

Abstract
Transient forebrain ischemia induces delayed death of the hippocampal pyramidal neurons, particularly in the CA2 and medial CA1 area. Early pharmacological inhibition of inflammatory response can ameliorate neuronal death, but it also inhibits processes leading to tissue regeneration. Therefore, research efforts are now directed to modulation of post-ischemic inflammation, with the aim to promote beneficial effects of inflammation and limit adverse effects. Transcription factor NF-κB plays a key role in the inflammation and cell survival/apoptosis pathways. In the brain, NF-κB is predominantly found in the form of a heterodimer of p65 (RelA) and p50 subunit, where p65 has a transactivation domain while p50 is chiefly involved in DNA binding. In this study, we subjected middle-aged Nfkb1 knockout mice (lacking p50 subunit) and wild-type controls of both sexs to 17 min of transient forebrain ischemia and assessed mouse performance in a panel of behavioral tests after two weeks of post-operative recovery. We found that ischemia failed to induce clear memory and motor deficits, but affected spontaneous locomotion in genotype- and sex-specific way. We also show that both the lack of the NF-κB p50 subunit and female sex independently protected CA2 hippocampal neurons from ischemia-induced cell death. Additionally, the NF-κB p50 subunit deficiency significantly reduced ischemia-induced microgliosis, astrogliosis, and neurogenesis. Lower levels of hippocampal microgliosis significantly correlated with faster spatial learning. We conclude that NF-κB regulates the outcome of transient forebrain ischemia in middle-aged subjects in a sex-specific way, having an impact not only on neuronal death but also specific inflammatory responses and neurogenesis.
Keywords: cerebrovascular disease, neuroinflammation, neurogenesis, memory, transgenic mice
A transient forebrain ischemia is defined as a relatively homogeneous temporary reduction in the blood supply to the forebrain leading to depletion of oxygen and glucose in the brain tissue. In humans, this is often caused by cardiac arrest with subsequent cardiopulmonary resuscitation or occurs during cardiac bypass surgery. Transient forebrain ischemia is modeled by bilateral common carotid artery occlusion (BCCAo) in rodents. Similar to the clinical situation, BCCAo induces the death of selectively vulnerable neuronal populations, especially pyramidal neurons of the hippocampus and medium spiny neurons of the striatum, in a time-dependent manner [1-5].
The inflammatory response to ischemia contributes to delayed hippocampal neuronal death [6-10]. However, the proper inflammatory response is now also considered to be necessary for tissue regeneration, including neurogenesis and angiogenesis [11]. Proliferating endogenous microglia can limit ischemic damage by clearing tissue debris and cytotoxic neutrophils, and by secreting neurotrophic factors [12-15]. Pharmacological inhibition of inflammatory response reduces stroke-induced proliferation of neural progenitor cells [16, 17].
NF-κB is a pleiotropic transcription factor playing a key role in inflammation and cell survival/apoptosis pathways [18]. The most common form of NF-κB in the brain is a heterodimer consisting of p65 and p50 subunits [19] with p65 exhibiting transactivation activity and p50 (encoded by the Nfkb1 gene) being involved in DNA binding [18, 20]. In a rat transient forebrain ischemia model, strong transient upregulation of NF-κB has been reported throughout the hippocampal area with maximum activity at 48h [21], while prolonged NF-κB upregulation has been selectively observed in degenerating hippocampal neurons [21, 22]. Pharmacological inhibition of ischemia-induced NF-κB activation confers neuroprotection in a gerbil model of transient forebrain ischemia [23]. Also, genetic deletion of p50 subunit has been shown to be protective in rodent focal ischemia models [24, 25]. However, earlier studies concentrated on short-term outcomes and did not investigate the effect of modulation of NF-κB activity on ischemia-induced inflammation and post-ischemic functional recovery. Furthermore, to date no studies have assessed the impact of genetic deletion of p50 subunit on ischemia-induced inflammatory responses and functional recovery in BCCAo.
Here we show that both the lack of the NF-κB p50 subunit encoded by the Nfkb1 gene and female sex independently protected CA2 hippocampal neurons from transient forebrain ischemia-induced cell death. Nfkb1 gene deficiency also significantly reduced ischemia-induced microgliosis, astrogliosis, and neurogenesis. On the individual level, low hippocampal microgliosis significantly correlated with spatial learning.
MATERIALS AND METHODS
Animals
Nfkb1-/- (p50/p105 knockout (ko), hereafter referred as Nfkb1-ko) mice [26] were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and back-crossed to the C57BL/6J strain for 6 generations and maintained as homozygotes. Wild-type (Nfkb1-wt) C57BL/6J mice of the same age and sex were used as controls. Both male and female mice were included in the study. Experimental setup is summarized in Fig. 1. The similarity in the structure of the circle of Willis between the groups was verified by magnetic resonance angiography in a cohort of mice operated for short-term follow-up (Fig. 1 Cohort 2; Supplementary table 1).
Figure 1.
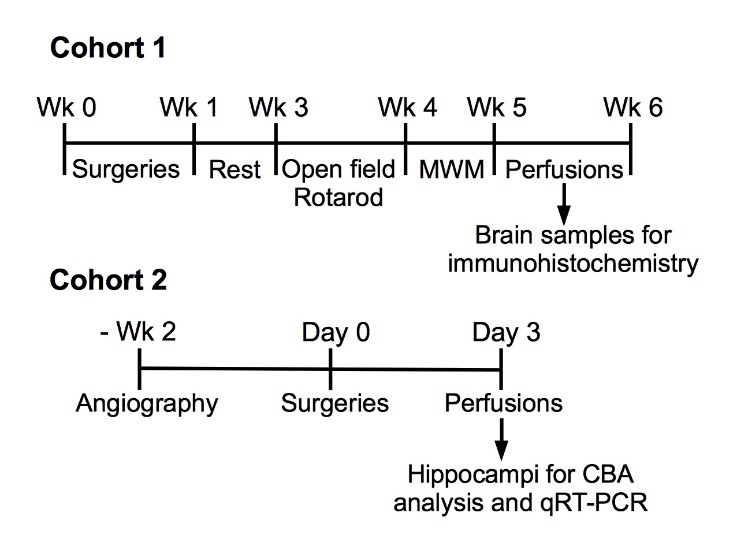
Experimental setup. Wk, week; MWM, Morris Water Maze (swim task).
The animals were kept in the Laboratory Animal Center at the University of Eastern Finland on a 12 h light/dark cycle and provided with standard laboratory chow and water ad libitum. All behavioral tests were conducted during the light phase. All animal work was done according to the national regulations and the Council of Europe (Directive 86/609) of the usage and welfare of laboratory animals and approved by the Animal Experiment Board of Finland.
Magnetic resonance angiography
Mice were imaged in vivo using 1.0 - 1.5% isoflurane (Baxter Oy, Helsinki, Finland) in 80% N2O / 20% O2 as carrier gas mixture for anesthesia two weeks prior to the surgeries. Imaging was conducted with a 9.4 T vertical magnet (Oxford Instruments PLC, Abingdon, UK) interfaced to a Varian DirectDrive console (Varian Inc, Palo Alto, CA, USA). A quadrature volume RF coil (diameter 20 mm; Rapid Biomedical GmbH, Rimpar, Germany) was used for transmission and reception. The MR angiography was conducted with 3D gradient recalled echo pulse sequence with the following parameters: TR = 40 ms, TE = 2.6 ms, flip angle = 30, matrix size = 100 x 150 x 150 and field of view = 10 x 15 x 15 mm3, giving an isotropic resolution of 100 μm, and scan time = 15 min. Blood vessels were further highlighted with a magnetization transfer preparation using a 3 ms sinc RF pulse with an amplitude of 925 Hz applied -2500 Hz off-resonance.
Surger
Transient forebrain ischemia as modelled by bilateral common carotid artery occlusion (BCCAo) was induced in 10- to 11-month-old mice targeted for long-term follow-up (Fig. 1 Cohort 1) or 12- to 14-month-old mice for short-term follow-up (Fig. 1 Cohort 2) as described [27, 28]. Anesthesia was induced with 3% halothane and maintained with 1-1.5% halothane (Nicholas Piramal India Ltd., Mumbai, Maharashtra, India) in 70% N2O / 30% O2 as the carrier gas. Core body temperature was maintained at 37 ± 1 °C by using a thermostatically controlled rectal probe connected to a homeothermic blanket system. Midline incision was made in the neck and both common carotid arteries were carefully isolated from vagus nerve using 6.0 silk suture and compressed for 17 min with atraumatic miniature aneurysm clips (Codman & Shurtleff, Inc., Raynham, MA, USA). The length of BCCAo was chosen based on our previous studies [27, 28]. In sham-operated mice, the arteries were dissected out but not compressed. Surgery time was kept constant at about 30-35 min. Artery occlusion and subsequent restoration of blood flow were confirmed visually. After the surgery the mice were given 0.5 ml saline ip and placed for overnight into the heated chamber set at 31-32 °C, after which the mice were returned to their home cages. The mice were sacrificed before the final time-point if they exhibited ≥ 20% weight loss and/or immobility.
The mice from Cohort 1 were randomly assigned to ischemia or sham groups. Altogether 50 males (wt ischemia n = 15, ko ischemia n = 17, wt sham n = 9, ko sham n = 9) and 53 females (wt ischemia n = 19, ko ischemia n = 13, wt sham n = 12, ko sham n = 9) were successfully operated. Mortality rate was overall low: 16% in the wt ischemic male group, 14% in the ko ischemic male group, 0% in female ischemic groups. Cohort 2 was operated for short-term follow-up (3 days) without including sham-operated animals: wt males, n = 7, ko males, n = 9, wt females, n = 10, ko females, n = 7. Only one animal from wt male group had to be sacrificed prematurely.
Behavioral tests
After full two weeks of post-operative recovery, the mice underwent a neurological test battery for two weeks (Fig. 1 Cohort 1). During the first week, spontaneous locomotor activity was assessed in the open field, and motor coordination and balance using the rotarod test. During the second week, spatial learning and memory were assessed by Morris swim task.
Open field test
Mice were tested in a dimly lit and quiet room using the white wading pool (120 cm in diameter) aimed for the Morris swim task but without water. The mice had 10 min to freely explore the arena. The movement was recorded by a ceiling camera, and tracked and analyzed by Ethovision XT 7 system (Noldus, Wageningen, Netherlands) including the following parameters: total distance moved, time in the arena center (1/3 of the area) and rearings. For the first test cohort, rearing was analyzed both by visual inspection and Ethovision to validate the automatic analysis. Since both methods matched well, only automatic analysis was used for all groups. Also, the fecal bullets were counted at the end of the 10 min test.
Rotarod test
Motor coordination and balance was tested with the Rota-Rod® apparatus (Ugo Basile, Comerio, Italy). The test was carried out in a quiet and well-lit room. The mouse was gently placed on the round rod (2 cm diameter) facing away from the investigator and allowed to adapt to the stationary rod for 30 s and to the rotating rod (5 rpm) for another 30 s. Then, for the next 8 min the speed of rotation was increased stepwise up to 30 rpm. The latency to fall off (or spin two full rounds around the rod) after the 1 min adaptation period was recorded until an 8-min cut-off time. The apparatus was cleaned with 70% ethanol before each mouse.
Morris swim task (water maze)
Spatial learning and memory were tested with Morris swim navigation task. The test was carried out in a quiet and well-lit room. The apparatus consisted of a white plastic pool (120 cm in diameter) and a transparent platform (10 cm × 10 cm) submerged 1.0 cm below the water surface. Water temperature was kept at 20 ± 0.5 °C throughout the testing, and an 8-10 min recovery period in a warmed cage was allowed between all trials. Before actual navigation task the mice were pretrained for two days to find and climb onto the submerged platform, aided by a guiding alley (1 m × 14 cm × 25 cm) with opaque walls preventing any spatial clues to be seen. In the learning phase (days 1-4), five 60-s trials per day were conducted with the hidden platform. The platform location was kept constant, and the starting position varied between 4 constant locations at the pool rim, with all mice starting from the same position and nose pointing toward the wall in any single trial. If a mouse failed to find the escape platform within 60 s, it was placed on the platform for 10 s by the experimenter. Also when mice found the platform independently they were allowed to stay on the platform for 10 s. On day 5, the first and the last trials were run without the platform to test memory retention after 24 h and 15 min, respectively. The experimenter marked the start of each trial using a remote controller and the trial ended automatically when a mouse landed on the platform. Swim paths and other parameters were tracked and analyzed by Ethovision XT 7.
Histochemistry
The mice were anesthetized with an overdose of tribromoethanol (Avertin, Sigma) diluted in tert-amylalcohol and flushed transcardially with heparinized saline (2.5 IU/ml). The brains were removed and postfixed in 4% paraformaldehyde at 4 °C for 21-22 h. The fixed brains were cryoprotected in 30% sucrose for two days, snap frozen in liquid nitrogen and cut with a cryostat into 20-µm coronal sections at the dorsal hippocampal level starting at -1.3 mm from bregma. Five to six sections at 200-μm intervals per animal were analysed immunohistochemically with the following antibodies: anti-NeuN (hexaribonucleotide binding protein-3, a marker of neuronal nuclei; 1:200; Millipore, MA, USA) to assess neuronal survival, GFAP (glial fibrillary acidic protein; 1:500; Dako, Glostrup, Denmark) to assess astrogliosis, Iba1 (ionized calcium-binding adapter molecule 1; 1:200; Wako Chemicals GmbH, Neuss, Germany) and CD (cluster of differentiation) 45 (1:100; Serotec, Oxford, UK) to assess microglial activation and proliferation, and doublecortin (DCX; 1:200; Millipore) to assess neurogenesis. After overnight incubation with the primary antibody, brain sections were incubated with appropriate Alexa Fluor 568-conjugated secondary antibodies (1:200; Molecular Probes, Eugene, OR, USA) or appropriate biotinylated secondary antibodies (Vector Laboratories, Burlingame, CA, USA) followed by Vectastain ABC peroxidase system (Vector) with Ni-enhanced 3,3'-diaminobenzidine development. In each experiment, a negative control was included, in which incubation with primary antibody was omitted.
For the double immunostaining, the sections were first stained for CD45, and the immunoreactivity was visualized by using tyramide-based signal amplification (TSA kit; PerkinElmer, Waltham, MA) according to the manufacturer’s instructions. Anti-CD45 staining was followed by anti-Iba1 staining visualized with Alexa Fluor 488-conjugated secondary antibody (1:200 dilution; Molecular Probes).
Cell counting, quantification and image analysis
Immunostained cells were visualised under appropriate filter sets with Olympus AX70 microscope (Olympus, NY, USA) connected to ColorView camera (Soft Imaging System, Münster, Germany) running an Analysis Software (Soft Imaging System). Left and right hippocampi were analyzed separately.
NeuN immunoreactivity in pyramidal cell layer was evaluated in two regions of interest (220 x 180 μm) per hippocampus per section - cornus ammonis (CA) 2 (as shown in Fig. 4) and medial CA1 - by using ImageJ Software (National Institutes of Health, USA) from five consecutive hippocampal sections at 200-μm intervals starting at approximately -1.3 mm from bregma. Images taken at 20x magnification were used for the analysis. The results are presented as the percentage of immunoreactive area per whole area of interest and expressed as mean NeuN immunoreactivity per section ± standard error of the mean (SEM).
Figure 4.

Neuronal loss in the hippocampus was predominantly observed in the CA2 area (A, rectangle) and CA1 area (A, arrow) five weeks after the BCCAo. (A) A representative image of NeuN immunostaining in the hippocampal area of ischemic mouse, scale bar = 500 μm; (C-H), representative higher magnification images of NeuN staining in CA2 area in male (C-E) and female (F-H) ischemic Nfkb1-wt (C, F), ischemic Nfkb1-ko (D, G) and sham Nfkb1-wt (E, H) animals, scale bar = 100 μm. (B) NeuN immunoreactivity in the CA2 area as delineated with a rectangle in images (C-H) and expressed as the percentage of the average value in the wt sham group (females and males normalized separately). P values are derived from three-way ANOVA followed by pairwise comparisons with Bonferroni’s adjustment; * p < 0.05 in comparison to the ischemic Nfkb1-wt animals; # p < 0.05, ## p < 0.01 in comparison to the corresponding sham-treated controls.
GFAP, Iba1 and CD45 immunoreactivity was evaluated in the whole hippocampal area outlined as described [29] by using ImagePro Plus Software (Media Cybernetics, Silver Spring, MD, USA). Additionally, GFAP immunoreactivity was evaluated specifically in the dentate gyrus region outlined as shown in Fig. 5 A. Images taken at 4x magnification were used for the analysis. Immunoreactive signal within the area of interest was segmented by applying constant threshold. Data are expressed as the percentage of immunoreactive area per whole area of interest and shown in the graphs as mean immunoreactivity per section ± SEM.
Figure 5.

Astrocytosis was evaluated by immunostaining with anti-GFAP antibody five weeks after the BCCAo. Astrocytosis was evaluated by immunostaining with anti-GFAP antibody and was observed predominantly in the CA1, CA2 and dentate gyrus (DG) regions of ischemic hippocampus (A) as compared to sham-operated animals (B), scale bar = 500 μm. GFAP immunoreactivity was evaluated in the whole hippocampal area (C) delineated by solid boundary line in (A) and expressed as the percentage of the whole area of interest. P values are derived from three-way ANOVA followed by pairwise comparisons with Bonferroni’s adjustment; * p < 0.05 in comparison to the Nfkb1-wt animals; # p < 0.05, ## p < 0.01, ### p < 0.001 in comparison to the sham-treated controls. Panel (D-I) representative images of GFAP immunoreactivity in the CA2 area of ischemic Nfkb1-wt and ko animals and wt sham controls, scale bar = 100 μm.
The number of round cells expressing high levels of CD45 was quantified with ImageJ Software from hippocampal images taken at 4x magnification by using the following parameters: particle size 5-30 pixels, circularity 0.6-1.0, threshold constant (high) and region of interest whole hippocampal area. The data are presented as the mean number of cells per section ± SEM.
DCX-positive cells in the dentate gyrus were counted directly under the microscope, and the data are presented as the mean number of cells per section ± SEM.
Cytokine analysis and quantitative Real-Time PCR
The mice were anesthetized with an overdose of tribromoethanol (Avertin, Sigma) diluted in tert-amylalcohol and flushed transcardially with heparinized saline (2.5 IU/ml). Hippocampi were dissected out, snap-frozen in liquid nitrogen and stored at -80 °C until further use.
To isolate both protein and RNA fraction, hippocampi were homogenized manually in 160 µl of the homogenization buffer containing sterile-filtered 20 mM Tris-HCl (pH 7.4), 0.25 M sucrose, 0.5 mM EDTA, 0.5 mM EGTA and protease inhibitor mixture (Complete; Roche Applied Science, Mannheim, Germany). The homogenate was centrifuged at 5,000 x g at 4 °C for 10 min. 30 µl aliquot was taken for protein analysis and frozen at -70 °C. 1 ml of TRIzol reagent (Life Technologies, Carlsbad, CA, USA) was added to the rest of the samples for later isolation of RNA. Also this fraction was frozen at -70 °C.
Protein concentration in the protein fraction was determined by using Bio-Rad Protein Assay (Bio-Rad Laboratories; Hercules, CA, USA). The amount of tumor necrosis factor (TNF) α, interferon (IFN) γ, interleukin (IL)-6, IL-10, monocyte chemoattractant protein (MCP)-1 and IL-12 p70 protein in the protein fraction was determined by using Cytometric Bead Array (CBA) Mouse Inflammation Kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions and expressed as pg of target protein per mg of whole protein content in the sample.
RNA was extracted using TRIzol reagent according to manufacturer’s instructions. Shortly, 0.2 ml of chloroform was added to the homogenates containing 1 ml of TRIzol. Samples were shaken manually, vortexed and centrifuged at 15,500 x g for 15 min at 4°C. Upper aqueous phase containing RNA was removed, and 0.5 ml of isopropyl alcohol was added to it. The samples were vortexed, incubated at room temperature for 10 min and then centrifuged at 15,000 x g for 10 min at 4°C. The pellet was washed twice with 75 % ethanol, air-dried and resuspended in 40 µl of DEPC water (Sigma-Aldrich St. Louis, MO, USA) containing RNAase inhibitor (Ribolock, Thermo Fisher Scientific, Waltham, MA, USA). RNA concentration and purity was measured with Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized from 500 ng of total RNA using random hexamer primers as a template and Maxima reverse transcriptase (all reagents from Fermentas, Fermentas Finland, Helsinki, Finland). The relative expression levels of mRNA encoding mouse Aif1 (allograft inflammatory factor 1 gene encoding for Iba1), Igf1 (insulin-like growth factor 1), Il1b (interleukin 1 β), Nos2 (inducible nitric oxide synthase) Bcl2 and Casp3 (caspase 3) were measured with quantitative RT-PCR StepOnePlus machine (Applied Biosystems, Life Technologies) by using specific assays-on-demand (Applied Biosystems) target mixes according to manufacturer’s instructions. The expression levels were normalized to glyceraldehyde 3-phosphate dehydrogenase gene and presented as fold change in the expression.
Statistical analysis
The data are expressed as mean ± SEM. The data were analyzed with SPSS software (SPSS Inc. Chicago, IL, USA). The main means of analysis was two-way ANOVA with genotype (Nfkb1-ko vs. wt) and operation (transient forebrain ischemia vs. sham) as between-subject factors or three-way ANOVA with genotype, operation and sex (female vs. male) as between-subject factors. When several time points were included as a within-subject factor, ANOVA for repeated measures was used with the same genotype and operation factors. The difference between data sets was considered statistically significant if p < 0.05.
RESULTS
Transient forebrain ischemia failed to induce significant memory and motor deficits
To investigate whether Nfkb1 gene deficiency has an effect on neurological outcome following transient forebrain ischemia, we subjected large cohorts of male and female Nfkb1-wt and Nfkb1-ko mice to 17 min of BCCAo. The time-point was chosen based on our previous studies [27, 28]. After two weeks of post-operative recovery we assessed the mouse performance in a panel of behavioral tests, including the open field test for spontaneous locomotor activity, rotarod test for motor coordination and balance, and Morris swim task (water maze) for spatial learning and memory (Fig. 1 Cohort 1).
The most notable finding in the Morris swim task was increased swimming speed of Nfkb1-ko male mice (F (1,43) = 5.5; p = 0.024) throughout the test and of females on days 4 and 5 (females: day x genotype F (4, 47) = 2.98; p = 0.028). As the mice get used to the test and gain confidence in finding the escape platform, they usually slow down toward later test days. The maintained high swim speed in Nfkb1-ko mice may indicate increased anxiety level or enhanced motivation to find an escape. In any event, it confounds the use of escape latency as a measure of learning. Therefore, we used path length as the primary outcome measure. Here we found no main effect of genotype on learning (Fig. 2 A-B; males: p = 0.10; females: p = 0.20). Both male (p = 0.029) and female (p = 0.024) Nfkb1-ko mice displayed reduced thigmotaxis (swimming in the vicinity of the wall), indicating that they abandoned the inefficient strategy to look for an escape through the wall sooner than did the wt mice. However, the probe test for search bias on day 5 (considered the most specific outcome measure for the hippocampal-dependent spatial memory) showed no genotype difference in the search bias (p > 0.13, Fig. 2 C). Overall, BCCAo did not strongly affect spatial learning and memory, suggesting a very mild neuronal loss in our ischemia model.
Figure 2.
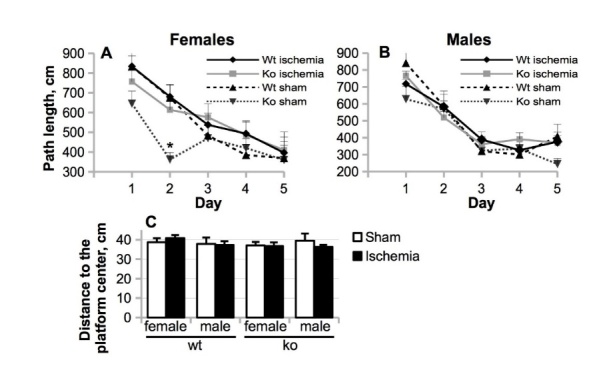
The effect of Nfkb1 gene deficiency on mouse spatial learning and memory as assessed by Morris swim task during the week 4 after the BCCAo. (A-B) The length of a path to the hidden platform was assessed over five test days in female (A) and male mice (B). (C) 15 min memory retention expressed as the mean distance to the center of the previous platform location. The data are shown as mean ± SEM. P values are derived from two-way repeated measures or simple two-way ANOVA; * p < 0.05 in comparison to the Nfkb1-wt animals.
Motor balance and coordination as assessed by the rotarod test did not reveal any effect of genotype (males: F (1,43) = 0.3, p = 0.61; females: F (1,50) = 2.8, p = 0.10) or BCCAo (males: p = 0.40; females: p = 0.85). However, we detected sex and Nfkb1 genotype-specific effects on spontaneous locomotion in the open field. BCCAo caused a dramatic reduction in spontaneous locomotion of Nfkb1-ko female mice but not in the other groups (Fig. 3 A) resulting in a significant genotype x BCCAo interaction (p = 0.006). Furthermore, irrespective of the genotype, BCCAo decreased the time spent in the center of the open field by female mice, while increasing the time in the center spent by male mice (Fig. 3 B; operation x sex F (1, 93) = 4.49; p = 0.037), suggesting that transient forebrain ischemia was anxiogenic in the female but not in the male. In support of this, there was a trend towards a higher number of fecal bullets in the open field by ischemic female mice (Fig. 3 C; main effect of operation F (1, 50) = 3.65; p = 0.062). In contrast, Nfkb1 deficiency dramatically decreased the average number of rearings in the open field irrespective of the operation and sex (Fig. 3 D; females: main genotype effect p < 0.001; males: main genotype effect p < 0.001). Thus, the decreased rearing in the open field was highly specific for Nfkb1-ko animals.
Figure 3.

The effects of Nfkb1 gene deficiency and female sex on activity in the open field test assessed during the week 3 after the BCCAo. Total distance moved (A), time spent in the center of the open field (B), the number of fecal pellets produced (C) and the number of rearings (D) during 10 min of free exploration of the novel arena. P values are derived from three-way ANOVA followed by pairwise comparisons with Bonferroni’s adjustment; # p < 0.05, ### p < 0.001 in comparison to the corresponding sham-operated controls; ** p < 0.01, *** p < 0.001 in comparison to the Nfkb1-wt animals.
Nfkb1 deficiency had marginal protective effect on the hippocampal neurons against the BCCAo-induced death
To investigate the effect of Nfkb1 gene deficiency on transient forebrain ischemia-induced neuronal loss, the mice were sacrificed five weeks post-BCCAo, and histological samples were collected for the immunostaining (Fig. 1 Cohort 1). Immunostaining with anti-NeuN antibody labeling neuronal nuclei showed that 17 min of BCCAo caused mild neuronal damage in the hippocampus, chiefly in the pyramidal cell layer of cornus ammonis (CA) 2 and the medial portion of CA1 (Fig. 4 A; Supplementary table 2). Two-way ANOVA (treatment x genotype) revealed a significant loss of NeuN immunoreactivity in the CA2 area in both males and females (Fig. 4 B, Supplementary table 2; males: main effect of ischemia F (1, 46) = 19.42, p = 0.009; females: main effect of ischemia F (1, 49) = 13.46, p = 0.015) and in the medial CA1 region in the male (p = 0.01), but not female mice (ischemia vs. sham p = 0.69). Three-way ANOVA model with Bonferroni’s adjusted post hoc pairwise comparisons, which included the data from both sexes, revealed that Nfkb1 genotype marginally, but significantly, increased NeuN immunoreactivity in the CA2 area of ischemic mice when compared to the wt mice (F (1, 95) = 4.83, p = 0.030; Fig. 4 C-H). Thus, Nfkb1 gene deficiency exhibited only a mild neuroprotection in our ischemia model. Also, female mice exhibited a significantly higher level of preservation of NeuN immunoreactivity in the CA2 area as compared to the male mice (p = 0.046). No significant interaction between the genotype and sex was observed.
Nfkb1 gene deficiency reduced BCCAo-induced astrogliosis
Since the transcription factor NF-κB is a crucial mediator of inflammation, and virtually any form of neuronal damage causes inflammatory response and gliosis in the brain, we further analyzed if the Nfkb1 gene deficiency had any effect on astrogliosis and microgliosis at five weeks post-BCCAo. As expected, 17 min of BCCAo induced significant astrogliosis in the hippocampus as revealed by anti-GFAP immunostaining (Fig. 5). Increased GFAP immunoreactivity was chiefly observed in the CA2 area accompanying the neuronal loss, as well as in the dentate gyrus region (DG) (Fig. 5 A-B). GFAP immunoreactivity was analyzed in the whole hippocampal area outlined with solid line in Fig. 5 A, and the quantification data are presented in Fig. 5 C. Significant increase in GFAP immunoreactivity in the whole hippocampal area in comparison to the sham-operated animals was detected in the wt male group (on average 2.0-fold upregulation, p = 0.001), Nfkb1-ko male group (1.6-fold upregulation, p = 0.033), wt female group (2.3-fold upregulation, p < 0.001), but not Nfkb1-ko female group (p = 0.139). Representative images of GFAP immunostaining in the CA2 area in different mouse groups are shown in Fig. 5 D-I, respectively. Three-way ANOVA model with Bonferroni’s adjusted post hoc pairwise comparisons, which included the data from both sexes, revealed that the Nfkb1 gene deficiency mildly, but significantly, reduced GFAP immunoreactivity in ischemic mice (p = 0.043). The same trends were observed when only GFAP immunoreactivity within the dentate gyrus region was quantified (data not shown).
Nfkb1 deficiency strongly reduced BCCAo-induced microgliosis
We further analyzed the effect of Nfkb1 gene deficiency on the BCCAo-induced microgliosis in the hippocampus at 5-week time-point by using anti-CD45 and anti-Iba1 immunostaining. CD45, a receptor type protein tyrosine phosphatase, is expressed at high level in circulating blood leukocytes and also can be upregulated in endogenous brain microglia in response to an inflammatory stimulus or cellular damage. BCCAo dramatically upregulated total CD45 immunoreactivity in the hippocampal area in all animal groups tested (p ≤ 0.007, Fig. 6 A), predominantly in the areas of neuronal damage, especially CA2 (Fig. 6 B-C). Total CD45 was induced on average 4.8-fold in the wt male group, 2.5-fold in the Nfkb1-ko male group, 3.8-fold in the wt female group and 2.2-fold in the Nfkb1-ko female group. Three-way ANOVA model with Bonferroni’s adjusted post hoc pairwise comparisons, which included the data from both sexes, demonstrated that Nfkb1 gene deficiency strongly decreased CD45 immunoreactivity in the hippocampus of ischemic mice (p < 0.001, Fig. 6 A).
Figure 6.
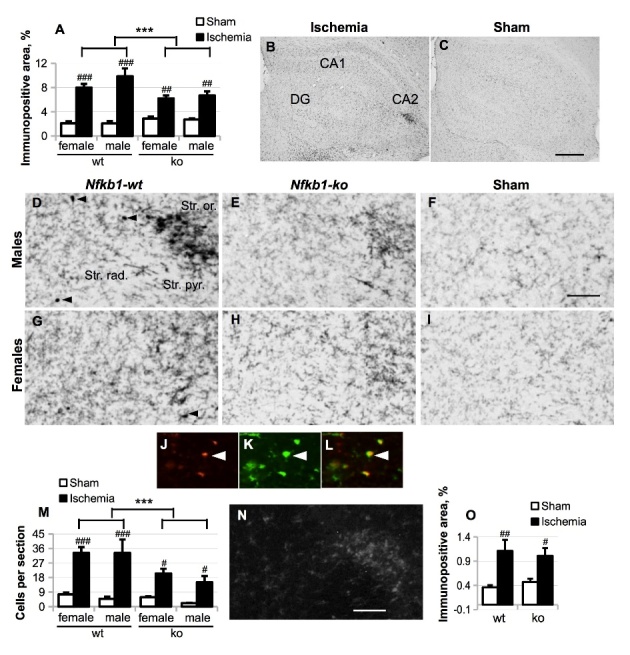
Microgliosis was evaluated by anti-CD45 immunostaining five weeks after the BCCAo. (A), total CD45 immunoreactivity was evaluated in the whole hippocampal area (as delineated in Figure 5 A) and expressed as the percentage of the area of interest. Panel (B-I), representative images of CD45 immunostaining in the whole hippocampal area (B, C) or only CA2 area in the Nfkb1-wt ischemic mice (B, D, G), Nfkb1-ko ischemic mice (E, H) and Nfkb1-wt sham-operated mice (C, F, I). Scale bar in (B-C) equals 500 μm; scale bar in (D-I) equals 100 μm; DG, dentate gyrus. (J-L), CD45 immunoreactivity in CD45high-expressing cells (J; red) co-localized with Iba1-immunoreactivity (K, L; green). (M), the average number of CD45high small round cells (shown in (K-L)) per section. (N-O), a representative picture of Iba1 staining in the CA2 area of the ischemic hippocampus, scale bar equals 100 μm (N) and quantification of Iba1 immunoreactivity in the whole hippocampal area in male mice (O). P values are derived from three-way ANOVA followed by pairwise comparisons with Bonferroni’s adjustment. *** p < 0.001 in comparison to the Nfkb1-wt animals; # p < 0.05, ## p < 0.01, ### p < 0.001 in comparison to the sham-treated controls.
Representative images of CD45 immunoreactivity in the CA2 area in different mouse groups are shown in Fig. 6 D-I. While the cells expressing intermediate level of CD45 exhibited normal ramified microglia-like morphology, cells expressing high level of CD45 (CD45high) were round in shape with little or no processes (Fig. 6 D,G, J-L). Double-staining with anti-Iba1 antibody specific for cells of monocyte/macrophage/microglia origin demonstrated that CD45high round cells were also Iba1-positive, as expected (Fig. 6 J-L). Since in our previous study we detected an extremely low number of infiltrated leukocytes in BCCAo model [27], these cells are likely to be migrating microglia [30, 31]. Counting of these CD45high round cells showed that, similarly to total CD45 immunoreactivity, the average number of CD45high round cells per section was significantly increased by BCCAo, but the Nfkb1 gene deficiency strongly reduced this increase (Fig. 6 M). However, Nfkb1 deficiency did not inhibit ischemia-induced upregulation of other microglial marker Iba1 (Fig. 6 N-O).
Interestingly, the lower level of CD45, but not Iba1, immunoreactivity in the hippocampus of ischemic mice was associated with better mouse performance in Morris swim task significantly correlating with mean path length (nonparametric Spearman’s rho 0.39, p = 0.002, n = 60) over 5 acquisition days.
Nfkb1 deficiency slightly, but significantly, inhibited BCCAo-induced neurogenesis in male mice
Since ischemia-induced inflammation [32-34] and hippocampal astrocytic activation [35] have the ability to modulate adult neurogenesis, and we clearly detected increased astrogliosis within the dentate gyrus region of ischemic mice, we further evaluated DCX immunoreactivity in our BCCAo mice at five weeks post-insult. DCX is transiently expressed in proliferating progenitor cells and newly generated neuroblasts for about two weeks before they differentiate into mature neurons [36]. This fact makes DCX a good marker of adult neurogenesis. As expected, we saw two types of DCX-positive (DCX+) cells in the dentate gyrus region: amorphous and short-branched cells in the subgranular zone characteristic of neuronal progenitors and early neuroblasts, and later-stage neuroblasts located at the base of the granule cell layer with processes reaching tangentially into the molecular cell layer (Fig. 7 A-F). Occasionally, DCX+ cells were observed in the middle of the granular cell layer (Fig. 7 D-E, arrowheads), predominantly in ischemic animals. To detect the differences in hippocampal neurogenesis, DCX+ cell profiles were counted manually, and the results are presented as the average number of DCX+ cell profiles per section (Fig. 7 G). BCCAo dramatically increased the number of DCX+ cell profiles in all groups in comparison to sham-operated animals (p < 0.001). Overall, the average numbers of DCX+ cell profiles, including both ischemic and sham groups, were lower in the female mice as compared to the male mice (three-way ANOVA main effect of sex F (1, 95) = 11.64, p = 0.001). Nfkb1 deficiency slightly, but significantly, decreased the average number of DCX+ cell profiles in ischemic mice (p = 0.033).
Figure 7.

Neurogenesis in the dentate gyrus area was evaluated by anti-DCX (doublecortin) immunostaining five weeks after the BCCAo. (A-F) Representative images of DCX immunoreactivity in ischemic animals (A, B, D, E), and sham-treated controls (C, F). (D-F) Higher magnification images of the areas delineated with a rectangle in (A-C). Str. gr., stratum granulare (the granular layer); str. mol., stratum moleculare (the molecular layer); arrowheads show post-mitotic immature neurons. Scale bar in (A-C) equals 200 μm; scale bar in (D-F) equals 80 μm. (G) The average number of DCX-positive cell profiles per section. P values are derived from three-way ANOVA followed by pairwise comparisons with Bonferroni’s adjustment; * p < 0.05 in comparison to the Nfkb1-wt animals, ## p < 0.01 main sex effect. (H, I) Correlations between the average number of DCX+ cells and total CD45 immunoreactivity in the hippocampus (two hippocampi per animal analyzed separately) in female (H) and male animals (I). Spearman’s rho (nonparametric) correlation coefficient R was calculated separately for hippocampi with CD45 immunoreactivity < 9.5% (< 10% in the case of females) shown on the left side of the correlation plot and CD45 immunoreactivity > 9.5% shown on the right.
Interestingly, there was a U-shaped inverse correlation between total CD45 immunoreactivity in the hippocampus and the number of DCX+ cell profiles in the dentate gyrus with the maximum increase in the number of DCX+ cell profiles achieved when CD45 immunoreactivity was approx. 10% of the hippocampal area (Fig. 7 H, J). These results suggest that while modest hippocampal inflammation may promote neurogenesis as a part of the reparative response, high level of inflammation may be detrimental for the generation/survival of DCX+ cells. However, since most hippocampi had CD45 immunoreactivity level below the critical value, a lower level of microglial activation in the Nfkb1-ko animals resulted in a slight decrease in neurogenesis as compared to the wt animals.
Nfkb1 genotype did not significantly affect BCCAo-induced expression of inflammatory markers
To dissect out the link between the Nfkb1 gene deficiency and a reduction in the level of BCCAo-induced inflammation and neurogenesis, we subjected a new cohort of mice to 17 min of BCCAo and collected hippocampal samples to evaluate protein and mRNA levels of inflammatory cytokines and neurogenesis markers. We measured protein level of six common inflammatory cytokines (TNFα, IFNγ, IL-6, IL-10, IL-12p70 and MCP-1) by using CBA Mouse Inflammation Kit and mRNA levels of Aif1 (encoding for Iba1), Nos2, Il1b, Igf1, Bcl2 and Casp3 genes by quantitative reverse-transcription RT-PCR. While protein expression of IL-6, IL-10, IL-12p70, TNFα and IFNγ was too low for reliable detection, Nfkb1 genotype strongly tended to decrease MCP-1 levels (p = 0.053) and the expression of Aif1 (p = 0.055), the two markers that were more robustly expressed in the hippocampus. Nfkb1 gene deficiency did not have an effect on Nos2, Il1b, Igf1, Bcl2 and Casp3 gene expression (data not shown).
DISCUSSION
In this study, we show for the first time that Nfkb1 gene deficiency protected CA2 hippocampal neurons from forebrain ischemia-induced death in middle-aged mice when using long-term follow-up. The ischemic damage seen in our model was rather mild with statistically significant cell loss observed only in the hippocampal CA2 area among female mice. However, microgliosis, astrogliosis and neurogenesis response were clearly detectable. Our current study serves as an extension of earlier studies conducted on young male mice showing a neuroprotective effect of Nfkb1 gene deficiency in focal ischemia models [24, 25]. The main novel features of the current study were the use of BCCAo as a model of ischemia, more relevant subjects (aged mice of both sexes), concentration on inflammatory response and long-term follow-up and behavioral assessment. Importantly, we observed that female sex was a significant independent protective factor in our ischemia model.
The p50/p105 subunit encoded by the Nfkb1 gene is known to play a dual role in the activation of NF-κB. P50 together with NF-κB p65 subunit form a canonical heterodimer, and its absence in the Nfkb1-ko mice strongly impairs NF-κB DNA-binding activity induced by ischemia [25], LPS and amyloid pathology in the brain [29]. Genetic inhibition of the canonical NF-κB pathway specifically in neurons has shown protective effect in focal ischemia models [37, 38], and therefore, the neuroprotective effect seen in this study was likely due to direct modulation of cell death/survival pathways in neurons as suggested previously [23-25]. However, the inflammatory response can exacerbate neuronal death in the hippocampus [6-9]. Microglia undergoing the so-called M1-type pro-inflammatory activation can produce a number of cytotoxic mediators [39], and it has also been recently shown that microglia can phagocytose otherwise viable neurons and thus promote delayed neuronal death [10]. Modulation of microglial activation from the pro-inflammatory M1-like mode to pro-regenerative M2-like mode has been suggested as a therapeutic strategy in neuroinflammatory conditions [40, 41]. In vitro and in vivo studies largely using LPS as classical inflammatory stimulus have demonstrated that Nfkb1 gene deficiency in macrophages and microglia impairs the shift from M1 to M2-type activation and thus, timely resolution of the inflammation [42, 43]. This effect has been attributed to the inhibitory function of p50/p50 homodimers and p105 precursor protein. However, our study demonstrates that in the forebrain ischemia model Nfkb1 gene deficiency protects hippocampal neurons from apoptosis, even when injury is assessed five weeks post-ischemia, and reduces both microgliosis and astrogliosis in the brain as secondary outcome measures.
At five weeks post-ischemia, we analyzed immunoreactivity of GFAP, the most widely used marker of astrocytosis, and CD45 and Iba1, the markers for activated microglia/macrophages, in the ischemic hippocampus. CD45 expression in microglia is normally lower than in blood-derived macrophages. However, microglia do upregulate CD45 in inflammatory conditions, and extremely low number of blood-derived cells if any are present in the hippocampus following BCCAo [27] as the damage to the blood-brain barrier is negligible in this model. Therefore, we further refer to CD45+ cells in our model as ‘activated’ microglia. In contrast, Iba1 is considered as a general marker for microglial cells binding to both ‘resting’ and ‘activated’ microglia, including proliferating microglia. It has been shown that Iba1 and CD45 antibodies can mark different populations of microglial cells in the inflamed brain [44]. Importantly, hippocampal CD45, but not Iba1 immunoreactivity, exhibited significant direct correlation with the path to the platform in the Morris swim task, suggesting that ischemia-induced microglial activation reflected as increased CD45 immunoreactivity might be associated with impaired cognitive functions as far as the path length is considered a relevant outcome measure.
NF-κB p50/p105 subunit has also been implicated in the aging-associated cellular senescence. Genetic deletion of p50/p105 can lead to premature aging [45], age-related neuronal degeneration [46], accelerated noise-induced hearing loss [47], and the development of spontaneous optic neuropathy [48, 49]. Importantly, Bernal and coworkers [45] have shown that aged Nfkb1-ko mice display a decrease in spontaneous apoptosis in the brain, increase in cellular DNA damage and increased astrogliosis. Thus, we cannot exclude the possibility that despite the higher level of neuronal preservation in the hippocampus, Nfkb1-ko mice in our study displayed higher level of cellular senescence not detected by our immunohistochemical methods. Interestingly, we found that the number of rearings in the open field was decreased in the Nfkb1-deficient mice independent of the sex or BCCAo. BCCAo also induced a reduction in spontaneous locomotion specifically in the Nfkb1-deficient female mice. Similarly, a strain of senescence-accelerated OXYS rats has been reported to exhibit a dramatic decrease in open field rearing [50].
Previous studies investigating the effect of Nfkb1 gene deletion on anxiety and learning have suggested it is highly dependent on genetic background [51-53]. The mechanisms behind this effect of genetic background are not yet clear, although the study by Lehmann and coworkers [53] reported enhanced activation of the stress axis in the Nfkb1-ko male mice on C57Bl/6J background. Inbred mouse strains differ in their activity in the open field [54]. Transgenic mouse lines, including Nfkb1-ko, are usually created in one of the 129 substrains and then back-crossed to the C57BL/6J strain for several generations to get a mouse strain on pure C57BL/6J background. However, there have been several reports that even after back-crossing to the C57BL/6J strain for six generations, there can still be a certain amount of genetic material from the 129 substrain remaining, which can affect mouse performance in behavioral tests [55, 56]. Increase in the hippocampal neurogenesis is well documented in the post-ischemic brain [27, 32-34, 57-60]. Enhanced neurogenesis is believed to aim at tissue regeneration, and can be modulated by both NF-κB activity and inflammatory mediators. Aging is associated with a dramatic decline in the level of neurogenesis [61, 62]. Accordingly, our 11-month-old sham-operated animals exhibited a low basal level of hippocampal neurogenesis, and we found significantly enhanced neurogenesis in all ischemic groups as compared to sham-operated animals at five weeks post-ischemia. Proper inflammatory response is thought to be necessary for the induction of neurogenesis by ischemic episode [11, 16, 17, 33]. Indeed, we observed that the correlation between the number of neuronal precursors and hippocampal inflammation expressed as total CD45 immunoreactivity followed an inverted U-shape, suggesting that while moderate neuroinflammation may promote compensatory neurogenesis, excessive inflammation may inhibit it. Since Nfkb1 gene deficiency decreased neuronal loss and dramatically reduced CD45 immunoreactivity in the hippocampus, it is not surprising that it also slightly reduced ischemia-induced neurogenesis at five weeks post-ischemia.
Neuronal cell loss and inflammation in the hippocampus have previously been shown to significantly impair synaptic plasticity and spatial learning [28, 63-66]. In our current study, we did not detect a clear effect of BCCAo on spatial learning and memory most likely due to both small average lesion size and advanced age of the animals (11-month-old on average). Aging is associated with both progressive decrease in neurogenesis and specific impairments in the acquisition of spatial memory [61, 62], and dramatically increased neurogenesis in ischemic mice might have partially compensated for the neuronal loss. However, we did detect a significant correlation between the CD45 immunoreactivity in the hippocampus and the average path length to the platform, especially in the wt male group, suggesting an inverse relationship between neuroinflammation and spatial learning.
Similar to the Morris swim task, BCCAo had only mild effects on mouse locomotion in the open field likely reflecting small ischemia-induced damage. It is also important to note that open field test was done two weeks before animal sacrifice, and inflammation and neurogenesis status had likely changed by the time the histological samples were taken. Indeed, the effect of transient forebrain ischemia on rat behavior in the open field has been shown to be time-point dependent [67].
In summary, our study indicates that transcription factor NF-κB has a crucial role in the brain response when an ischemic insult hits hippocampal structures. As NF-κB, including its p50 subunit, is expressed in all cell types of the brain, we cannot judge from the data to which extent the reduced gliosis and inflammation in Nfkb1-deficient mice contributes to the neuroprotection we observed after global brain ischemia. Our finding that sex is a significant contributory factor when evaluating molecular and behavioral responses to a brain insult in general, as well as in relation to specific genetic manipulation, calls for including both male and female animals in experiments modelling brain diseases.
Supplementary table 1.
Average body weight and the number of posterior communicating arteries (PcommAs) per animal in the wt and Nfkb1-ko male and female mice. Data are shown as mean ± SEM. P values are derived from Student’s t-test; *** p < 0.001 in comparison to the wt female animals.
| Gender | Genotype | Body weight, g | PcommAs per animal |
|---|---|---|---|
| Male | Nfkb1-wt | 34.5 ± 0.6 | 1.3 ± 0.2 |
| Nfkb1-ko | 35.4 ± 0.7 | 1.4 ± 0.1 | |
| Female | Nfkb1-wt | 26.1 ± 0.6 | 1.5 ± 0.1 |
| Nfkb1-ko | 23.4 ± 0.3*** | 1.3 ± 0.2 |
Supplementary table 2.
NeuN immunoreactivity in medial CA1 and CA2 areas (delineated in Fig. 3) expressed as the percentage of average immunoreactivity in wt sham animals (females and males normalised separately). Data are shown as mean ± SEM. P values are derived from two-way ANOVA followed by pairwise comparisons with Bonferroni’s adjustment and are shown in brackets.
| Gender | Genotype | Treatment | Area of the hippocampus
|
|
|---|---|---|---|---|
| Medial CA1 | CA2 | |||
| Male | Nfkb1-wt | Ischemia | 88.1 ± 6.0 (p = 0.068) | 75.4 ± 5.3 (p = 0.003) |
| Sham | 100.0 ± 2.3 | 100.0 ± 3.6 | ||
| Nfkb1-ko | Ischemia | 91.2 ± 2.7 (p = 0.056) | 82.5 ± 4.1 (p = 0.014) | |
| Sham | 103.4 ± 2.9 | 102.1 ± 2.4 | ||
| Female | Nfkb1-wt | Ischemia | 101.2 ± 2.0 (p = 0.692) | 81.8 ± 4.4 (p = 0.005) |
| Sham | 100.0 ± 2.0 | 100.0 ± 3.0 | ||
| Nfkb1-ko | Ischemia | 101.2 ± 2.2 (p = 0.391) | 91.5 ± 3.5 (p = 0.061) | |
| Sham | 104.2 ± 2.7 | 104.5 ± 2.6 | ||
Acknowledgements
This study was partially supported by the Sigrid Juselius Foundation. We thank Hanna-Maija Lahtinen for performing the behavioral tests, and Sisko Juutinen, Mirka Tikkanen and Ekaterina Savchenko for their expert technical assistance with preparation of histological samples, immunohistochemistry, and cell counting.
References
- [1].Petito CK, Feldmann E, Pulsinelli WA, Plum F (1987). Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology, 37: 1281-1286 [DOI] [PubMed] [Google Scholar]
- [2].Pulsinelli WA, Brierley JB (1979). A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke, 10: 267-272 [DOI] [PubMed] [Google Scholar]
- [3].Kelly S, McCulloch J, Horsburgh K (2001). Minimal ischaemic neuronal damage and HSP70 expression in MF1 strain mice following bilateral common carotid artery occlusion. Brain Res, 914: 185-195 [DOI] [PubMed] [Google Scholar]
- [4].Fujii M, Hara H, Meng W, Vonsattel JP, Huang Z, Moskowitz MA (1997). Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57black/6 mice. Stroke, 28: 1805-1810. discussion 1811 [DOI] [PubMed] [Google Scholar]
- [5].Yang G, Kitagawa K, Matsushita K, Mabuchi T, Yagita Y, Yanagihara T, et al. (1997). C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res, 752: 209-218 [DOI] [PubMed] [Google Scholar]
- [6].Hamby AM, Suh SW, Kauppinen TM, Swanson RA (2007). Use of a poly(ADP-ribose) polymerase inhibitor to suppress inflammation and neuronal death after cerebral ischemia-reperfusion. Stroke, 38: 632-636 [DOI] [PubMed] [Google Scholar]
- [7].Candelario-Jalil E, Alvarez D, Gonzalez-Falcon A, Garcia-Cabrera M, Martinez-Sanchez G, Merino N, et al. (2002). Neuroprotective efficacy of nimesulide against hippocampal neuronal damage following transient forebrain ischemia. Eur J Pharmacol, 453: 189-195 [DOI] [PubMed] [Google Scholar]
- [8].Kauppinen TM, Suh SW, Berman AE, Hamby AM, Swanson RA (2009). Inhibition of poly(ADP-ribose) polymerase suppresses inflammation and promotes recovery after ischemic injury. J Cereb Blood Flow Metab, 29: 820-829 [DOI] [PubMed] [Google Scholar]
- [9].Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J (1998). Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A, 95: 15769-15774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Neher JJ, Emmrich JV, Fricker M, Mander PK, Thery C, Brown GC (2013). Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A, 110: E4098-4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ekdahl CT, Kokaia Z, Lindvall O (2009). Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience, 158: 1021-1029 [DOI] [PubMed] [Google Scholar]
- [12].Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, et al. (2007). Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab, 27: 1941-1953 [DOI] [PubMed] [Google Scholar]
- [13].Neumann H, Kotter MR, Franklin RJ (2009). Debris clearance by microglia: an essential link between degeneration and regeneration. Brain, 132: 288-295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Neumann J, Sauerzweig S, Ronicke R, Gunzer F, Dinkel K, Ullrich O, et al. (2008). Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci, 28: 5965-5975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J (2007). Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci, 27: 2596-2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kim BJ, Kim MJ, Park JM, Lee SH, Kim YJ, Ryu S, et al. (2009). Reduced neurogenesis after suppressed inflammation by minocycline in transient cerebral ischemia in rat. J Neurol Sci, 279: 70-75 [DOI] [PubMed] [Google Scholar]
- [17].Sasaki T, Kitagawa K, Sugiura S, Omura-Matsuoka E, Tanaka S, Yagita Y, et al. (2003). Implication of cyclooxygenase-2 on enhanced proliferation of neural progenitor cells in the adult mouse hippocampus after ischemia. J Neurosci Res, 72: 461-471 [DOI] [PubMed] [Google Scholar]
- [18].Baldwin AS Jr. (1996). The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol, 14: 649-683 [DOI] [PubMed] [Google Scholar]
- [19].Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA (1994). Constitutive NF-kappa B activity in neurons. Mol Cell Biol, 14: 3981-3992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kunsch C, Ruben SM, Rosen CA (1992). Selection of optimal kappa B/Rel DNA-binding motifs: interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol Cell Biol, 12: 4412-4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Clemens JA, Stephenson DT, Dixon EP, Smalstig EB, Mincy RE, Rash KS, et al. (1997). Global cerebral ischemia activates nuclear factor-kappa B prior to evidence of DNA fragmentation. Brain Res Mol Brain Res, 48: 187-196 [DOI] [PubMed] [Google Scholar]
- [22].Clemens JA, Stephenson DT, Smalstig EB, Dixon EP, Little SP (1997). Global ischemia activates nuclear factor-kappa B in forebrain neurons of rats. Stroke, 28: 1073-1080; discussion 1080-1071 [DOI] [PubMed] [Google Scholar]
- [23].Nurmi A, Vartiainen N, Pihlaja R, Goldsteins G, Yrjanheikki J, Koistinaho J (2004). Pyrrolidine dithiocarbamate inhibits translocation of nuclear factor kappa-B in neurons and protects against brain ischaemia with a wide therapeutic time window. J Neurochem, 91: 755-765 [DOI] [PubMed] [Google Scholar]
- [24].Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, et al. (2004). Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke, 35: 987-991 [DOI] [PubMed] [Google Scholar]
- [25].Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M (1999). NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med, 5: 554-559 [DOI] [PubMed] [Google Scholar]
- [26].Sha WC, Liou HC, Tuomanen EI, Baltimore D (1995). Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell, 80: 321-330 [DOI] [PubMed] [Google Scholar]
- [27].Heikkinen R, Malm T, Heikkila J, Muona A, Tanila H, Koistinaho M, et al. (2014). Susceptibility to focal and global brain ischemia of Alzheimer mice displaying abeta deposits: effect of immunoglobulin. Aging Dis, 5: 76-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kemppainen S, Hamalainen E, Miettinen PO, Koistinaho J, Tanila H (2014). Behavioral and neuropathological consequences of transient global ischemia in APP/PS1 Alzheimer model mice. Behav Brain Res, 275C: 15-26 [DOI] [PubMed] [Google Scholar]
- [29].Rolova T, Puli L, Magga J, Dhungana H, Kanninen K, Wojciehowski S, et al. (2014). Complex regulation of acute and chronic neuroinflammatory responses in mouse models deficient for nuclear factor kappa B p50 subunit. Neurobiol Dis, 64: 16-29 [DOI] [PubMed] [Google Scholar]
- [30].Neubrand VE, Pedreno M, Caro M, Forte-Lago I, Delgado M, Gonzalez-Rey E (2014). Mesenchymal stem cells induce the ramification of microglia via the small RhoGTPases Cdc42 and Rac1. Glia, 62: 1932-1942 [DOI] [PubMed] [Google Scholar]
- [31].Dibaj P, Nadrigny F, Steffens H, Scheller A, Hirrlinger J, Schomburg ED, et al. (2010). NO mediates microglial response to acute spinal cord injury under ATP control in vivo. Glia, 58: 1133-1144 [DOI] [PubMed] [Google Scholar]
- [32].Kim DH, Kim JM, Park SJ, Lee S, Yoon BH, Ryu JH (2010). Early-activated microglia play a role in transient forebrain ischemia-induced neural precursor proliferation in the dentate gyrus of mice. Neurosci Lett, 475: 74-79 [DOI] [PubMed] [Google Scholar]
- [33].Kim DH, Lee HE, Kwon KJ, Park SJ, Heo H, Lee Y, et al. (2015). Early immature neuronal death initiates cerebral ischemia-induced neurogenesis in the dentate gyrus. Neuroscience, 284: 42-54. [DOI] [PubMed] [Google Scholar]
- [34].Liu J, Solway K, Messing RO, Sharp FR (1998). Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci, 18: 7768-7778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Song H, Stevens CF, Gage FH (2002). Astroglia induce neurogenesis from adult neural stem cells. Nature, 417: 39-44 [DOI] [PubMed] [Google Scholar]
- [36].Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG (2003). Transient expression of doublecortin during adult neurogenesis. J Comp Neurol, 467: 1-10 [DOI] [PubMed] [Google Scholar]
- [37].Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, et al. (2005). IKK mediates ischemia-induced neuronal death. Nat Med, 11: 1322-1329 [DOI] [PubMed] [Google Scholar]
- [38].Zhang W, Potrovita I, Tarabin V, Herrmann O, Beer V, Weih F, et al. (2005). Neuronal activation of NF-kappaB contributes to cell death in cerebral ischemia. J Cereb Blood Flow Metab, 25: 30-40 [DOI] [PubMed] [Google Scholar]
- [39].Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011). Physiology of microglia. Physiol Rev, 91: 461-553 [DOI] [PubMed] [Google Scholar]
- [40].Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. (2015). Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol, 11: 56-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liao B, Zhao W, Beers DR, Henkel JS, Appel SH (2012). Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol, 237: 147-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. (2009). Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A, 106: 14978-14983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Taetzsch T, Levesque S, McGraw C, Brookins S, Luqa R, Bonini MG, et al. (2015). Redox regulation of NF-kappaB p50 and M1 polarization in microglia. Glia, 63: 423-440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Puli L, Pomeshchik Y, Olas K, Malm T, Koistinaho J, Tanila H (2012). Effects of human intravenous immunoglobulin on amyloid pathology and neuroinflammation in a mouse model of Alzheimer's disease. J Neuroinflammation, 9: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bernal GM, Wahlstrom JS, Crawley CD, Cahill KE, Pytel P, Liang H, Kang S, Weichselbaum RR (2014). Loss of Nfkb1 leads to early onset aging. Aging, 6: 931-943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lu ZY, Yu SP, Wei JF, Wei L (2006). Age-related neural degeneration in nuclear-factor kappaB p50 knockout mice. Neuroscience, 139: 965-978 [DOI] [PubMed] [Google Scholar]
- [47].Lang H, Schulte BA, Zhou D, Smythe N, Spicer SS, Schmiedt RA (2006). Nuclear factor kappaB deficiency is associated with auditory nerve degeneration and increased noise-induced hearing loss. J Neurosci, 26: 3541-3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nakamura-Yanagidaira T, Takahashi Y, Sano K, Murata T, Hayashi T (2011). Development of spontaneous neuropathy in NF-kappaBp50-deficient mice by calcineurin-signal involving impaired NF-kappaB activation. Molecular vision, 17: 2157-2170 [PMC free article] [PubMed] [Google Scholar]
- [49].Takahashi Y, Katai N, Murata T, Taniguchi SI, Hayashi T (2007). Development of spontaneous optic neuropathy in NF-kappaBetap50-deficient mice: requirement for NF-kappaBetap50 in ganglion cell survival. Neuropathol Appl Neurobiol, 33: 692-705 [DOI] [PubMed] [Google Scholar]
- [50].Kolosova NG, Vitovtov AO, Muraleva NA, Akulov AE, Stefanova NA, Blagosklonny MV (2013). Rapamycin suppresses brain aging in senescence-accelerated OXYS rats. Aging, 5: 474-484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kassed CA, Herkenham M (2004). NF-kappaB p50-deficient mice show reduced anxiety-like behaviors in tests of exploratory drive and anxiety. Behav Brain Res, 154: 577-584 [DOI] [PubMed] [Google Scholar]
- [52].Denis-Donini S, Dellarole A, Crociara P, Francese MT, Bortolotto V, Quadrato G, et al. (2008). Impaired adult neurogenesis associated with short-term memory defects in NF-kappaB p50-deficient mice. J Neurosci, 28: 3911-3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lehmann ML, Brachman RA, Listwak SJ, Herkenham M (2010). NF-kappaB activity affects learning in aversive tasks: possible actions via modulation of the stress axis. Brain Behav Immun, 24: 1008-1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Crawley JN, Belknap JK, Collins A, Crabbe JC, Frankel W, Henderson N, et al. (1997). Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology, 132: 107-124 [DOI] [PubMed] [Google Scholar]
- [55].Eisener-Dorman AF, Lawrence DA, Bolivar VJ (2010). Behavioral and genetic investigations of low exploratory behavior in Il18r1(-/-) mice: we can't always blame it on the targeted gene. Brain Behav Immun, 24: 1116-1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].de Ledesma AM, Desai AN, Bolivar VJ, Symula DJ, Flaherty L (2006). Two new behavioral QTLs, Emo4 and Reb1, map to mouse Chromosome 1: Congenic strains and candidate gene identification studies. Mamm Genome, 17: 111-118 [DOI] [PubMed] [Google Scholar]
- [57].Tanaka R, Yamashiro K, Mochizuki H, Cho N, Onodera M, Mizuno Y, et al. (2004). Neurogenesis after transient global ischemia in the adult hippocampus visualized by improved retroviral vector. Stroke, 35: 1454-1459 [DOI] [PubMed] [Google Scholar]
- [58].Raber J, Fan Y, Matsumori Y, Liu Z, Weinstein PR, Fike JR, et al. (2004). Irradiation attenuates neurogenesis and exacerbates ischemia-induced deficits. Ann Neurol, 55: 381-389 [DOI] [PubMed] [Google Scholar]
- [59].Ortega FJ, Jolkkonen J, Mahy N, Rodriguez MJ (2013). Glibenclamide enhances neurogenesis and improves long-term functional recovery after transient focal cerebral ischemia. J Cereb Blood Flow Metab, 33: 356-364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rodriguez-Grande B, Varghese L, Molina-Holgado F, Rajkovic O, Garlanda C, Denes A, et al. (2015). Pentraxin 3 mediates neurogenesis and angiogenesis after cerebral ischaemia. J Neuroinflammation, 12: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kuhn HG, Dickinson-Anson H, Gage FH (1996). Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci, 16: 2027-2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gil-Mohapel J, Brocardo PS, Choquette W, Gothard R, Simpson JM, Christie BR (2013). Hippocampal neurogenesis levels predict WATERMAZE search strategies in the aging brain. PLoS One, 8: e75125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bellinger FP, Madamba S, Siggins GR (1993). Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res, 628: 227-234 [DOI] [PubMed] [Google Scholar]
- [64].Bellinger FP, Madamba SG, Campbell IL, Siggins GR (1995). Reduced long-term potentiation in the dentate gyrus of transgenic mice with cerebral overexpression of interleukin-6. Neurosci Lett, 198: 95-98 [DOI] [PubMed] [Google Scholar]
- [65].Butler MP, O'Connor JJ, Moynagh PN (2004). Dissection of tumor-necrosis factor-alpha inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early-but not late-phase LTP. Neuroscience, 124: 319-326 [DOI] [PubMed] [Google Scholar]
- [66].Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. (2011). CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci, 31: 16241-16250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Milot MR, Plamondon H (2009). Time-dependent effects of global cerebral ischemia on anxiety, locomotion, and habituation in rats. Behav Brain Res, 200: 173-80 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1.
Average body weight and the number of posterior communicating arteries (PcommAs) per animal in the wt and Nfkb1-ko male and female mice. Data are shown as mean ± SEM. P values are derived from Student’s t-test; *** p < 0.001 in comparison to the wt female animals.
| Gender | Genotype | Body weight, g | PcommAs per animal |
|---|---|---|---|
| Male | Nfkb1-wt | 34.5 ± 0.6 | 1.3 ± 0.2 |
| Nfkb1-ko | 35.4 ± 0.7 | 1.4 ± 0.1 | |
| Female | Nfkb1-wt | 26.1 ± 0.6 | 1.5 ± 0.1 |
| Nfkb1-ko | 23.4 ± 0.3*** | 1.3 ± 0.2 |
Supplementary table 2.
NeuN immunoreactivity in medial CA1 and CA2 areas (delineated in Fig. 3) expressed as the percentage of average immunoreactivity in wt sham animals (females and males normalised separately). Data are shown as mean ± SEM. P values are derived from two-way ANOVA followed by pairwise comparisons with Bonferroni’s adjustment and are shown in brackets.
| Gender | Genotype | Treatment | Area of the hippocampus
|
|
|---|---|---|---|---|
| Medial CA1 | CA2 | |||
| Male | Nfkb1-wt | Ischemia | 88.1 ± 6.0 (p = 0.068) | 75.4 ± 5.3 (p = 0.003) |
| Sham | 100.0 ± 2.3 | 100.0 ± 3.6 | ||
| Nfkb1-ko | Ischemia | 91.2 ± 2.7 (p = 0.056) | 82.5 ± 4.1 (p = 0.014) | |
| Sham | 103.4 ± 2.9 | 102.1 ± 2.4 | ||
| Female | Nfkb1-wt | Ischemia | 101.2 ± 2.0 (p = 0.692) | 81.8 ± 4.4 (p = 0.005) |
| Sham | 100.0 ± 2.0 | 100.0 ± 3.0 | ||
| Nfkb1-ko | Ischemia | 101.2 ± 2.2 (p = 0.391) | 91.5 ± 3.5 (p = 0.061) | |
| Sham | 104.2 ± 2.7 | 104.5 ± 2.6 | ||