these cancer cells in both stromal environments. The effective concentration for 50% maximal biologic effect (EC50) was reduced for both stromal and cancer cells in HS-27A compared with HS-5 co-cultures. These data suggest that unlike HS-5 cells, HS-27A cells produce a compensatory shift toward the pentose phosphate pathway within spheroids.
We previously suggested that selective elimination of quiescent and growing MDA-MB-231 cells by inhibition of constitutively active K-Ras (MEK inhibitor, PD0325901) could be attributed to nonproliferation-based mechanisms (27) (Figure 5F). To test the metabolic effects of MEK inhibition, we pretreated MDA-MB-231 cells overnight before measuring extracellular flux in the Seahorse system. Metabolic activity was suppressed for glutaminolysis, glycolysis, and maximal glycolytic capacity (Figure 6). These data further suggest that MEK inhibition functions in part through a noncell cycle-based mechanism and suppresses both glycolytic and oxidative metabolism.
Figure 6.
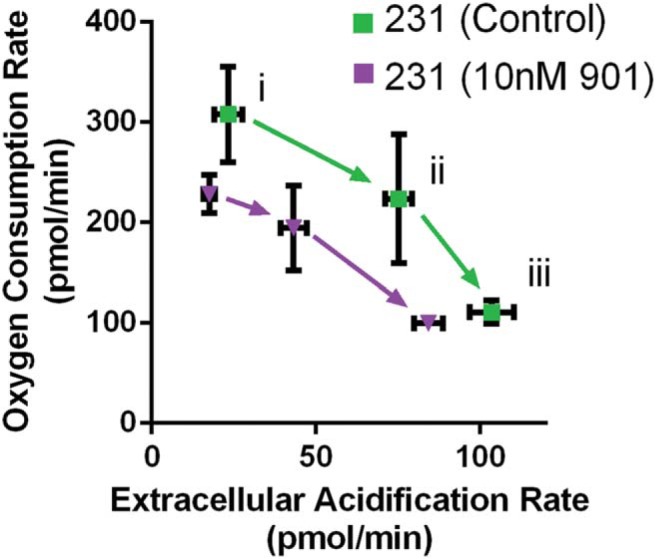
Drug inhibition of MEK suppresses metabolic activity of MDA-MB-231 cells. The graph shows mean values ± SEM (N = 6 per condition) of extracellular acidification and oxygen consumption rates of MDA-MB-231 cancer cells untreated or pretreated with PD0325901; higher extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) indicate greater metabolic activity. i, baseline; ii, after glucose injection; and iii, after oligomycin injection.
We also tested metabolic inhibitors 2-deoxy-glucose, dichloroacetate, oligomycin, and DHEA on spheroids composed of T47D breast cancer cells and HS-5 or HS-27A stromal cells. By bioluminescence imaging, T47D breast cancer cells in HS-5 spheroids were selectively eliminated using dichloroacetate, oligomycin, and DHEA, but not 2-deoxy-glucose (likely because of our earlier finding that HS-5 cells are highly dependent on glycolytic metabolism) (Figure 7A-D). T47D breast cancer cells in HS-27A spheroids were selectively eliminated by treatment with all 4 metabolic inhibitors (Figure 7A-D). Overall, these data show that targeting multiple metabolic pathways preferentially kills 2 different breast cancer cell lines in 3D models of bone marrow metastases.
Figure 7.
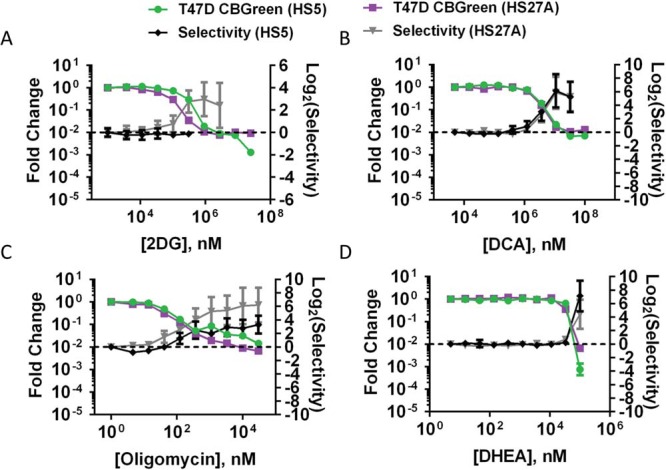
Metabolic inhibition selectively eliminates T47D cancer cells. Graphs show mean values ± SEM (N = 7 per condition) of fold-change in cancer cell bioluminescence and drug selectivity in spheroids composed of T47D cancer cells and HS-5 or HS-27A stromal cells treated with 2-deoxy-glucose (A), dichloroacetate (B), oligomycin (C), or DHEA (D).
Discussion
Recent commentaries highlight potential advantages of therapies targeting cancer metabolism relative to standard cytotoxic drugs (38, 39). These advantages include reduced toxicity to other rapidly proliferating cells affected by standard chemotherapy drugs, including hematopoietic stem cells and mucosa of the gastrointestinal tract. Metabolic cancer therapy offers the potential to eliminate quiescent cancer cells that are refractory to standard therapies and contribute to relapse, which is the primary focus of the current manuscript. Here we build upon our previous co-culture model of bone marrow metastases to analyze metabolic phenotypes in quiescent and growing cancer cells (27). Using this system, we discovered that glycolytic HS-5 spheroids promote cancer cell quiescence, and the basally less metabolically active but more metabolically responsive HS-27A spheroids promote cancer growth. Our complementary strategies to measure and perturb metabolic status of growing and quiescent cells may provide a platform to screen therapies that limit delayed recurrences of breast cancer.
Despite derivation from the same MSC population, our results reveal that HS-5 and HS-27A cells represent 2 metabolically distinct stromal populations (40). Our measured redox ratios, extracellular flux and glucose consumption analyses, basal NADH fluorescent lifetimes, and radial NADH phase lifetime gradients describe HS-5 cells as more glycolytic and less metabolically responsive than HS-27A cells. These results, previous results of HS-27A cells expressing a marker of multipotency associated with the hematopoietic niche (40), and description of metabolically flexible MSCs (13) suggest that HS-27A are less differentiated and more common to the hematopoietic stem cell niche in vivo than HS-5 cells. Furthermore, our results suggest that HS-5 spheroids contain a larger reservoir of glycolytic cancer cells that are resistant to inhibition of oxidative metabolism compared with HS-27A co-cultures. These data, combined with localization of cancer cells in co-culture spheroids and minimal responsiveness of NADH fluorescence lifetime, suggest that HS-5 co-cultures may model rigid hypoxic-like, glycolytic phenotypes compared with more metabolically responsive HS-27A spheroids. In contrast, in HS-27A spheroids, cancer sensitivity to inhibition of oxidative metabolism, glycolysis, and the pentose phosphate pathway, taken with cancer cell distribution on NADH lifetime profile, all suggest these stromal cells to be more metabolically responsive.
Although it is difficult to assign cause and effect with these outcomes, it is clear that the more metabolically responsive HS-27A spheroids provide protection from glucose–glutamine withdrawal for breast cancer cell lines that are representative of 2 different molecular subtypes (basal MDA-MB-231 and estrogen receptor-positive T47D). Using optical imaging methods, we discovered that MDA-MB-231 cells were highly dependent on both glucose and glutamine in spheroids with stromal cell types, which may reflect a previous report of combined dependence of K-Ras-transformed cells on both glucose and glutamine (41). In this work, they describe that K-Ras-transformed cells, such as MDA-MB-231, rely on glycolysis for adenosine triphosphate and glutamine for anaplerosis. In addition, we found that the inhibition of MEK within MDA-MB-231 cells suppressed both oxidative metabolism and glycolysis, resulting in selective cancer cell elimination in both spheroid environments. For these cancer cells, mimicking combined oxidative and glycolytic inhibition may provide a strategy for eliminating metastases similar to normalization of metabolism to reverse the symptoms of systemic lupus erythematosus (35). In comparison, estrogen receptor-positive T47D cells showed extreme sensitivity toward glucose deprivation, consistent with a previous report linking increased expression of hormone receptors in breast cancer to glycolysis (34).
Overall, this research provides a platform to link optical readouts of cell metabolism with metabolic flux parameters and response to treatment in 3D bone marrow environments. In particular, we demonstrate how these integrated research methods can be applied to identify metabolic perturbations and therapies that preferentially target proliferating and/or quiescent cancer cells relative to bone marrow stroma. This technology platform expands upon prior research using optimal imaging to measure metabolism of cancer cells and response to therapy in 2D and 3D environments without considering the effects of the surrounding stromal environment (31, 34, 42). To better define mechanisms for each of these cancer cell lines, we propose measuring cell cycle status, NADH lifetime, and drug-dependent outcomes for the cancer cells in each stromal environment. Using these strategies to screen and develop compounds that target metabolism of cancer cells may provide novel alternatives to cytotoxic therapies that would better eliminate quiescent cancer cells in bone marrow and prevent late recurrences of breast cancer. These data could be linked with clinical imaging methods such as positron emission tomography with F18-fluorodeoxyglucose or potentially hyperpolarized magnetic resonance imaging techniques to analyze metabolic therapies in patients.
Materials and Methods
Reagents
Unless otherwise stated, we purchased all cell culture reagents from Life Technologies (Carlsbad, California, USA). We obtained drugs and compounds from the following sources: PD0325901 from SelleckChem (Houston, Texas, USA); D-glucose, 2-deoxy-D-glucose, rotenone, and sodium dichloroacetate from Sigma Aldrich (St. Louis, Missouri, USA); and oligomycin from Seahorse Biosciences (Agilent Technologies, Santa Clara, California, USA). We prepared a rotenone stock at 10mM in ethanol. We prepared all other compounds in formulations supplied or specified by manufacturers.
Cell Culture and Spheroid Assays
We grew HS-5, HS-27A, MDA-MB-231, and T47D cells as described previously (27). In brief, we maintained all cells in 10% fetal bovine serum (FBS) (HyClone, Thermo Fisher Scientific, Waltham, Massachusetts, USA) Dulbecco's Modified Eagle's Medium (DMEM) (#11995, Thermo Fisher Scientific) supplemented with penicillin, streptomycin, and glutamine (Thermo Fisher Scientific). Cells were maintained at subconfluence and passaged every 2 to 4 days. Stromal and cancer cells express CBRed and CBGreen, respectively. Cancer cells also expressed a fluorescent ubiquitin-based cell cycle indicator (43). We used human bone marrow MSCs from Lonza (Basel, Switzerland).
We detailed our spheroid assays previously (27). In brief, 3 × 103 cells were distributed as a suspension in 25 μL per well to black 384 shallow, round-bottom well plates (Corning Incorporated #3676, Corning, New York, USA). For cancer cell growth and dose–response assays, we composed spheroids of 1% MDA-MB-231 cells with the balance of stromal cells. For experiments looking at the distribution of cancer cells throughout spheroids, we seeded 3.3 × 103 total cells with the additional 10% MDA-MB-231 cells. For optical metabolic imaging in different-sized spheroids, we seeded 300, 1000, and 3000 MSCs. For comparison of metabolic gradients in different stromal cell types, we seeded spheroids with 3 × 103 HS-5, HS-27A, and MSCs. For optical metabolic imaging in 2 dimensions, we seeded 1 × 106 HS-5 or HS-27A cells in 35-mm dishes in spheroid media with 10% FBS. Once cells were firmly adhered, we replaced medium with that used for spheroids.
The culture conditions of all spheroids were consistent unless otherwise stated. We seeded and maintained all spheroids in phenol red free DMEM (#31053, Thermo Fisher Scientific), supplemented with 1% FBS (HyClone), penicillin/streptomycin/glutamine (Thermo Fisher Scientific), and pyruvate (Thermo Fisher Scientific). For experiments modulating the glucose and glutamine levels, the base medium was replaced with glucose- and glutamine-free DMEM, supplemented with glutamine-free penicillin/streptomycin (Thermo Fisher Scientific), and pyruvate. We then supplemented D-glucose (Sigma Aldrich) and glutamine (Thermo Fisher Scientific) at listed concentrations. We exchanged ∼80% of the volume of media every 2nd day for the entirety of each assay.
The timing of each assay was consistent unless otherwise stated. For growth assays, we measured bioluminescence flux at 2 and 10 days, changing media with fresh spheroid media every 2 days. For dose-response curves, after spheroids formed for 2 days, we initialized treatment by exchanging media on the 2nd day and every 2 days thereafter for a total assay time of 10 days. For optical metabolic imaging in 2 dimensions and spheroids and for microscopy of cancer cell distribution, we imaged spheroids 24 hours after seeding.
Bioluminescence Imaging
We previously described CBGreen and CBRed imaging in detail (27).
Microscopy
For microscopy of 2D and 3D cultures, we used an Olympus MPE 1000 system using a 25X NIR corrected objective (XLPLN25XWMP, NA = 1.05, Olympus, Tokyo, Japan). We facilitated stable imaging of spheroids by transferring spheroids to a custom spheroid imaging plate that we described previously (44).
Intensity-Based Metabolic Imaging
For intensity-based optical metabolic imaging, we used 2 paired excitation and emission wavelengths for NADH and FAD imaging similar to previous descriptions (34). For NADH intensities, we excited with 740 nm light from a Spectra-Physics Mai Tai DeepSee femtosecond pulsed laser (Mountain View, California, USA) and collected light with a band-pass filter (420-460 nm). For FAD intensities, we excited with 890 nm light and collected emitted light with a band-pass filter (495-540 nm). We maintained constant imaging parameters within each experimental run, including laser power, detector voltage, and offset. To quantify image intensities and ratios, we exported pixel-by-pixel intensities from Fluoview (Olympus, Tokyo, Japan) into Microsoft Excel (Seattle, Washington, USA), where we removed below background pixels (determined from cell-free regions) and performed additional calculations. We also used Excel to determine frequency distributions and NADH to FAD ratios.
Frequency-Domain Fluorescence Imaging and Data Analysis
For lifetime-based measure of metabolic status, we used an ISS FastFLIM system (ISS, Champaign, Illinois, USA), containing a Spartan 6 processor, coupled with our Olympus MPE microscope. We calibrated the FLIM system to a fluorescein dye slide, using open filters and 740-nm excitation and an expected single component lifetime of 4.1 nanosecond. Using an excitation of 740 nm with a 2-photon laser, we collected NADH emission using a 511-LP dichroic and 460/25-nm band-pass filters. We also measured FAD lifetime simultaneously using the long-pass dichroic cut and 530/11-nm band-pass filter. The caveat with the FAD measurement with 740-nm excitation is that there is a small spectral overlap with NADH (45). Using excitation of 890 eliminates NADH excitation but would require recalibration of the FLIM system with each laser wavelength change. We used a 256 × 256 view field and 100 μs dwell time for all frequency-domain images. We captured between 15 and 25 frames and adjusted laser intensity to maximize pixels above our count threshold. To image size-based gradients in spheroids, we used smaller view fields for smaller spheroids to maximize the view area occupied by the spheroid.
To analyze the data for phase lifetimes, we exported the phase lifetime and counts from the VistaVision software (ISS) into Excel. In Excel, we removed lifetime and count values that fell outside 0.5-5 nanoseconds for phase lifetime and below 5 for counts. We then applied a count-weighted average for imaging segments as described in Supplemental Figure 1. For images of stromal cells cultured in 2D, we used the count-weighted average for the entire frame. For spheroids, we used radial cuts in 4 directions for each spheroid consisting of 1 × 50 pixel vectors that progressed from exterior to interior of each spheroid. Aligning the spheroid edge for each of 4 radial cross sections for 4 spheroids from 2 independent setups, we used the average lifetime across the radius profile. To analyze FLIM data for S and G values of the phasor plot corresponding to lifetime, we eliminated noise by setting the minimum count threshold at 10 counts and used the smoothing operation. We then used the VistaVision “Zoom” operation to select regions of interest, and the “Go to Peak” operation to objectively determine the average phasor values for individual cells.
Glucose Measurements
To measure bulk glucose from cell culture supernatants, we used a commercial blood glucose meter Accu-chek Aviva Plus (Roche Diagnostics, Risch-Rotkreuz, Switzerland). Samples were diluted 50% in phosphate-buffered saline containing 20 wt% Probumin (EMD Millipore, Billerica, Massachusetts, USA) to bypass blood quality errors. All values were benchmarked against high glucose measurements in spheroid culture medium (25mM, 450 mg/dL). We measured supernatants pooled from 4 spheroid wells and from 20 separate spheroids, totaling N = 5 measurements for each stromal combination.
Extracellular Flux Assays
We performed extracellular flux assays using an XFe96 Extracellular Flux Analyzer (Agilent Technologies). We performed the XF Glycolysis Stress test per manufacturer specifications. We seeded cells overnight at a density of 1.5 × 104 cells per well. Injections were performed sequentially with 25mM D-glucose, 1 μM oligomycin, and 100mM 2-deoxy-D-glucose. For pretreatment with PD0325901, we treated cells with 10 nM overnight and during the 1-hour incubation in unbuffered assay medium. Data are plotted for average of N = 6 wells over 3 measurement cycles.
Statistical Analysis and Plotting
We processed bioluminescence and optical metabolic data in Microsoft Excel (Seattle, Washington, USA) before plotting data using GraphPad Prism (San Diego, California, USA). All graphs display mean values ± standard error of the mean (SEM) unless stated otherwise. We propagated errors for all data normalized to experimental measurements.
We described our strategy for measuring selective elimination of cancer cells but not stromal cells using dual-colored CB luciferase previously (27). For growth curves, we divided average bioluminescence flux for red and green signals at day 10 by their corresponding value at day 2. For dose–response curves, we divide background subtracted flux values by their corresponding untreated condition for both green and red signals. To visualize the selectivity of cancer cell elimination, we plotted the log-2 ratio of the red signal divided by the green signal and subtracted the value of the untreated control. Selectivity for the untreated control is zero, and positive selectivity corresponds to relatively lower cancer (green) signals compared with stromal (red) signals.
Supplemental Materials
Acknowledgments
This work was supported by United States National Institutes of Health grant R01CA170198. SPC was supported by a NSF predoctoral fellowship F031543 and the Advanced Proteome Informatics of Cancer Training Grant T32 CA140044. We thank the University of Michigan Biointerfaces Institute for access to the Seahorse Extracellular Flux Analyzer. We also thank Ulas Coskun from ISS for assistance on operation and analysis of frequency-domain FLIM data and Mark Meyerhoff and Bruce Cohan for help with glucose measurements.
Conflict of Interest: The authors declare no competing financial interests.
Footnotes
- 3D
- 3-dimensional
- NADH
- nicotinamide adenine dinucleotide
- MSCs
- mesenchymal stem cells
- FLIM
- fluorescence lifetime imaging
- 2D
- 2-dimensional
- FAD
- flavin adenine dinucleotide
- CB
- click beetle
- DHEA
- dehydroepiandrosterone
- FBS
- fetal bovine serum
- DMEM
- Dulbecco's Modified Eagle's Medium
- SEM
- standard error of the mean
- SEM
- standard error of the mean
References
- 1. Demicheli R, Terenziani M, Bonadonna G. Estimate of tumor growth time for breast cancer local recurrences: rapid growth after wake-up? Breast Cancer Res Treat. 1998;51(2):133–137. [DOI] [PubMed] [Google Scholar]
- 2. Demicheli R. Tumour dormancy: findings and hypotheses from clinical research on breast cancer. Semin Cancer Biol. 2001;11(4):297–306. [DOI] [PubMed] [Google Scholar]
- 3. Brackstone M, Townson J, Chambers A. Tumour dormancy in breast cancer: an update. Breast Cancer Res. 2007;9(3):208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mitra A, Mishra L, Shulin L. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6(13):10697–10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pantel K, Alix-Panabieres C. Bone marrow as a reservoir for disseminated tumor cells: a special source for liquid biopsy in cancer patients. Bonekey Rep. 2014;3:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boulais P, Frenette P. Making sense of hematopoietic stem cell niches. Blood. 2015;125(17):2621–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, Milner LA, Kronenberg HM, Scadden DT. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–846. [DOI] [PubMed] [Google Scholar]
- 8. Morrison S, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, Harris S, Wiedemann LM, Mishina Y, Li L. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425(6960):836–841. [DOI] [PubMed] [Google Scholar]
- 10. Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, Zaher W, Mortensen LJ, Alt C, Turcotte R, Yusuf R, Côté D, Vinogradov SA, Scadden DT, Lin CP. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508(7495):269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15(4):243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Robey I, Lien A, Welsh S, Baggett B, Gillies R. Hypoxia-inducible factor-1α and the glycolytic phenotype in tumors. Neoplasia. 2005;7(4):324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pattappa G, Heywood H, de Bruijn J, Lee DA. The metabolism of human mesenchymal stem cells during proliferation and differentiation. J Cell Physiol. 2011;226(10):2562–2570. [DOI] [PubMed] [Google Scholar]
- 14. Reyes J, Fermanian S, Yang F, Zhou S, Herretes S, Murphy D, Elisseeff JH, Chuck RS. Metabolic changes in mesenchymal stem cells in osteogenic medium measured by autofluorescence spectroscopy. Stem Cells. 2006;25(5):1213–1217. [DOI] [PubMed] [Google Scholar]
- 15. Arranz L, Urbano-Ispizua A, Mendez-Ferrer S. Mitochondria underlie different metabolism of hematopoietic stem and progenitor cells. Haematologica. 2013;98(7):993–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prigione A, Adiaye J. Modulation of mitochondrial biogenesis and bioenergetic metabolism upon in vitro and in vivo differentiation of human ES and iPS cells. Int J Dev Biol. 2010;54(11-12):1729–1741. [DOI] [PubMed] [Google Scholar]
- 17. Chiche J, Rouleau M, Gounon P, Brahimi-Horn M, Pouyssegur J, Mazure NM. Hypoxic enlarged mitochondria protect cancer cells from apoptotic stimuli. J Cell Physiol. 2010;222(3):648–657. [DOI] [PubMed] [Google Scholar]
- 18. Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, Lu XJ, Ledergor G, Kollet O, Lapidot T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid Redox Signal. 2014;21(11):1605–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shao L, Li H, Pazhamisamy S, Meng A, Wang Y, Zhou D. Reactive oxygen species and hematopoietic stem cell senescence. Int J Hematol. 2011;94(1):24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kleffel S, Schatton T. Tumor dormancy and cancer stem cells: two sides of the same coin? Adv Exp Med Biol. 2013;734:145–179. [DOI] [PubMed] [Google Scholar]
- 21. Roecklein B, Torok-Storb B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood. 1995;85:997–1005. [PubMed] [Google Scholar]
- 22. Graf L, Iwata M, Torok-Storb B. Gene expression profiling of the functionally distinct human bone marrow stromal cell lines HS-5 and HS-27a. Blood. 2002;100(4):1509–1511. [DOI] [PubMed] [Google Scholar]
- 23. Weisberg E, Wright R, McMillin D, Mitsiades C, Ray A, Barrett R, Adamia S, Stone R, Galinsky I, Kung AL, Griffin JD. Stromal-mediated protection of tyrosine kinase inhibitor-treated BCR-ABL-expressing leukemia cells. Mol Cancer Ther. 2008;7(5):1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fiaschi T, Marini A, Giannoni E, Taddei M, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P, Chiarugi P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72(19):5130–5140. [DOI] [PubMed] [Google Scholar]
- 25. Ko Y, Lin Z, Flomenberg NP, RG, Howell A, Sotgia F, Lisanti M, Martinez-Outschoorn UE. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: implications for preventing chemotherapy resistance. Cancer Biol Ther. 2011;12(12):1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stringari C, Nourse J, Flanagan L, Gratton E. Phasor fluorescence lifetime microscopy of free and protein-bound NADH reveals neural stem cell differentiation potential. PLoS One. 2012;7(11):e48014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cavnar S, Rickelmann A, Meguiar K, Xiao A, Dosch J, Leung B, Cai Lesher-Perez S, Chitta S, Luker KE, Takayama S, Luker GD. Modeling selective elimination of quiescent cancer cells from bone marrow. Neoplasia. 2015;17:625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Digman M, Caiolfa V, Zamai M, Gratton E. The phasor approach to fluorescence lifetime imaging analysis. Biophys J. 2008;94(2):L14–L16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Longati P, Jia X, Eimer J, Wagman A, Witt M, Rehnmark S, Verbeke C, Toftgård R, Löhr M, Heuchel RL. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer. 2013;13:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mayevsky A, Rogatsky GG. Mitochondrial function in vivo evaluated by NADH fluorescence: from animal models to human studies. Am J Physiol Cell Physiol. 2007;292(2):C615–C640. [DOI] [PubMed] [Google Scholar]
- 31. Walsh A, Cook R, Sanders M, Aurisicchio L, Ciliberto G, Arteaga C, Skala MC. Quantitative optical imaging of primary tumor organoid metabolism predicts drug response in breast cancer. Cancer Res. 2014;74(18):5184–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsai A, Friesenecker B, Mazzoni M, Kerger H, Buerk D, Johnson P, Intaglietta M. Microvascular and tissue oxygen gradients in the rat mesentery. Proc Natl Acad Sci U S A. 1998;95(12):6590–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bertout J, Patel S, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8(12):967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Walsh A, Cook R, Manning H, Hicks D, Lafontant A, Arteaga C, Skala MC. Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer. Cancer Res. 2013;73(20):6164–6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yin Y, Choi S, Xu Z, Perry D, Seay H, Croker B, Sobel ES, Brusko TM, Morel L. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7(274):274ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lemons J, Feng X, Bennett B, Legesse-Miller A, Johnson E, Raitman I, Pollina EA, Rabitz HA, Rabinowitz JD, Coller HA. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 2010;8(10):e1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jimenez P, Frolova A, Chi M, Grindler N, Willcockson, Reynolds KA, Zhao Q, Moley KH. DHEA-mediated inhibition of the pentose phosphate pathway alters oocyte lipid metabolism in mice. Endocrinology. 2013;154(12):4835–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Galluzzi L, Kepp O, Vander Heiden M, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12:829–846. [DOI] [PubMed] [Google Scholar]
- 39. Martinez-Outschoorn U, Peiris-Pages M, Pestell R, Sotgia F, Lisanti M. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2016;May 4 doi: 10.1038/nrclinonc.2016.60 [Epub ahead of print]. [DOI] [Google Scholar]
- 40. Iwata M, Sandstrom R, Delrow J, Stamatoyannopoulos J, Torok-Storb B. Functionally and phenotypically distinct subpopulations of marrow stromal cells are fibroblast in origin and induce different fates in peripheral blood monocytes. Stem Cells Dev. 2014;23(7):729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gaglio D, Metallo C, Gameiro P, Hiller K, Danna L, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Varone A, Xylas J, Quinn K, Pouli D, Sridharan G, McLaughlin-Drubin M, Alonzo C, Lee K, Münger K, Georgakoudi I. Endogenous two-photon fluorescence imaging elucidates metabolic changes related to enhanced glycolysis and glutamine consumption in precancerous epithelial tissues. Cancer Res. 2014;74(11):3067–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miyawaki A. Visualization of the spatial and temporal dynamics of intracellular signaling. Dev Cell. 2003;4(3):295–303. [DOI] [PubMed] [Google Scholar]
- 44. Cavnar S, Salomonsson E, Luker K, Luker G, Takayama S. Transfer, imaging, and analysis plate for facile handling of 384 hanging drop 3D tissue spheroids. J Lab Autom. 2014;19(2):208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang S, Heikal A, Webb WW. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J. 2002;82(5):2811–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.