

Abstract
The extracellular matrix (ECM) is an environment that has various enzymes attended in regeneration and restoration processes which is very important to sustain physiological and biological functions of central nervous system (CNS). One of the participating enzyme systems in ECM turnover is matrix metalloproteinases. A disintegrin-like and metalloproteinase with thrombospondin type 1 motifs (ADAMTS) is a unique family of ECM proteases found in mammals. Components of this family may be distinguished from the ADAM (A Disintegrin and Metalloproteinase) family based on the multiple copies of thrombospondin 1-like repeats. The considerable role of the ADAMTS in the CNS continues to develop. Evidences indicate that ADAMTS play an important role in neuroplasticity as well as nervous system pathologies such as Alzheimer’s disease (AD). It is hopeful and possible that ADAMTS family members may be utilized to develop therapies for CNS pathologies, ischemic injuries, neurodegenerative and neurological diseases. To understand and provide definitive data on ADAMTS to improve structural and functional recovery in CNS injury and diseases, this review aimed to enlighten the subject extensively to reach certain information on metalloproteinases and related molecules/enzymes. It will be interesting to examine how ADAMTS expression and action would affect the initiation/progression of above-mentioned clinical situations, especially AD.
Keywords: matrix metalloproteinases, ADAM, ADAMTS, Alzheimer’s disease, neurodegeneration
A glance for ADAMTS genes and proteins
Alzheimer’s disease (AD) is a progressive neurodegenerative illness affecting the elderly population and the most common cause of dementia. The neuropathological characteristics of AD contain extracellular deposition of β-amyloid (Aβ), which is originated by proteolytic cleavage from the Aβ precursor protein (APP) and intraneuronal accumulation of aberrant forms of hyperphosphorylated tau as well as synapse dysfunction [1]. Following the identification of the ADAM family members, a new group of ADAM-associated protein being shown by Kuno et al. [2], for the first time. This new cDNA clone, recognized as a cachexigenic tumor selective gene, encodes a cysteine rich protein, which displays a sequence resemblance to that of thrombospondins and metalloproteinases of snake venoms. They named this cDNA clone as ‘A disintegrin-like and metalloproteinase with thrombospondin type 1 motifs’ (ADAMTS) which consists of six domains, including a metalloproteinase, a disintegrin-like, a thrombospondin (TSP) homologous domain containing TSP type I motif, a spacer region, and COOH-terminal TSP sub-motifs [2]. Also, a few ADAMTS have a protease and lacunin (PLAC) domain [3] and a complement C1r/C1s-urchin epidermal growth factor-bone morphogenetic protein-1 (CUB) domain at the C-terminal [4].
ADAM(s) include either an epidermal growth factor-like or transmembrane domain. In contrast to the ADAM family members, none of them (ADAMTS) are located in the cell membrane. ADAMTS1 is a putative secretory protein that has a spacer region in its COOH-terminal half-region and TSP type 1 motifs [2]. TSP was discovered the first angiogenesis inhibitor which is associated with platelet membranes [5] and released from platelets in response to activation by thrombin [6]. Instead of these domains, ADAMTS1 was represented a new type of ADAM family protein [2]. Nineteen members of this new protease family were revealed by the complete sequence of the human genome [7].
The roles of metalloproteinases including MMPs, ADAMs, and ADAMTS are known in the process of extracellular matrix (ECM) damage and repair. ADAMTS family is a subgroup of newly identified zinc metalloproteases play a key role in adhesion, cell fusion, signalling, proteolysis and ECM degredation [8]. ADAMTS, a member of the M12 Metallopeptidase family [9], is involved in many physiological processes that associated with ECM proteolysis such as morphogenesis, angiogenesis, ovulation [7] as well as tissue deterioration in diseases such as AD, arthritis, cancer, and a range of inflammatory circumstances [10]. Also, ADAMTS has functions in maturation of procollagen and von Willebrand factor (vWF) [7].
Clarification of the relationship with the diseases and ADAMTS which is degraded proteoglycans at the ECM, are expected to make significant contributions either diagnosis or treatment, in the near future. Determining the factors of ADAMTS inhibition and the signal transduction pathways will enhance the clinical uses of ADAMTS. Some proteoglycan fragments that occur as a result of ADAMTS proteolytic activity are estimated to be used the development of new drugs [11].
The functions of tau proteins
Tau protein is a neuronal microtubule-associated protein (MAP) that is able of producing some features of an axonal shape and an axon-like organization of the cytoskeleton of cell. It was discovered in the Alzheimer neurofibrillary tangles [12].
Tau proteins consist of a family of proteins, which associate with microtubules in vivo and are induced during neurite outgrowth. Tau is coded by a single gene on chromosome 17 that are generated by substitute splicing of its mRNA. Previous studies show that tau is a tripartite molecule composed of a C-terminal tubulin binding region with different numbers of repeats, a constant middle domain, and variable N-terminal domains. The heterogeneity of the N-terminal domains has implications for the role of tau in mediating interactions with other components in the cell [13].
Tau takes role as a potent promoter of tubulin construction in vitro [14]. The essence of the connection between tau and microtubules is ionic, as is evident from the many basic residues within the tau binding domain and the communicating of this domain with a glutamic acid-rich region at the carboxy terminus of tubulin [12]. The normal tau protein is highly enriched in neurons as noted by both immunohistochemical investigations [15]. Highly elastic paracrystals that become rigid when tau is phosphorylated by calcium/calmodulin-dependent protein kinase have been pronounced in pure tau preparations [16]. Study has shown that tau phosphorylation regulates the microtubule dynamics and microtubule affinity of the protein [17]. Phosphorylation is the regulation of its binding to microtubules that is the post-translational modification of tau [12]. Extremely phosphorylated tau assembles in the somatodendritic part of neurons, aggregates and ultimately forms neurofibrillary tangles [18].
The mechanisms of accumulation of β-amyloid in the brain
The Aβ collection is believed to be a primary phenomenon leading to eventual cognitive and motor dysfunction in AD. Formation of Aβ demands proteolytic cleavage of a large type-1 transmembrane protein, the APP [19]. APP is cut out by three types of proteases, which are designated α-, β- and γ-secretases. Processing by β- and γ-secretase cleaves on the N- and C-terminal ends of the Aβ region respectively, liberating Aβ, whereas α-secretase cleaves within the Aβ sequence. Gama-secretase cleaves at a number of neighboring sites to yield Aβ species containing 39-43 amino acid residues [20]. Aβ occurs in two predominant forms with different COOH-termini, Aβ40 and Aβ42. Overproduction of Aβ42 has been proposed to be the cause of familial early-onset AD [21].
APP genes are located on the long arm of chromosome 21 in humans. α-secretases cleave APP within the amyloid sequences, whereas β- and γ-secretases cleave on the N- and C-terminal ends, respectively. The transmembrane aspartyl protease BACE has been recognized as β-secretase and several proteases (ADAM10, TACE, PC7) may be α-secretases [22]. APP is proteolyzed to the monomer Aβ and sAPP by β- and γ- secretases. The monomeric Aβ forms oligomeric and fibrillar Aβ caused by the obstruction of the dismissal transport and peptidolytic mechanism of Aβ. The collection of neuronal Aβ seriously provides to tau hyperphosphorylation, and may produce a series of neuronal signal transduction events. Intraneuronal Aβ peptides are the critical factor to start neurodegeneration and form senile plaques. The accumulation of intraneuronal Aβ peptides manifests earlier than extracellular Aβ peptides. According to several researches, Aβ peptides are secreted from the neuronal membrane by β- and γ-secretases, eventually Aβ peptides are derived from the membrane and go into the extracellular fluid [23].
Normal healthy human body has clearance mechanisms for Aβ peptides available, and the created Aβ peptides are identical to the cleared Aβ peptides. Clearance can be achieved by way of two major pathways: proteolytic cleavage and receptor-mediated transport from the brain [24]. Amongst the diverse Aβ species with variable C-terminal lengths, Aβ42 is regarded to be the most neurotoxic and aggregation-prone species [25]. An inadequated removal of neuronal Aβ manifests in the sporadic forms of AD, and the production and removal of Aβ peptides are out of equilibrium. A lot of Aβ peptides will gather and form senile plaques in neurons [23]. In the exterior of the cell, the Aβ peptide aggregates into clumps called oligomers, which gather and produce deposits called amyloid plaques [25].
Mature APP is metabolized by two competing pathways, the α-secretase pathway that causes sAPPα and C83, and the β-secretase pathway that resulting in sAPPβ and C99. Some β-secretase digestion is displaced by 10 amino acid residues and induces sAPPβ and C89. All carboxyterminal fragments [C83, C99, and C89] are substrates for γ-secretase, generating the APP intracellular domain (AICD) and, respectively, the secreted peptides p3, Aβ, and Glu11Aβ. Aβ aggregates into tiny multimers known as oligomers. Oligomers appear to be the strongest neurotoxins, while the end stage senile plaque is comparatively inert [26].
Genetic proofs demonstrate that accumulation of Aβ in some biophysical form is damaging to the brain. Metabolism and trafficking of APP are firmly controlled events, and it is completely possible that surplus Aβ42 production and/or accumulation is an preliminary symptom of some other primary problem that is itself independently neurotoxic. In such a framework, it remains possible that Aβ gathering is a second, parallel pathway, which is accepted as toxic. Dissection of the genesis of the Aβ accumulation phenotype is important in order to point the way to any upstream, primary lesions. The development of effective anti-amyloid treatments prevails a key goal, the attainment of which is needed in order to illuminate the role-played by cerebral accumulation of toxic Aβ oligomers in the clinical picture of AD [26].
Biochemical enzymatic degradations in extracellular space that ADAMTS can afford
The roles of metalloproteinases including MMPs, ADAMs, and ADAMTS are known in the process of ECM damage and repair. As discussed before, ADAMTS family is a subgroup of newly identified zinc metalloproteases plays a key role in adhesion, cell fusion, signaling, proteolysis and ECM degradation [8]. ADAMTS proteases consist of a protease domain and an ancillary domain by which second one provide substrate-binding specificity, and first one provides cleavage site specificity [7]. Thrombospondins bind to cell surfaces and matrix macromolecules such as heparan sulfate, proteoglycans, fibronectin, laminin and collagen [27]. The structural proteins of the ECM such as collagen, versican and aggrecan are degraded by ADAMTS proteases, and tissue inhibitors of metalloproteinase inhibit ADAMTS proteases [28].
Some members of the ADAMTS (1, 4, 5, 8, 9, 15, 16 and 18) are classified as aggrecanases [29] having the ability to degrade aggrecan. It is also known as cartilage-specific proteoglycan core protein that is a major component of cartilage [7]. ADAMTS4 [Aggrecanase-1) was named for the first time in 1999, and then ADAMTS5 (aggrecanase-2) [30] was found and also named as ADAMTS11. Their implication in aggrecan degradation in arthritic diseases has been reported [30]. ADAMTS2, ADAMTS3 and ADAMTS14 known as procollagen restriction enzymes [31] translate procollagen to collagen. Additionally, ADAMTS2 mutations cause Ehlers-Danlos syndrome [32].
Aggrecanases also degrade other lecticanes such as versican and brevican. They are involved in the pathogenesis of musculoskeletal disorders such as osteoarthritis [7, 31, 33]. Aggrecanase levels increase in osteoarthritis. ADAMTS5 deficiency in fibrous tissues results in a limited repair response due to the cumulation of aggrecan in the pericellular matrix of fibroblast progenitor cells. Tissue repair signaling suggests a critical role for ADAMTS5 that apart from other members of the ADAMTS family of proteases [34]. Cartilage oligomeric matrix protein (COMP) is a matrix glycoprotein that has influence the structural integrity of cartilage and the interactions with other ECM molecules. COMP levels were significantly higher in patients with aggressive arthritis. ADAMTS7 and ADAMTS12 degrade COMP in vitro, and they are significantly overexpressed in the cartilage and synovium of patients with rheumatoid arthritis [35].
ADAMTS9 and ADAMTS20 constitute a subset provided their strong resemblance to the GON-1 protease [7]. GON is a metalloproteinase that expressed in the embryonic development. GON-1 is required for morphogenesis of the gonadal somatic structures [36]. A large adhesion molecule vWF allows the adhesion of platelets that expressed of the endothelial cells and megakaryocytes [37]. ADAMTS13 cleaves the peptide bond between tyrosine and methionine amino acids in the central A2 domain of the VWF molecule, and smaller vWF occurs which is necessary for coagulation [38]. ADAMTS13 is the molecular mechanism responsible for TTP, and it is in charge enzyme for physiologic proteolysis of vWF [39], which is expressed in hepatic stellate cells [40]. ADAMTS1 and ADAMTS4 also have the ability to degrade versican in human aorta [41], whereas ADAMTS4 is responsible for brevican degradation in glioma cells [42].
The ECM is important for structural and functional development and maintenance of CNS. In studies, CSPGs degradation by ADAMT4 showed that ADAMTS4 overcomes inhibition of axonal regeneration and promotes neurite outgrowth through a proteolytic mechanism [43]. ADAMTS4 caused a decrease in phosphocan accumulation [44], and ADAMTS4 increased the neurite outgrowth on rat neuron culture [45].
Another study, which conducted by matrix events in CNS injury, suggested that ADAMTS1 and ADAMTS4 transcription and protein expression are stimulated in astrocyte culture in response to IL-1. Therefore, essential aggrecanese ADAMTS1 and ADAMTS4 are candidates responsible for the increased proteoglycan degradation in early phase of injury [46].
The proposed functions of ADAMTS in CNS
Unlike ADAMs protease enzymes, none of the ADAMTS proteases are integral membrane proteins. Currently 19 mammalian ADAMTS proteainases have identified in Fig. 1. One of the biological functions of ADAMTS is proteolytic activity against ECM proteins, including proteoglycans (aggrecan, versican, neurocan and brevican) [7]. As a member of matrix metalloproteinases, ADAMTS play a critical role in the degradation/repairing of ECM. The essential substrates of the ADAMTS peptidase enzymes are the aggregating chondroitin sulphate proteoglycans (CSPGs), including brevican, versican, and aggrecan, which are known to be the total integral components of ECM of the CNS [47, 48]. Proteolysis of proteoglycans by ADAMTS enzymes can have destructive effects, producing matrices with disrupted cell-matrix communication, matrices with impaired fibrillary networks, or matrices with altered biomechanical properties [11]. After numerous comprehensive studies, ADAMTS expression has been found in the CNS [49-60] and is known to be changed in disease conditions [44, 51, 53].
Figure 1.

Domain structure of ADAMTS proteins (It was adapted from Apte SS [7] and Stanton et al. [11])
ADAMTS are expressed in several CNS structures, including hippocampus, temporal lobe, frontal cortex, cortex, striatum and spinal cords detected by direct immunohistochemistry, Western blot, RT-PCR, and by using ADAMTS-specific neoepitope antibodies [33, 50, 53, 54, 56, 57, 60]. Roles of ADAMTS proteoglycanases in CNS are suggested to be both physiological and pathological such as neuroplasticity, inflammation, regeneration, remyelination, neurorepair (CSPGs degradation), and angiogenesis [61].
Recent studies indicate that expression level of ADAMTS and CPGs may be change after spinal cord injury in animal models [58, 59, 60]. Demircan et al. [60] reported that ADAMTS1, -5 and -9 expression levels were found to be upregulated following spinal cord injury (SCI) in mouse. However, expression level of ADAMTS4 was not altered in this report. Another study demonstrated both increased ADAMTS4 protein and aggrecan cleavage following SCI [58]. ADAMTS were detected in the astrocytes implying its cellular source in SCI by means of immunohistochemistry. Total aggrecan amount was shown to be decreased and aggrecanase-generated versican and brevican fragmentation was also observed in SCI in mouse [60]. Normally, ADAMTS13 protease cleaves vWF. Another study indicated that ADAMTS13 was increased in astrocytes and microglia following SCI. They cannot detect any expression of ADAMTS13 in intact spinal cords. The proteolytic activity of ADAMTS13 was increased after SCI. Therefore it may have a critical role in the CNS, particularly after neuronal injuries. It was hypothesized that increased ADAMTS13 degrades vWF and regulates blood-brain barrier (BBB) permeability to reduce secondary inflammation after SCI [59].
Cross et al. [54] examined expression of ADAMTS1, -4, -5 and tissue inhibitor of metalloproteinase (TIMP)-3, by Western blotting, immunocytochemistry, and real-time RT-PCR in spinal cord from animals’ subsequent experimental autoimmune encephalomyelitis at varying periods of disease progression. They revealed a decline in ADAMTS4 mRNA and protein expression. TIMP-3 was declined at the mRNA level although protein amounts were elevated in the animals that have disease. They displayed changes in the expression levels of ADAMTS and TIMP-3. These results may be significant in tissue damage during inflammation as well as in tissue remodeling and repair. They consider a decrease in ADAMTS resulting in decreased degradation of CSPGs and leading to accumulation of ECM proteins and formation of glial scars [54]. Matrix damage is seen during in human immunodeficiency virus (HIV) encephalitis and simian immunodeficiency virus (SIV) neuroinfection and lentiviral infection induced microglial and macrophage expression of ADAMTS1 and -4 in brains of 12 macaques infected with SIV. Authors suggest interventions to keep brain ECM proteoglycans might prevent or delay retroviral-induced neurodegeneration [51]. Levels of TIMP-3, ADAMTS1 and -5 mRNA were decreased in multiple sclerosis (MS) cases. Protein levels of ADAMTS4 were notably elevated in MS cases. Additionally, expression of ADAMTS4 was associated predominantly with astrocytes manifesting increased expression within MS lesions detected by immunohistochemical methods. Authors suggested that ADAMTS4 might have a role in the pathogenesis of MS [44].
Ajmo et al. [55] described the distribution and typical immunoreactivity for the ADAMTS-directed cleavage of brevican, and compare this with Wisteria floribunda agglutinin (WFA) binding in the rodent CNS. They noticed a considerable discordance between the two, with the great width of allocation of the ADAMTS-originated brevican fragment being much expansive than that of WFA reactivity, which is an ordinary reagent needed to identify perineuronal nets of undamaged matrix and a marker which is thought to be a hallmark for regions of relative neural stability [55]. Yuan et al. [50] reported the evidence of the increased expression of ADAMTS1 and 4. Therefore, ADAMTS-cleaved brevican fragments in these regions of kainate-induced seizures is pretty high. ADAMTS1and -4 mRNAs are elevated after excitotoxic damage in kainate-sensitive brain areas, and the secreted protease(s) degrade brevican potentially in perisynaptic regions in response to injury. Additionally, a loss of synaptic density in the dentate gyrus 120 h after intraperitoneal injection of kainite was found. Results suggest that the perisynaptic localization of brevican may be important to the short -and long-term structural plasticity that takes place in reaction to kainic acid [50]. Another study indicates that ADAMTS activity in the dentate outer molecular layer was examined for 2, 7 and 30 days after lesion. Proteolytic processing of brevican appears to be a significant extracellular event in the remodeling of the dentate after entorhinal cortex lesion, and ADAMTS may modulate the process of developing and/or synaptogenesis and the recovery and repair after neuronal and/or synaptic loss [52]. Recently, Howell and collaborators [56] has been explored role of CSPGs/ADAMTS proteoglycanases to regulate plasticity. Results demonstrate that the synaptosomal-associated protein 25 (SNAP-25), postsynaptic density protein 95 (PSD-95) and synaptophysin were declined in the developing frontal cortex of ADAMTS-1 deficient female mice, but not in male mice, suggesting in vivo ADAMTS1 knockout leads to sexual dimorphism in frontal cortex synaptic protein levels. However, the decline in expression of synaptic proteins was not attended by deficits of learning and memory in the adult female ADAMTS1 null mice. They were thought that ADAMTS1 might play a role in regulating neural plasticity [56].
The proven functions of ADAM proteases in AD
A type I transmembrane glycoprotein, APP, is processed by three types of proteases, which are designated α-, β-, and γ-secretases. The APP proteolytically processed by β-, and γ-secretases occurs neurotoxic Aβ peptide [22]. Cleavage of APP by α-secretase releases a large, soluble N-terminal ectodomain, soluble α-secretase-released N-terminal APP domain (sAPPα), into the extracellular space [62]. Several studies have reported that α secretases exerted proteolytic effects on APP preventing production, and accumulation of Aβ proteins in AD [22, 62-65]. The important roles of ADAM9, -10, and -17 in these processes such as APP amyloid cascade, functioning of pathogenetic processes, and subtypes of amyloid proteins are already known [22, 63, 64]. ADAM group of enzymes are termed as α group secretases. These proteases prevent production, accumulation of Aβ proteins from APP, and antagonize the activities of amyloids via various mechanisms with potential trophic effects on nerve tissue [22, 64, 66]. Additionally, several groups have been reported that the levels of ADAM10, which is especially effective on proteolytic processes involving APP, decreased in AD [65, 67, 68]. An increase in ADAM10 levels in a mouse model corresponded to a decrease in Aβ plaque levels [69]. APP is processed by β-, and γ-secretases to produce Aβ in platelets [67, 68]. Alternatively, platelets can be cleavage by α secretase within the β-amyloid domain [67, 68, 70]. Tang et al. [68] investigated blood platelet levels of Aβ peptide, β-secretase, α-secretase (ADAM10), and APP isoform ratios from AD patients and control subjects. They found elevated amounts of Aβ4, escalated activation of β-secretase, diminished activation of α-secretase (ADAM10) and reduced APP ratios in AD patients. Authors thought that APP processing might be a useful biomarker for diagnosis of AD, and for monitoring drug responses in clinical trials [68]. Rosenberg and collaborators [70] reported that APP isoforms were quantitated with the use of Western blot methods in the blood from control and patients with AD. The mean ratio of the 120-130 kD APP isoform to the 110 kD APP isoform in blood platelets in patients with AD is significantly lower than that of control subjects in whom genotyping of apolipoprotein E was performed [70]. Colliaghi and collaborators [67] reported that levels of sAPPα and ADAM10 are decrease in platelets and in cerebrospinal fluid (CSF) in cases with AD patients by means of Western blot with a specific antibody. This substance (sAPPα) could be used as a marker in cases with AD. This results show that the correlation between peripheral and central compartments is extremely important. Additionally, they demonstrated a decrease in the levels of ADAM10 plays an important in vivo role in the molecular pathogenesis of AD, and asserted that if its levels could be increased then important advances could be achieved in the treatment of AD [67]. Another study has been reported a positive association between cerebrospinal fluid levels of sAPPα and cognitive performance in rats. Therefore, sAPPα may be involved in the spatial learning and memory [71]. Using immunohistochemical techniques, ADAM10 brain immunostaining is lower in different neocortical areas of the AD patients compared with control subjects [72]. In a transgenic AD mouse model, overexpression of ADAM10 was also demonstrated. Increased sAPPα concentrations by a non-amyloidogenic pathway, which results in a reduced the formation of Aβ peptide was noticed. Aβ deposition in plaque and cognitive defects of transgenic AD mouse was restored by overexpression of ADAM10. Activation of ADAM10 may be particularly promising for treatment of the neurodegeneration in AD. [69]. We examined tissue samples taken from temporal brain regions, included 2 cases with AD, and 7 control cases for histochemical analysis of β-APP, ADAM9, ADAM10, and ADAM17. In the cases with AD, extensity scores of immunohistochemical staining for β-APP (51-75%vs51-75%), ADAM9 (50-75%vs26-50%), ADAM10 (76-100% vs 0-25%), and ADAM17 (76-100%vs 0%) were detected as indicated in parentheses [unpublished observations). Bekris et al [65] reported elevated CSF sAPPα levels in cognitively normal subjects compared with patients with AD and higher hippocampus ADAM10 protein levels in subjects with a low neuritic plaque score compared with those with a high neuritic plaque score. ADAM10 mRNA expression is higher in AD patients compared with control subjects in cerebellum, but not in hippocampus [65]. Similar results were reported by Gatta et al. in which ADAM10 mRNA expression was higher in AD cerebellum and hippocampus [73]. Bekris et al. demonstrated genetic variation within the ADAM10 promoter associated with CSF sAPPα levels in AD patients compared with cognitively normal control subjects. As a result, they proposed the role of ADAM10 in the pathogenesis of AD [74]. Thus, taken together, these studies demonstrate that ADAM10 expression and sAPPα levels may be affected by an ADAM10 regulatory structure, promoter genetic context, and microenvironment. The effect of ADAM10 genetic variation on ADAM10 expression may help to better define the role of pathogenesis of AD, specific marker for the diagnosis of AD, and for treatment of AD [65, 73, 74].
The possible functions of ADAMTS proteases in AD
Little studies are available on the ADAMTS in neurodegenerative disorders although numerous studies have been present on ADAMs in neurodegenerative disorders such as AD. Thus, effect(s) of ADAMTS and their products in neurodegenerative disorders are currently under investigation. Recent studies performed in the CNS highlight the pathophysiological relevance of ADAMTS gene; the determination of alterations in expression profiles of ADAMTS family genes in AD patients may contribute to the explanation of pathogenesis and also new ideas for remedial approaches.
The class aggrecanases of MMPs have been known to have members ADAMTS1, -4 (aggrecanase-1), -5 (aggrecanase-2), -8, -9, -15 and -16 [7]. Clark and collaborators reported a cloned small fragment of rat ADAMTS9 from a β amyloid-treated rat astrocyte cDNA library [75]. Another study demonstrate that ADAMTS4 mRNA level is induced in rat cultured astrocytes by Aβ treatment [49]. These proteinases may be played a role in the pathogenesis of AD [49, 75] and this process may contribute development of chronic neurodegenerative disorders [49]. Miguel et al. [53] reported that postmortem brain samples were obtained from frontal cortex region with AD, Down syndrome (DS), and Pick’s disease (PD) for western blotting using antibodies against ADAMTS1 and ADAMTS5. Study results show the manifold increase of metalloproteinase ADAMTS1 but not ADAMTS5 in brain neurodegeneration such as AD. This finding may represent specific marker for the diagnosis of AD, and for the treatment of AD. Pehlivan et al. [76] demonstrates that ADAMTS4 and 5 is slightly under-expressed in the brains from autopsied AD cases compared to control brains and suggests that ECM degradation is not promoted in AD brain. On the other hand, ADAMTS9 and 15 aggrecanases were not found to be expressed in brain sections of AD and control cases.
Furthermore, using immunohistochemical methods, ADAMTS4 expression was found to be related predominantly with astrocytes with elevated expression in the lesions of MS. It was then suggested ADAMTS4 to be an important factor in the pathogenesis of MS [44]. By using Western blot analyses, Ajmo et al. [55] reported the localization and typical immunoreactivity for the ADAMTS-degraded piece of brevican, and compare this with Wisteria floribunda agglutinin (WFA) binding in the rodent CNS. They found a noticeable discordance between the two, with the width of dispersal of the ADAMTS-originated brevican fragment being much wider that of WFA reactivity, which is a common reagent used to detect perineuronal nets of intact matrix and a marker which is thought to label regions of relative neural stability.
Reelin has been known to be a glycoprotein crucial for brain development and functions that is largely expressed in brain [77] and is mandatory for neuronal functions included in learning and memory [78, 79]. Decreased Reelin activity is associated with the onset of neuropsychiatric diseases such as AD [80]. Reduced expression of Reelin has a significant effect on amyloidogenic APP processing and in synaptic dysfunction associated with amyloid-β deposition, sufficient to enhance Tau phosphorylation and tangle formation in the hippocampal formation in aged Reelin-deficient transgenic AD mice [80]. Botella-López et al. [81] reported an increased proteolytic Reelin fragments in the CSF of AD patients compare to controls subjects by Western blot. ADAMTS4 (aggrecanase-1) and -5 (aggrecanase-1) cut Reelin at both the C- and N-terminal cleavage site in transgenic mice with AD [57]. Another study reported that ADAMTS4 cleaves Reelin in an isoform-specific manner. Among ADAMTS4 isoforms, p50 cleaves the N-terminal site only, while p75 cleaves both sites [82]. These results demonstrate that the lack of Reelin is associated with increased phosphorylation of Tau and widespread formation of NFTs (Fig. 2) [83]. Further studies, including in human brain AD, will be needed to better comprehend the regulation of these proteases and their influence on Reelin-mediated signaling in the neurodegenerative disorders [57, 82]. ADAMTS family members may be utilized to develop therapies for neurodegenerative and neurological disorders.
Figure 2.
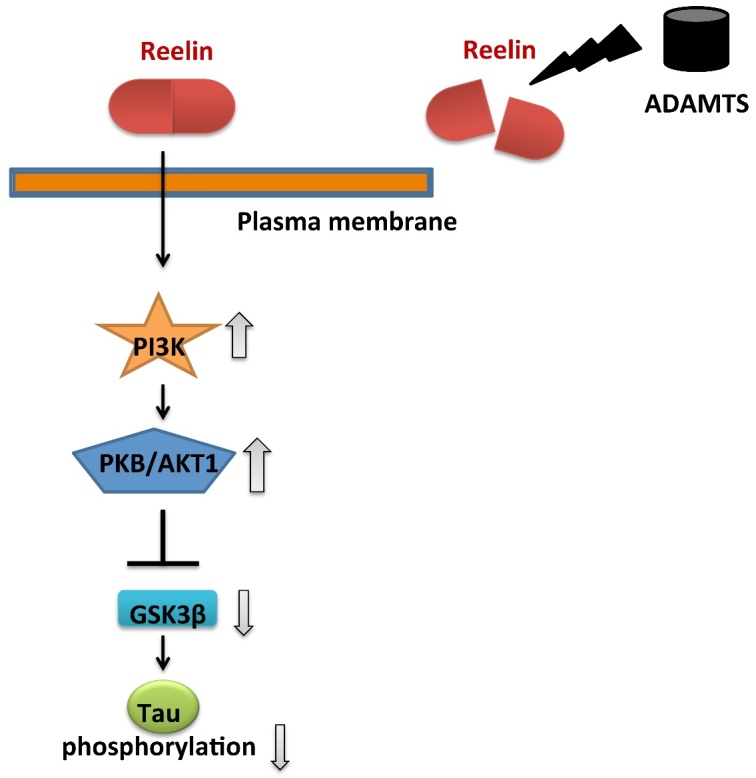
The representation of the effects of ADAMTS on Tau production. Reelin binds to its receptors and activates phosphatidylinositol-3-kinase (PI3K) and protein kinase B (PKB/Akt). It leads a remarkable inhibition in glycogen synthase kinase 3β (GSK3β), which is an enzyme that regulates phosphorylation of the microtubule-stabilizing protein tau. After ADAMTS digest Reelin, depressed reelin level may in turn increase tau phosphorylation at the end of the signaling pathway. (It was adapted from the resources Krstic D [57], Hisanaga A [82], and Yu NN [83]).
Recently, Ve´gh and collaborators [84] reported that increased ECM proteins, including brevican, hyaluronan and proteoglycan link protein 1, neurocan and tenascin-R, were found at an early age in Alzheimer model mice associated with a loss of contextual fear memory and long-term potentiation (LTP) defect. The behavioral plasticity (fear conditioning) and physiological plasticity (LTP) are early affected in APP/PS1 mice and that treatment with chondroitinase ABC reverses these early deficits. Thus, Gottschall and Howell [85] think that increased expression of ADAMTS can produce the same effects as treatment with chondroitinase ABC in such models.
Conclusion and future perspectives
Finally, the up-regulation of ADAMTS proteins in CNS diseases suggests that dysregulated expression of members of the ADAMTS family may be involved in AD. In view of their enzymatic nature, they would serve to brain cells not just in terms of their physiological functions, but also in neurodegenerative processes. It would also be of interest to see if some of the future ADAMTS gene knockouts become less susceptible to AD, like some of the MMP knockouts.
Although growing evidences indicate that ADAMTS family enzymes are metalloproteinases with multiple functions, the exact expression profiling, regulation and function of ADAMTS in various pathophysiological processes of CNS, especially the signaling pathways and molecular events, remain to be delineated. Further studies would helpful scientists to understand better of the function and regulation of ADAMTS on CNS functioning.
As we fleetingly analyzed and discussed herein, much work on the transcriptional regulation of metalloproteinases including ADAMTS for regeneration and restoration processes of CNS tissue in the past 10 years was focused on individual transcription patterns of ADAMTS, and some of these studies relied solely on alters in phenotypes of the knockout mice to assess the function of different transcription patterns. The molecular target genes of a couple of transcription factors in CNS are largely unknown. As we understand from previous great studies, ADAMTS might be an important pioneer molecule group for neurodegenerative diseases. Especially, the abnormal deposition of Aβ proteins might be connected with dysfunction of metalloproteinases including ADAMTS. From this point of view, one can be hypothesized that curative protocols that have an impact on the activities/amounts of these proteins will help us to cope with neurodegenerative diseases including AD.
References
- [1].Hardy J, Selkoe DJ (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science, 297: 353-356. [DOI] [PubMed] [Google Scholar]
- [2].Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, Matsushima K (1997). Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J Biol Chem, 272: 556-562. [DOI] [PubMed] [Google Scholar]
- [3].Nardi JB, Martos R, Walden KK, Lampe DJ, Robertson HM (1999). Expression of lacunin, a large multidomain extracellular matrix protein, accompanies morphogenesis of epithelial monolayers in Manduca sexta. Insect Biochem Mol Biol, 29: 883-897. [DOI] [PubMed] [Google Scholar]
- [4].Bork P, Beckmann G (1993). The CUB domain. A widespread module in developmentally regulated proteins. J Mol Biol, 231: 539-545. [DOI] [PubMed] [Google Scholar]
- [5].Baenziger NL, Brodie GN, Majerus PW (1971). A thrombin-sensitive protein of human platelet membrane. Proc Natl Acad Sci USA, 68: 240-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lawler JW, Slayter HS, Coligan JE (1978). Isolation and characterization of a high molecular weight glycoprotein from human blood platelets. J Biol Chem, 253: 8609-8616. [PubMed] [Google Scholar]
- [7].Apte SS (2009). A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem, 284: 31493-31497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Porter S, Clark IM, Kevorkian L, Edwards DR (2005). The ADAMTS metalloproteinases. Biochem J, 386: 15-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rawlings ND, O'Brien E, Barrett AJ (2002). MEROPS: the protease database. Nucleic Acids Res, 30: 343-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Werb Z (1997). ECM and cell surface proteolysis: regulating cellular ecology. Cell, 91: 439-442. [DOI] [PubMed] [Google Scholar]
- [11].Stanton H, Melrose J, Little CB, Fosang AJ (2011). Proteoglycan degradation by the ADAMTS family of proteinases. Biochim Biophys Acta, 1812: 1616-1629. [DOI] [PubMed] [Google Scholar]
- [12].Kosik KS (1993). The molecular and cellular biology of tau. Brain Pathol, 3: 39-43. [DOI] [PubMed] [Google Scholar]
- [13].Himmler A, Drechsel D, Kirschner MW, Martin DW Jr (1989). Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol, 9: 1381-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cleveland DW, Hwo SY, Kirschner MW (1977). Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol, 116: 207-225. [DOI] [PubMed] [Google Scholar]
- [15].Kowall NW, Kosik KS (1987). Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer's disease. Ann Neurol, 22: 639-643. [DOI] [PubMed] [Google Scholar]
- [16].Lichtenberg B, Mandelkow EM, Hagestedt T, Mandelkow E (1988). Structure and elasticity of microtubule-associated protein tau. Nature, 34: 359-362. [DOI] [PubMed] [Google Scholar]
- [17].Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM (1999). Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry, 38: 3549-3558. [DOI] [PubMed] [Google Scholar]
- [18].Götz J, Probst A, Spillantini MG, Schäfer T, Jakes R, Bürki K, et al. (1995). Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J, 14: 1304-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, (1987). The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature, 325: 733-736. [DOI] [PubMed] [Google Scholar]
- [20].Mills J, Reiner PB (1999). Regulation of amyloid precursor protein cleavage. J Neurochem, 72: 443-460. [DOI] [PubMed] [Google Scholar]
- [21].Younkin SG (1998). The role of A beta 42 in Alzheimer's disease. J Physiol Paris, 92: 289-292. [DOI] [PubMed] [Google Scholar]
- [22].Nunan J, Small DH (2000). Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett, 483: 6-10. [DOI] [PubMed] [Google Scholar]
- [23].Huang HC, Jiang ZF (2009). Accumulated amyloid-beta peptide and hyperphosphorylated tau protein: relationship and links in Alzheimer's disease. J Alzheimers Dis, 16: 15-27. [DOI] [PubMed] [Google Scholar]
- [24].Tanzi RE, Moir RD, Wagner SL (2004). Clearance of Alzheimer's Abeta peptide: the many roads to perdition. Neuron, 43: 605-608. [DOI] [PubMed] [Google Scholar]
- [25].Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M et al. (2010). Neurotoxicity of Alzheimer's disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J, 29: 3408-3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gandy S (2005). The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest, 115: 1121-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bornstein P (1992). Thrombospondins: structure and regulation of expression. FASEB J, 6: 3290-3299. [DOI] [PubMed] [Google Scholar]
- [28].Levy GG, Motto DG, Ginsburg D (2005). ADAMTS13 turns 3. Blood, 106: 11-17. [DOI] [PubMed] [Google Scholar]
- [29].Fosang AJ, Little CB (2008). Drug insight: aggrecanases as therapeutic targets for osteoarthritis. Nat Clin Pract, 4: 420-427. [DOI] [PubMed] [Google Scholar]
- [30].Tortorella MD, Burn TC, Pratta MA, Abbaszade I, Hollis JM, Liu R et al. (1999). Purification and cloning of aggrecanase-1: a member of the ADAMTS family of proteins. Science, 284: 1664-1666. [DOI] [PubMed] [Google Scholar]
- [31].Tortorella MD, Malfait F, Barve RA, Shieh HS, Malfait AM (2009). A review of the ADAMTS family, pharmaceutical targets of the future. Curr Pharm Des, 15: 2359-2374. [DOI] [PubMed] [Google Scholar]
- [32].Colige A, Sieron AL, Li SW, Schwarze U, Petty E, Wertelecki W et al. (1999). Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am J Hum Genet, 65: 308-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jungers KA, Le Goff C, Somerville RP, Apte SS (2005). ADAMTS9 is widely expressed during mouse embryo development. Gene Expr Patterns, 5: 609-617. [DOI] [PubMed] [Google Scholar]
- [34].Velasco J, Li J, DiPietro L, Stepp MA, Sandy JD, Plaas A (2011). ADAMTS5 deletion blocks murine dermal repair through CD44-mediated aggrecan accumulation and modulation of transforming growth factor β1 (TGFβ1) signaling. J Biol Chem, 286: 26016-26027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu CJ (2009). The role of ADAMTS-7 and ADAMTS-12 in the pathogenesis of arthritis. Nat Clin Pract Rheumatol, 5: 38-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Blelloch R, Anna-Arriola SS, Gao D, Li Y, Hodgkin J, Kimble J (1999). The gon-1 gene is required for gonadal morphogenesis in Caenorhabditis elegans. Dev Biol, 216: 382-393. [DOI] [PubMed] [Google Scholar]
- [37].Furlan M, Robles R, Lammle B (1996). Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood, 87: 4223-4234. [PubMed] [Google Scholar]
- [38].Lancellotti S, De Cristofaro R (2011). Structure and proteolytic properties of ADAMTS13, a metalloprotease involved in the pathogenesis of thrombotic microangiopathies. Prog Mol Biol Transl Sci, 99: 105-144. [DOI] [PubMed] [Google Scholar]
- [39].Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM et al. (2001). Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature, 413: 488-494. [DOI] [PubMed] [Google Scholar]
- [40].Zhou W, Inada M, Lee TP, Benten D, Lyubsky S, Bouhassira EE et al. (2005). ADAMTS13 is expressed in hepatic stellate cells. Lab Invest, 85: 780-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sandy JD, Westling J, Kenagy RD, Iruela-Arispe ML, Verscharen C, Rodriguez-Mazaneque JC et al. (2001). Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem, 276: 13372-13378. [DOI] [PubMed] [Google Scholar]
- [42].Matthews RT, Gary SC, Zerillo C, Pratta M, Solomon K, Arner EC et al. (2000). Brain-enriched hyaluronan binding (BEHAB)/brevican cleavage in a glioma cell line is mediated by a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) family member. J Biol Chem, 275: 22695-22703. [DOI] [PubMed] [Google Scholar]
- [43].Cua RC, Lau LW, Keough MB, Midha R, Apte SS, Yong VW (2013). Overcoming neurite-inhibitory chondroitin sulfate proteoglycans in the astrocyte matrix. Glia, 61: 972-984. [DOI] [PubMed] [Google Scholar]
- [44].Haddock G, Cross AK, Plumb J, Surr J, Buttle DJ, Bunning RA et al. (2006). Expression of ADAMTS-1, -4, -5 and TIMP-3 in normal and multiple sclerosis CNS white matter. Mult Scler, 12: 386-396. [DOI] [PubMed] [Google Scholar]
- [45].Hamel MG, Ajmo JM, Leonardo CC, Zuo F, Sandy JD, Gottschall PE (2008). Multimodal signaling by the ADAMTSs (a disintegrin and metalloproteinase with thrombospondin motifs) promotes neurite extension. Exp Neurol, 210: 428-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Abali O, Gokce EC, Cemil B, Erdogan B, Yonezawa T, Demircan K (2014). Early induction of ADAMTS 1, 4, 5 and 9 in IL-stimulated mouse astrocytes. Turk Neurosurg, 24: 519-524. [DOI] [PubMed] [Google Scholar]
- [47].Yamaguchi Y (2000). Lecticans: organizers of the brain extracellular matrix. Cell Mol Life Sci, 57: 276-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Galtrey CM, Fawcett JW (2007). The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res Rev, 54: 1-18. [DOI] [PubMed] [Google Scholar]
- [49].Satoh K, Suzuki N, Yokota H (2000). ADAMTS-4 (a disintegrin and metalloproteinase with thrombospondin motifs) is transcriptionally induced in beta-amyloid treated rat astrocytes. Neurosci Lett, 289: 177-180. [DOI] [PubMed] [Google Scholar]
- [50].Yuan W, Matthews RT, Sandy JD. Gottschall PE (2002). Association between protease-specific proteolytic cleavage of brevican and synaptic loss in the dentate gyrus of kainate-treated rats. Neuroscience, 114: 1091-1101. [DOI] [PubMed] [Google Scholar]
- [51].Medina-Flores R, Wang G, Bissel SJ, Murphey-Corb M, Wiley CA (2004). Destruction of extracellular matrix proteoglycans is pervasive in simian retroviral neuroinfection. Neurobiol Dis, 16: 604-616. [DOI] [PubMed] [Google Scholar]
- [52].Mayer J, Hamel MG, Gottschall PE (2005). Evidence for proteolytic cleavage of brevican by the ADAMTSs in the dentate gyrus after excitotoxic lesion of the mouse entorhinal cortex. BMC Neurosci, 6: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Miguel RF, Pollak A, Lubec G (2005). Metalloproteinase ADAMTS-1 but not ADAMTS-5 is manifold overexpressed in neurodegenerative disorders as Down syndrome, Alzheimer's and Pick's disease. Brain Res Mol Brain Res, 133: 1-5. [DOI] [PubMed] [Google Scholar]
- [54].Cross AK, Haddock G, Surr J, Plumb J, Bunning RA, Buttle DJ et al. (2006). Woodroofe MN. Differential expression of ADAMTS-1, -4, -5 and TIMP-3 in rat spinal cord at different stages of acute experimental autoimmune encephalomyelitis. J Autoimmun, 26: 16-23. [DOI] [PubMed] [Google Scholar]
- [55].Ajmo JM, Eakin AK, Hamel MG, Gottschall PE (2008). Discordant localization of WFA reactivity and brevican/ADAMTS-derived fragment in rodent brain. BMC Neurosci, 9: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Howell MD, Torres-Collado AX, Iruela-Arispe ML, Gottschall PE (2012). Selective decline of synaptic protein levels in the frontal cortex of female mice deficient in the extracellular metalloproteinase ADAMTS1. PLoS One, 7: e47226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Krstic D, Rodriguez M, Knuesel I (2012). Regulated proteolytic processing of Reelin through interplay of tissue plasminogen activator (tPA), ADAMTS- 4, ADAMTS-5, and their modulators. PLoS One, 7: e47793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tauchi R, Imagama S, Natori T, Ohgomori T, Muramoto A, Shinjo R et al. (2012). Matsuyama Y, Ishiguro N, Kadomatsu K. The endogenous proteoglycan-degrading enzyme ADAMTS-4 promotes functional recovery after spinal cord injury. J Neuroinflammation, 9: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tauchi R, Imagama S, Ohgomori T, Natori T, Shinjo R, Ishiguro N et al. (2012). ADAMTS-13 is produced by glial cells and upregulated after spinal cord injury. Neurosci Lett, 517: 1-6. [DOI] [PubMed] [Google Scholar]
- [60].Demircan K, Yonezawa T, Takigawa T, Topcu V, Erdogan S, Ucar F et al. (2013). ADAMTS1, ADAMTS5, ADAMTS9 and aggrecanase-generated proteoglycan fragments are induced following spinal cord injury in mouse. Neurosci Lett, 544: 25-30. [DOI] [PubMed] [Google Scholar]
- [61].Lemarchant S, Pruvost M, Montaner J, Emery E, Vivien D, Kanninen K et al. (2013). ADAMTS proteoglycanases in the physiological and pathological central nervous system. J Neuroinflammation, 10: 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lee HR, Shin HK, Park SY, Kim HY, Lee WS, Rhim BY et al. (2014). Hong KW, Kim CD. Cilostazol suppresses β-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-β. J Neurosci Res, 92: 1581-1590. [DOI] [PubMed] [Google Scholar]
- [63].Kojro E, Fahrenholz F (2005). The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem, 38: 105-127. [DOI] [PubMed] [Google Scholar]
- [64].Moss ML, Powell G, Miller MA, Edwards L, Qi B, Sang QX et al. (2011). ADAM9 inhibition increases membrane activity of ADAM10 and controls α-secretase processing of amyloid precursor protein. J Biol Chem, 286: 40443-40451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bekris LM, Lutz F, Li G, Galasko DR, Farlow MR, Quinn JF et al. (2012) ADAM10 expression and promoter haplotype in Alzheimer's disease. Neurobiol Aging, 33: 2229.e1-2229.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hata S, Fujishige S, Araki Y, Kato N, Araseki M, Nishimura M et al. (2009). Alcadein cleavages by amyloid beta-precursor protein (APP) alpha- and gamma-secretases generate small peptides, p3-Alcs, indicating Alzheimer disease-related gamma-secretase dysfunction. J Biol Chem, 284: 36024-36033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Colciaghi F, Borroni B, Pastorino L, Marcello E, Zimmermann M, Cattabeni F et al. (2002). α -Secretase ADAM 10 as well as APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol Med, 8: 67-74. [PMC free article] [PubMed] [Google Scholar]
- [68].Tang K, Hynan LS, Baskin F, Rosenberg RN (2006). Platelet amyloid precursor protein processing: a bio-marker for Alzheimer's disease. J Neurol Sci, 240: 53-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E et al. (2004). A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest, 113: 1456-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rosenberg RN, Baskin F, Fosmire JA, Risser R, Adams P, Svetlik D et al. (1997). Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch Neurol, 54: 139-144. [DOI] [PubMed] [Google Scholar]
- [71].Anderson JJ, Holtz G, Baskin PP, Wang R, Mazzarelli L, Wagner SL et al. (1999). Menzaghi F. Reduced cerebrospinal fluid levels of alpha-secretase-cleaved amyloid precursor protein in aged rats: correlation with spatial memory deficits. Neuroscience, 93: 1409-1420. [DOI] [PubMed] [Google Scholar]
- [72].Bernstein HG, Bukowska A, Krell D, Bogerts B, Ansorge S, Lendeckel U (2003). Comparative localization of ADAMs 10 and 15 in human cerebral cortex normal aging, Alzheimer disease and Down syndrome. J Neurocytol, 32: 153-160. [DOI] [PubMed] [Google Scholar]
- [73].Gatta LB, Albertini A, Ravid R, Finazzi D (2002). Levels of beta-secretase BACE and alpha-secretase ADAM10 mRNAs in Alzheimer hippocampus. Neuroreport, 13: 2031-2033. [DOI] [PubMed] [Google Scholar]
- [74].Bekris LM, Galloway NM, Millard S, Lockhart D, Li G, Galasko DR et al. (2011). Amyloid precursor protein (APP) processing genes and cerebrospinal fluid APP cleavage product levels in Alzheimer's disease. Neurobiol Aging, 32: 556.e13-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Clark ME, Kelner GS, Turbeville LA, Boyer A, Arden KC, Maki RA (2001). ADAMTS9, a novel member of the ADAM-TS/metallospondin gene family. Genomics, 67: 343-350. [DOI] [PubMed] [Google Scholar]
- [76].Pehlivan S, Fedakar R, Eren B, Akyol S, Eren F, Turkmen Inanır N et al. (2015). ADAMTS4, 5, 9, and 15 expressions in the autopsied brain of patients with Alzheimers Disease: a preliminary immünohistochemistry study. J Clin Psychopharmacol (in press). DOI: 10.5455/bcp.20150706034008 [DOI] [Google Scholar]
- [77].D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature, 374: 719-723. [DOI] [PubMed] [Google Scholar]
- [78].Herz J, Chen Y (2006). Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci, 7: 850-859. [DOI] [PubMed] [Google Scholar]
- [79].Rogers JT, Weeber EJ (2008). Reelin and apoE actions on signal transduction, synaptic function and memory formation. Neuron Glia Biol, 4: 259-270. [DOI] [PubMed] [Google Scholar]
- [80].Kocherhans S, Madhusudan A, Doehner J, Breu KS, Nitsch RM, Fritschy JM et al. (2010). Reduced Reelin expression accelerates amyloid-beta plaque formation and tau pathology in transgenic Alzheimer's disease mice. J Neurosci, 30: 9228-9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Botella-Lopez A, Burgaya F, Gavin R, Garcia-Ayllon MS, Gomez-Tortosa E, Pena-Casanova J et al. (2006). Reelin expression and glycosylation patterns are altered in Alzheimer's disease. Proc Natl Acad Sci USA, 103: 5573-5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Hisanaga A, Morishita S, Suzuki K, Sasaki K, Koie M, Kohno T et al. (2012). A disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS-4) cleaves Reelin in an isoform-dependent manner. FEBS Lett, 586: 3349-3353. [DOI] [PubMed] [Google Scholar]
- [83].Yu NN, Tan MS, Yu JT, Xie AM, Tan L (2015). The Role of Reelin Signaling in Alzheimer's Disease. Mol Neurobiol. (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- [84].Végh MJ, Heldring CM, Kamphuis W, Hijazi S, Timmerman AJ, Li KW et al. (2014). Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer's disease. Acta Neuropathol Commun. 2: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Gottschall PE, Howell MD (2015). ADAMTS expression and function in central nervous system injury and disorders. Matrix Biol. 44-46: 70-6. [DOI] [PMC free article] [PubMed] [Google Scholar]