

Abstract
Objective
Up-regulation of glucose metabolism has been implicated not only in tumor cell growth but also in immune cells upon activation. However, little is known about the metabolite profile in rheumatoid arthritis (RA), particularly in fibroblast-like synoviocytes (FLS). This study was undertaken to evaluate whether changes in glucose metabolism in RA FLS could play a role in inflammation and joint damage.
Methods
Synovium and FLS were obtained from patients with RA and patients with osteoarthritis (OA). The rate of glycolysis after stimulation of FLS with lipopolysaccharide and platelet-derived growth factor BB was measured using glycolysis stress test technology. FLS function was evaluated using a glycolysis inhibitor, 2-deoxy-D-glucose (2-DG). After stimulation of the FLS, a migration scratch assay, MTT assay, and enzyme-linked immunosorbent assay were performed to measure the effect of 2-DG on FLS migration, viability of the FLS, and cytokine secretion, respectively. IRDye 800CW 2-DG was used to assess glucose uptake in the arthritic joints and stromal cells of mice after K/BxN mouse serum transfer. The mice were injected daily, intraperitoneally, with 3-bromopyruvate (BrPa; 5 mg/kg) to assess the effect of inhibition of glycolysis in vivo.
Results
Compared to human OA FLS, the balance between glycolysis and oxidative phosphorylation was shifted toward glycolysis in RA FLS. Glucose transporter 1 (GLUT1) messenger RNA (mRNA) expression correlated with baseline functions of the RA FLS. Glucose deprivation or incubation of the FLS with glycolytic inhibitors impaired cytokine secretion and decreased the rate of proliferation and migration of the cells. In a mouse model of inflammatory arthritis, GLUT1 mRNA expression in the synovial lining cells was observed, and increased levels of glucose uptake and glycolytic gene expression were detected in the stromal compartment of the arthritic mouse joints. Inhibition of glycolysis by BrPa, administered in vivo, significantly decreased the severity of arthritis in this mousemodel.
Conclusion
Targeting metabolic pathways is a novel approach to understanding the mechanisms of disease. Inhibition of glycolysis may directly modulate synoviocyte-mediated inflammatory functions and could be an effective treatment strategy for arthritis.
Rheumatoid arthritis (RA) is a systemic autoimmune disease that mainly affects the joint synovium, leading to chronic inflammation, joint destruction, and loss of function (1). Pathogenesis of the disease involves a complex interaction between the innate and adaptive arms of the immune system, in concert with resident fibroblast-like synoviocytes (FLS). These cells are critical in RA pathogenesis (2,3). They display an aggressive/transformed phenotype that contributes to synovial inflammation and cartilage damage by producing inflammatory mediators. The autonomous ability of RA FLS to survive in an environment enriched with oxygen radicals, nitric oxide, and cytokines resembles the phenotype of some tumors (2,3). Although treatment of RA has improved, the currently available disease-modifying drugs do not directly target FLS dysfunction. Thus, new, rationally designed disease-modifying agents are needed to replace or complement current therapies.
Many factors in the cell microenvironment activate key signaling pathways that converge on cellular metabolism to support cell growth and survival (4,5). These changes in metabolism may be best described in tumor cells. However, there is increasing evidence of metabolic changes in stromal and immune cells (6–8) and in autoimmune diseases (9–11). Recently, we showed that choline metabolism is activated in RA FLS under proinflammatory conditions, and that selectively blocking the enzyme choline kinase α might be beneficial in inflammatory arthritis by suppressing the functions of FLS (12). Thus, the study of metabolic changes in these cells could potentially lead to identification of novel activated signaling pathways and therapeutic agents (13).
One of the best-characterized metabolic phenotypes in tumor cells is a shift toward increased glycolytic turnover (4,5). Historically, it was suggested that the persistent metabolism of glucose to lactate is an adaptation to the stressful and dynamic microenvironment of the solid tumor, where concentrations of crucial nutrients and oxygen are spatially and temporally heterogeneous (14,15). Cell populations with up-regulated glycolysis and acid resistance have a powerful growth advantage, which promotes proliferation and invasion. The microenvironment in inflamed joints is also characterized by hypoxia and low concentration of nutrients (16). The up-regulation of glycolysis, resulting in increased glucose consumption, can be observed with clinical imaging (17). The increased glucose uptake imaged with fluorodeoxyglucose–positron emission tomography (FDG-PET) is largely dependent on the rate of glycolysis, and several studies showed FDG accumulation in swollen joints. Both fibroblasts and activated macrophages contributed to a high level of FDG accumulation in the pannus, and hypoxia as well as cytokine stimulation significantly increased FDG uptake by these cells (18,19).
These considerations led us to assess whether RA FLS display an altered pattern of glucose metabolism and an intrinsic ability to employ glycolytic metabolism under conditions of metabolic stress in a manner different from other primary fibroblasts. We also evaluated whether glycolytic inhibition may be a disease-modifying strategy, by directly modulating the synoviocyte-mediated destruction of cartilage.
MATERIALS AND METHODS
Mice
KRN T cell receptor–transgenic mice were a gift from Drs. D. Mathis (Harvard Medical School, Boston, MA) and C. Benoist (Institut de Génétique et de Biologie Moléculaire et Cellulaire, Strasbourg, France). All mice used in these experiments were bred on the C57BL/6 background and were 8–12 weeks old. All protocols involving animals received prior approval from the Institutional Review Boards and followed the Guide for the Care and Use of Laboratory Animals from the Institute for Laboratory Animal Research (National Research Council).
Reagents
Lipopolysaccharide (LPS), 2-deoxy-D-glucose (2-DG), 3-bromopyruvate (BrPa), and all reagents for respirometry were obtained from Sigma-Aldrich, except for the XF plasma membrane permeabilizer (used in permeabilized cell experiments), which was obtained from Seahorse Bioscience. Platelet-derived growth factor BB (PDGF-BB) and tumor necrosis factor (TNF) were obtained from R&D Systems. IRDye 800CW 2-DG and IRDye 800CW carboxyl were purchased from Li-Cor Biosciences. MK-2206 (an Akt inhibitor) was obtained from Selleckchem.
Preparation of human synovium and synoviocytes
Synovium and FLS were obtained from patients with RA undergoing total joint replacement or synovectomy. All RA patients met the American College of Rheumatology 1987 revised criteria for seropositive RA (20). In addition, synovium and FLS were obtained from patients with osteoarthritis (OA), as previously described (21–23).
In vivo imaging
IRDye 800CW 2-DG (10 nmoles) or the dye control (IRDye 800CW carboxyl, 10 nmoles) was intravenously injected into mice. Twenty-four hours after the dye injection, the animals were imaged using a whole-body mouse imager (Maestro; CRI). The mice were killed by isoflurane overdose, followed by cervical dislocation. Imaging was performed using a whole-body mouse imager (Maestro; CRI). Spectral imaging was carried out by excitation of the IRDye 800CW at a mean±SD excitation wavelength of 730±42 nm, followed by measurement of emission at a wavelength of 800 nm long pass. Regions of interest were delineated and analyzed using ImageJ software.
Preparation of single cells from the joints of arthritic mice and nonarthritic control mice
Joints from arthritic mice and from mice that had not received arthritogenic serum transfer (nonarthritic controls) were digested using type IV collagenase (1 mg/ml) and DNase I (200 units/ml) in RPMI medium supplemented with 10% fetal calf serum (FCS). Floating cells were recovered from the digestion mix every 20 minutes, and the reaction was stopped by adding ice-cold phosphate buffered saline (PBS)−2% FCS–5mM EDTA. The cells were then spun down for 10 minutes at 1,200 revolutions per minute. Digestion was not carried out longer than 60 minutes.
After a single-cell suspension was obtained, cells were stained with fluorescence-labeled antibodies and then analyzed by flow cytometry. Alternatively, enrichment of CD45− cells that were positive for podoplanin (PDPN+) was achieved by first depleting CD45+ cells using anti-CD45 biotin-labeled antibodies, followed by use of anti-biotin magnetic beads (Miltenyi Biotec). PDPN+ cells were enriched within the population of negatively selected cells with the use of antiphycoerythrin (anti-PE) beads (Miltenyi Biotec) in combination with PDPN–PE labeling. Positively selected cells were harvested, and the purity of the CD45−PDPN+ cell population was assayed by flow cytometry, yielding a purity of ~90%. An LSRII cell analyzer (BD Biosciences) was used for the flow cytometry analyses, and FlowJo software (TreeStar) was used for data analysis.
Serum transfer and arthritis scoring in mice
Sera from adult, arthritic K/BxN mice were pooled for use in serum transfer. Recipient mice were injected intraperitoneally with 150 μl of the K/BxN mouse serum. Clinical arthritis scores were evaluated in the recipient mice after serum transfer, as described previously (24). Beginning on day 0 or day 4 after K/BxN mouse serum administration, BrPa (5 mg/kg daily) was injected intraperitoneally. On days 7 or 9 post–serum injection, the mice were killed and the paws were analyzed for histopathologic changes (histologic score range 0–5).
Statistical analysis
Statistical analysis was performed with Prism software (version 5; GraphPad). Results are expressed as the mean±SEM. Normality of the variables was assessed using the Kolmogorov-Smirnov and D’Agostino-Pearson normality tests. For comparison between 2 groups, Student’s 2-tailed t-tests or nonparametric Mann-Whitney tests were applied, depending on the normality of the distribution of the variables. For comparison between paired samples, paired t-tests or nonparametric Wilcoxon’s matched pairs tests were applied. We compared 3 or more groups with analysis of variance. Either Dunnett’s post hoc test or the Bonferroni test was chosen, depending on the homogeneity of variances. Pearson’s or nonparametric Spearman’s coefficients were used for assessment of correlations between 2 quantitative variables. Results were considered significant if the 2-sided P value was less than 0.05 (more details are provided in Supplementary Methods, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
RESULTS
FLS mitochondrial respiration at baseline
We first measured the rates of cellular respiration in cultured primary FLS to study whether RA FLS demonstrate an increase in the metabolism of glucose to lactate. Using a Seahorse XF analyzer, we measured the mitochondrial respiration rate (oxygen consumption rate [OCR]) and the rate of proton efflux (proton production rate [PPR]) in RA and OA FLS cell lines (Figures 1A and B). The ratio of PPR to OCR represents an estimate of the relative balance between glycolysis and oxidative phosphorylation (although the ratio does not directly reflect the relative ATP produced from either pathway; much less ATP is made from 1 mole of glucose incompletely oxidized to lactate as compared to complete oxidation by mitochondria). RA FLS showed a greater PPR:OCR ratio than that observed in OA FLS (mean±SEM 2.14±0.99 versus 1.14±0.3; P=0.03) (Figure 1C), suggesting that RA FLS are more dependent on glycolysis than are OA FLS.
Figure 1.
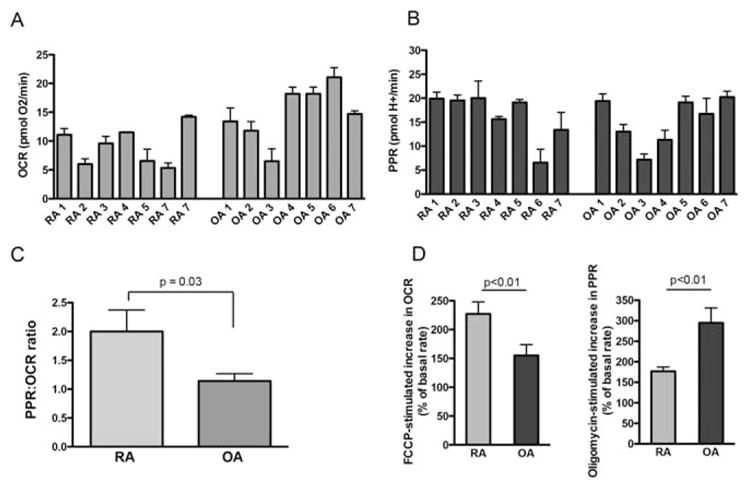
Baseline levels of mitochondrial respiration in fibroblast-like synoviocytes (FLS). A and B, Basal rates of mitochondrial respiration (oxygen consumption rate [OCR]) in 7 rheumatoid arthritis (RA) cell lines and 7 osteoarthritis (OA) cell lines (A), and the proton production rate (PPR) in the same cell lines (B). Values are the mean±SEM pooled results from 5 technical replicates (2–5 biologic replicates for each cell line, with 5 technical replicates for each cell line per experiment). C, PPR:OCR ratio in pooled RA FLS and OA FLS. D, FCCP-stimulated increase in the OCR (left) and oligomycin-stimulated increase in the PPR (right), expressed as a percentage increase from the basal rate, in pooled RA FLS and OA FLS. Values in C and D are the mean±SEM pooled results from the same cells as in A and B.
This altered balance between oxidative phosphorylation and glycolysis in RA FLS and OA FLS was not a direct function of limited capacities in either cell type. Compared to RA FLS, OA FLS respired at a rate closer to their maximal capacity. In contrast, RA FLS worked closer to their capacity than did OA FLS to meet the demand for ATP by glycolysis (Figure 1D).
To further determine whether the relatively lower respiratory rate and higher glycolytic rate in RA cells could be attributed to a decreased number of mitochondria, the protein levels of subunits of electron transport chain complexes were measured by Western blotting. The findings from Western blotting showed that the levels of the different components of the mitochondrial complex were not different between the 2 cell lines (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
Glucose transporter 1(GLUT1) expression and correlation with baseline FLS functions
We also measured the expression of selected genes related to the glycolytic pathway in FLS from 5 RA patients and 5 OA patients. The expression of messenger RNA (mRNA) for GLUT1, the main glucose transporter, was increased in RA FLS compared to OA FLS (Figure 2A), and the levels of GLUT1 mRNA strongly correlated with the levels of mRNA for hexokinase 2 (HK2), an enzyme that catalyzes the first step in glycolysis (Figures 2A and B).
Figure 2.

Correlation of glucose transporter 1 (GLUT1) mRNA expression with baseline FLS functions. A, Expression of select genes, as assessed by quantitative polymerase chain reaction, in RA FLS and OA FLS (n=5 cell lines per group), and migration and viability (as determined by MTT assay) of the FLS. Each symbol represents an individual sample; bars show the mean±SEM. B, Correlations of GLUT1 mRNA expression with the expression levels of select genes in the FLS and with migration and viability of the FLS. P values and Spearman’s correlation coefficients are shown. See Figure 1 for definitions.
The levels of other genes related to glucose metabolism and lactate production, such as lactate dehydrogenase A (LDHA) and pyruvate dehydrogenase kinase 1 (PDK1) genes, were not significantly different between OA FLS and RA FLS (see Supplementary Figure 2A, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract). However, the levels of these other genes were correlated with the levels of GLUT1 mRNA expression in the FLS (Figure 2B and Supplementary Figure 2C, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract). In addition, GLUT1 mRNA expression was also correlated with the baseline expression of matrix metalloproteinase 3 (MMP-3) mRNA, and with the baseline migration of these cell lines (Figures 2A and B and Supplementary Figure 2B, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract), suggesting that there is a correlation between the rate of glycolysis and certain FLS baseline functions. Of interest, findings from 6 RA and 6 OA FLS cell lines reported in the GEO data sets (array GSE7669; http://www.ncbi.nlm.nih.gov) also showed that the expression levels of GLUT1, HK2, and MMP mRNA were statistically significantly correlated in these cell lines (see Supplementary Figures 3A and B, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
Effect of inflammatory mediators on the rate of glycolysis in human FLS
To determine whether inflammatory mediators implicated in the FLS inflammatory response regulate the mitochondrial respiration and glycolysis rates, we stimulated OA FLS and RA FLS with LPS or PDGF-BB for 24 hours, a process that promotes FLS proliferation, FLS migration, and cytokine secretion. Stimulation of the FLS with LPS repressed the basal rate of respiration and stimulated glycolytic flux in both OA FLS and RA FLS (Figures 3A–C).
Figure 3.

FLS respiration following exposure to inflammatory mediators. A–C, RA FLS and OA FLS (n=7 cell lines per group) were stimulated with lipopolysaccharide (LPS; 1 μg/ml), or phosphate buffered saline (PBS) as vehicle control, for 24 hours, followed by measurement of the OCR (A), rate of proton efflux (PPR) (B), and PPR:OCR ratio (C). Values are the mean±SEM pooled results from 2–4 biologic replicates for each cell line, with 5 technical replicates for each cell line per experiment. D, The lactate concentration was measured in supernatants of cultured RA FLS (n=7 cell lines) after 24 hours of stimulation with LPS (1 μg/ml) or PBS vehicle control. E, The OCR and PPR were determined in human pulmonary fibroblasts (n=3 cell lines) after 24 hours of stimulation with LPS (1 μg/ml) or PBS vehicle control. Values are the mean±SEM pooled results from 1–3 biologic replicates, with 5 technical replicates for each cell line per experiment. F, The OCR and PPR were determined in RA FLS and OA FLS (n=6 cell lines per group) after 24 hours of stimulation with platelet-derived growth factor (PDGF; 10 ng/ml) or PBS vehicle control. Values are the mean±SEM pooled results from 5 technical replicates for each cell line. G, RA FLS (n=2 cell lines) were stimulated with either PDGF (10 ng/ml) or LPS (1 μg/ml) for 12, 24, or 48 hours, and glucose transporter 1 (GLUT-1) protein expression was determined by Western blotting. The control (C) comprised unstimulated FLS at 48 hours. H, RA FLS (n=3 cell lines) were incubated for 24 hours with LPS (1 μg/ml) or PBS vehicle control, and permeabilized cell respiration was then measured in the presence of either succinate plus rotenone (Succ/Rot) or pyruvate plus malate (Pyr/Mal) as substrates. In addition, the pooled rate of FCCP uncoupler–stimulated respiration was determined in 3 intact RA FLS cell lines following 24 hours of exposure to LPS. I, The PPR was determined in 4 different RA FLS cell lines after stimulation with LPS for 24 hours in the absence or presence of the Akt inhibitor MK-2206 (5 μM). Values in D, H, and I are the mean±SEM. NS=not significant (see Figure 1 for other definitions).
The drop in basal respiration in the FLS cell lines was accompanied by an increase in the PPR, suggesting that a shift from mitochondrial activity to glycolysis takes place in order to meet the energetic needs of the cells. Although the drop in basal mitochondrial respiration in response to LPS was greater in RA FLS than in OA FLS (mean±SEM decrease of 46.4±15% versus 25.9±37.9%; P=0.03), the increase in the PPR (199.8±82% in RA FLS versus 192.6±72.25% in OA FLS; P not significant) and increase in the PPR:OCR ratio (mean±SEM 3.9±1.6 in RA FLS versus 3.39± 2.4 in OA FLS; P not significant) upon LPS treatment did not differ significantly between the 2 cell lines.
The increase in extracellular PPR upon LPS stimulation was reinforced by an increase in the lactate levels measured in the culture medium, both in RA FLS and in OA FLS (Figure 3D and Supplementary Figure 4A, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract). The shift from mitochondrial respiration to glycolysis induced by LPS was not observed in primary human pulmonary fibroblasts, although the expression levels of Toll-like receptor 4 and mitochondrial content were similar between the cell lines (Figure 3E and Supplementary Figure 4B, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
In another experiment, we stimulated the FLS with PDGF-BB, which increased the mitochondrial respiration rate and the level of extracellular acidification in both RA FLS and OA FLS (Figure 3F). Interestingly, while the increase in mitochondrial respiration was similar between the RA and OA FLS cell lines, the RA FLS showed a greater increase in the glycolysis response after stimulation with PDGF-BB.
To further study the effect of LPS and PDGF-BB on glycolysis, we determined the expression of GLUT-1 by both Western blotting and quantitative polymerase chain reaction (qPCR) after stimulation of the cells with LPS or PDGF-BB. Addition of either stimulus increased the expression of GLUT-1 protein and GLUT1 mRNA in both RA FLS and OA FLS (Figure 2G and Supplementary Figure 4C, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
We then tested whether the shift from oxidative phosphorylation to glycolysis upon LPS stimulation could be attributed to mitochondrial dysfunction or whether it simply represented a shift toward whichever pathway was used to meet the ATP demand. To accomplish this, both RA and OA FLS were treated with LPS for 24 hours and subsequently permeabilized, allowing respiration to be measured in the absence of cytoplasmic signaling and control over ATP demand. In this manner, we could determine whether LPS treatment injured the mitochondria. Of note, in the permeabilized cell experiments using a Seahorse XF analyzer, recombinant perfringolysin O selectively permeabilizes the plasma membrane without damaging mitochondria (25). The inhibition in basal rates of respiration observed following LPS treatment in intact cells was not evident when permeabilized cells were treated with either succinate (plus rotenone) or pyruvate (plus malate) as oxidizable substrates (Figure 3H and Supplementary Figure 4D, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
To further prove that the drop in the OCR after stimulation of the cells with LPS could be attributed to a shift in the balance between oxidative phosphorylation and glycolysis, and not to mitochondrial dysfunction, we measured FCCP-stimulated respiration in whole cells. FCCP (with oligomycin) disengages mitochondrial function from regulation by the cellular ATP demand. Since protonophore-stimulated respiration did not change after LPS stimulation (Figure 3H and Supplementary Figure 4D, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract), it can be concluded that LPS does not damage mitochondrial substrate transport or oxidation.
As a potential mechanism for the LPS-induced depression of respiration, nitric oxide (NO) may act to depress mitochondrial respiration, similar to what has been previously observed in macrophages (26). However, we detected neither expression of inducible NO synthase, as assessed by qPCR, nor production of nitrate/nitrite, as measured by the Griess reaction, in LPS-stimulated FLS (data not shown).
Multiple other cellular pathways might lead to the glycolytic phenotype. One of these is the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway, which can be activated by hypoxia or several cytokines and growth factors, and this pathway regulates FLS functions in RA. However, incubation of the FLS with the Akt inhibitor MK-2206 (at a dose of 5 μM) did not prevent the increase in the rate of glycolysis after stimulation of the cells with LPS (Figure 3I and Supplementary Figure 4E, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
Effect of inhibition of glycolysis on FLS function
Since treatment of the cells with LPS and PDGF-BB stimulated glycolytic flux, we determined whether inhibition of glycolysis during LPS and PDGF-BB exposure could modify the response of the FLS to these factors. We used 2-DG, a glucose analog that is phosphorylated by HK to phospho–2-DG, but cannot be further metabolized by phospho–glucose isomerase (27). BrPa, for which a detailed mechanism of action remains to be fully elucidated, is believed to also inhibit HK, dampening the rate of glycolysis (27). As another approach to limiting glycolytic flux, we also tested the effects of culture of the FLS in medium lacking glucose (no glucose [NG] control medium). We first measured glucose consumption and lactate secretion after LPS stimulation in the presence or absence of both inhibitors. As might be expected, incubation of RA FLS with 2-DG (50 mM) and BrPa (25 μM) prevented the drop in glucose levels and increase in lactate levels in response to LPS (see Supplementary Figures 5A and B, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
Since glycolysis is involved in tumor cell proliferation and migration, we next determined whether inhibition of glycolysis may interfere with FLS cell growth and migration in vitro. RA FLS were pretreated with 2-DG, BrPa, or NG control medium and then cultured in the presence of PDGF-BB for 4 days, followed by an MTT assay. All 3 culture conditions significantly decreased the rate of cell proliferation as measured by MTT assay (Figure 4A). The 3 culture conditions also significantly reduced cell migration, as determined in a migration scratch assay, in both RA FLS and OA FLS (Figure 4B and Supplementary Figures 5C and D, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
Figure 4.

Effect of glycolytic inhibition on FLS functions. A, RA FLS were cultured in the presence of platelet-derived growth factor (PDGF; 10 ng/ml) or phosphate buffered saline (PBS) as vehicle control, with or without pretreatment with 2-deoxy-D-glucose (2-DG; 50 mM in PBS) or 3-bromopyruvate (BrPa; 25 μM in PBS) or no glucose (NG) medium. Cellular proliferation was determined on day 4 using an MTT assay. B, Migration of the RA FLS was assessed in the presence of PDGF (10 ng/ml) alone or PDGF (P) along with 2-DG, BrPA, or NG medium. C and D, RA FLS were cultured in the presence of lipopolysaccharide (LPS; 1 μg/ml) or PBS as vehicle control, with or without pretreatment with 2-DG (50 mM) or BrPa (25 μM). Supernatants from the RA cell cultures were prepared after 24 hours of LPS stimulation and were analyzed for secretion of interleukin-6 (IL-6) (C) and matrix metalloproteinase 3 (MMP-3) (D). Results in A–D are pooled from 4 different cell lines. E, Glucose oxidation was determined in RA FLS, OA FLS, or primary human pulmonary fibroblasts (PFs) in the presence or absence of 2-DG (50 mM). Results are pooled from 3 different cell lines assayed in duplicate. F, RA FLS, OA FLS, and PFs were stimulated with or without 2-DG (50 mM), PDGF (10 ng/ml), or LPS (1 μM) for a total of 8 hours, and purine synthesis was determined. Results are pooled from 3 different cell lines assayed in triplicate. Values are the mean±SEM. See Figure 1 for other definitions.
Pyruvate is transported into cells and provides oxidizable substrate to mitochondria, even in the presence of glycolytic inhibitors. Therefore, addition of pyruvate to cell cultures can allow us to determine whether the effect on FLS functions after inhibiting glycolysis can be rescued by support of mitochondrial metabolism. We tested the effect of these conditions in the migration scratch assay. The addition of pyruvate (1 mM) did not affect the inhibition of migration of the cells cultured in any of the 3 culture conditions (see Supplementary Figure 5E, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
Treatment with 2-DG and BrPa also decreased the secretion of interleukin-6 (IL-6) and MMP-3 in RA FLS (Figures 4C and D), and decreased the secretion of IL-6 in OA FLS (see Supplementary Figure 5F, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract). The addition of pyruvate to the medium did not reverse the inhibitory effect of 2-DG on IL-6 secretion (see Supplementary Figure 5G, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract), suggesting that the effects of glycolytic inhibitors are not simply attributable to a blockade of glycolytic provision of pyruvate to mitochondria. Treatment with 2-DG also decreased the expression of MMPs 1 and 3 and IL-6 after LPS stimulation in RA FLS (see Supplementary Figure 5H, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
In addition to inhibiting glycolysis, 2-DG would be expected to inhibit both the oxidative and nonoxidative arms of the pentose–phosphate (PP) pathway. We therefore measured [1-14C]glucose oxidation to 14CO2, which assesses carbon flow through the oxidative PP pathway, and also measured purine nucleotide synthesis, which assesses carbon flow through the nonoxidative PP pathway, because most ribose-for-purine nucleotide synthesis comes from the nonoxidative PP pathway (28,29). We found that treatment with 2-DG did not affect [1-14C]glucose oxidation or purine synthesis in either RA FLS or OA FLS, but that it did dramatically decrease purine synthesis and glucose oxidation in primary human pulmonary fibroblasts (Figures 4E and F), thus highlighting again the profound differences in glucose metabolism in FLS.
Glucose metabolite profile in human RA synovium
We then determined the glucose metabolite profile of whole synovium from patients with RA and patients with OA, using proton magnetic resonance spectroscopy (1H-MRS). One-dimensional 1H-MRS spectra of aqueous extracts revealed higher lactate accumulation in RA synovium compared to OA synovium (see Supplementary Figures 6A and B, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract). Levels of other metabolites related to glucose metabolism, as detected by 1H-MRS, were also higher in RA synovium compared to OA synovium (see Supplementary Figure 6B, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract), suggesting that RA synovium undergoes an increased rate of glycolysis. Given the heterogeneity observed in the RA samples, the metabolite profile of the RA synovium was not statistically significantly different from that of the OA synovium. Further studies with a higher number of samples are needed for a more detailed study of glucose metabolism in RA synovium.
Decreased severity of inflammatory arthritis in mice following inhibition of glycolysis
The effect of glycolytic inhibitors, observed as a decrease in the levels of inflammatory markers in FLS, led us to evaluate the role of this approach in the K/BxN mouse serum transfer model of arthritis. The K/BxN passive serum transfer model is FLS dependent (30) and requires only innate immunity (31). Since 2-DG has been shown to have significant toxic effects in animal studies (27), we used BrPa to evaluate glycolytic inhibition in vivo. This drug has been reported to be remarkably effective in animal models of solid tumor growth, without toxic effects (27,32–34).
We first determined whether glucose metabolism was increased in the mice, using IRDye 800CW 2-DG, a fluorescent optical imaging agent that assesses glucose uptake. IRDye 800CW 2-DG or the carboxyl dye control was intravenously injected after 6 days of arthritis induction by passive transfer of K/BxN mouse serum, while nonarthritic controls did not receive serum transfer. The mice were imaged 24 hours later. Direct IR800 fluorescence imaging of the mice injected with IRDye 800CW 2-DG showed a 2.3-fold higher emission in arthritic mouse joints compared to nonarthritic control mouse joints (mean±SEM 397.39±83.22 arbitrary units [AU] versus 170.4±29.8 AU [P<10−5]; n=3 mice per group). Moreover, IRDye 800 CW 2-DG–injected arthritic mouse joints showed a 1.9-fold higher emission than did control dye–injected arthritic mouse joints (mean±SEM 397 ± 83.22 AU versus 211.19±59.38 AU [P<10−5]; n=3 mice per group) (Figures 5A–C). These results are consistent with the findings from PET scans in humans.
Figure 5.

Glucose metabolism, as assessed using fluorescent 2-deoxy-D-glucose (2-DG) as a reporter, is increased in fibroblast-like synoviocytes from the joints of arthritic mice. A–C, Images of IRDye 800CW fluorescence intensity (mean±SD excitation 730±42 nm, emission 800 nm long pass) was assessed 24 hours after intravenous injection of IRDye 800CW carboxyl control dye in arthritic mice (A), IRDye 800CW 2-DG dye in nonarthritic control mice (B), or IRDye 800CW 2-DG dye in arthritic mice (C). D, Control and arthritic mice were injected with IRDye 800CW 2-DG on day 5 after arthritis induction, and 24 hours later, the joints were digested and stained with anti-CD45 and anti-podoplanin (anti-PDPN) antibodies. Plots show the 2-DG staining in the live CD45− cell population. E, Single cells were enriched for CD45−PDPN+ cells. Plots show the 2-DG staining in both the PDPN+ and PDPN− cell populations. F, Populations of CD45−PDPN+ and CD45−PDPN− cells obtained after enrichment were analyzed by quantitative polymerase chain reaction for the expression of selected genes. Values are the mean± SEM of 4 mice per group. G, Sections of the ankle joints of control nonarthritic mice and arthritic K/BxN mice were stained for GLUT1 expression on day 7 after arthritis induction. Representative results are shown. Original magnification × 100 in top panels; × 200 in bottom panels.
To further study glycolysis in FLS, we assessed glucose uptake in cells from the arthritic mouse joints, using fluorescence-activated cell sorting analysis. On day 5 after K/BxN serum injection, mice were injected with IRDye 800CW 2-DG, and 24 hours later, the joints were digested. After a single-cell suspension was obtained, the cells were stained with anti-CD45 and anti-PDPN antibodies. PDPN is a glycoprotein highly expressed in RA FLS (35). Although it is also expressed in other cell populations, such as human lymphatic endothelia, basal epithelial keratinocytes, myoepithelial cells, and myofibroblasts of certain glandular tissues, as well as follicular dendritic cells and fibroblastic reticular cells of lymphoid organs and alveolar type I cells, these cells should not be present or are underrepresented in our sample. In the population of CD45−PDPN+ cells from the arthritic mouse joints, we observed increased uptake of 2-DG, as shown in Figure 5D.
To further characterize this population, we enriched for CD45−PDPN+ cells, and within this population, we found that they incorporated more 2-DG (Figure 5E) and had higher expression of GLUT1 and LDHA mRNA (Figure 5F) than did the CD45−PDPN− cell population. Levels of other enzymes in the glycolytic pathway, such as enolase 1 and pyruvate kinase muscle isoenzyme 2, were also significantly increased in the CD45−PDPN+ cell population (Figure 5F).
We also determined the expression of GLUT1 by immunohistochemical staining of the joints of arthritic and nonarthritic mice. The results showed that GLUT1 mRNA was expressed in the synovial lining cells from the joints of normal (nonarthritic) mice, and was uniformly expressed in the invasive lining of the joints of arthritic mice (Figure 5G and Supplementary Figure 7A, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
We tested 2 doses of BrPa (5 mg/kg and 10 mg/kg) to study the effect of the drug on arthritis in this mouse model. The serum lactate concentration was slightly increased in mice injected with the higher dose of BrPa as compared to PBS-injected mice, but in mice injected with the lower dose of BrPa, the concentration was similar to that in PBS-injected mice (mean±SEM 15.3±1.9 nM in PBS-injected mice, compared to 16.7± 0.9 nM in low-dose BrPa–injected mice and 19.3±0.8 nM in high-dose BrPa–injected mice).
We then injected 5 mg/kg BrPa daily for 6 days after administration of the arthritogenic mouse serum. BrPa-treated mice had significantly lower clinical arthritis scores compared to PBS-injected mice, from day 2 onward (Figure 6A). Importantly, BrPa treatment not only prevented the onset of arthritis, but also successfully suppressed joint swelling in mice when treatment at the dose of 5 mg/kg was initiated in established disease (Figure 6B and Supplementary Figure 7B, http://onlinelibrary.wiley.com/doi/10.1002/art.39608/abstract).
Figure 6.

Glycolytic inhibition decreases histologic scores and levels of inflammatory mediators in the K/BxN mouse model of arthritis. A and B, Clinical arthritis scores were determined in phosphate buffered saline (PBS)–treated animals and 3-bromopyruvate (BrPa)–treated animals after injection of 150 μl of K/BxN mouse serum on day 0. Treatment with BrPa was administered every day from day 0 (arrow) at doses of 5 mg/kg or 10 mg/kg (A) or administered every day from day 4 (arrow) to day 9 at a dose of 5 mg/kg (B). Values are the mean±SEM of 5 mice per group. * =P<0.05; **=P<0.01; ***=P<0.001. C, Histologic scores were determined on day 7 after serum transfer in PBS-treated or BrPA-treated mice. D, Sections of the ankle joints of PBS-treated or BrPA-treated mice were stained with hematoxylin and eosin (H&E) or Safranin O on day 7 after arthritis induction. Black arrows indicate joint inflammation, and white arrows highlight cartilage damage, which was reduced in BrPa-treated mouse ankles. Representative results are shown. Original magnification × 200. E, Sections of the ankle joints of PBS-treated or BrPA-treated mice were stained for GLUT1 expression on day 7 after arthritis induction. Representative results are shown. Original magnification × 200. F, Mice were injected with K/BxN mouse serum to induce arthritis or injected with PBS alone as the nonarthritic control on day 0, and then treated with either PBS or BrPa (5 mg/kg). Levels of interleukin-6 (IL-6) and IL-1β in the mouse joints were analyzed by enzyme-linked immunosorbent assay on day 5 after serum transfer. Values in C and F are the mean±SEM in 5 mice per group.
Histopathologic analysis of the joints on day 7 of treatment showed markedly reduced inflammatory cell infiltration, joint destruction, and cartilage damage in BrPa-treated mice compared to PBS-treated controls (Figures 6C and D). In addition, GLUT1 mRNA expression was also dramatically decreased in BrPa-treated mice (Figure 6E).
Furthermore, to evaluate the influence of BrPa inhibition on the levels of synovial inflammatory mediators, we determined the relative levels of IL-6 and IL-1β in the joints from a separate group of mice on day 5 after serum transfer (day 5 clinical arthritis scores were a mean±SEM 1.8±0.83 in BrPA-treated mice versus 10.2±3.6 in PBS-injected controls; P<0.01). Analysis by enyme-linked immunosorbent assay confirmed that levels of IL-6 and IL-1β were reduced in BrPa-treated arthritic mouse joints (Figure 6F).
DISCUSSION
There are very few studies focused on targeting the metabolism in RA or assessing metabolic changes in synovial stromal cells such as FLS. However, FLS must meet the bioenergetic and biosynthetic demands of increased cell proliferation, and must adapt to inflammatory and hypoxic conditions, which are known to have profound effects on the cellular metabolism needed to support cell growth and survival. In this study, we showed that glucose metabolism is increased in stromal cells in a murine model of arthritis, and that glycolytic inhibition impaired FLS functions and also abrogated joint inflammation and damage in this murine model of RA. The observed correlation between the levels of glycolysis-related genes, such as GLUT1, and FLS functions, such as cell migration or MMP expression, although not causal, also suggest that there is a reliance of FLS on glucose metabolism. Moreover, RA FLS are working closer to their capacity than are OA FLS to meet the demand for ATP by glycolysis, suggesting that they undergo a shift in ATP generation from oxidative phosphorylation to glycolysis, which could be an adaptation to the joint microenvironment, similar to that seen in tumor cells.
After activation with PDGF-BB or LPS, both OA FLS and RA FLS displayed lower respiratory rates and increased glycolytic activity, indicating that this pathway is intrinsically important in FLS metabolism and is essential for FLS pathogenic functions under proinflammatory stimulation. Taken together, the data suggest that glucose metabolism is a potential therapeutic target in RA FLS that could contribute to the aggressive phenotype of these cells in vivo. We propose that inhibiting glucose metabolism could complement current RA therapies by suppressing pathogenic FLS behavior.
The molecular mechanisms leading to constitutive up-regulation of aerobic glycolysis or glycolytic switching after stimulation are not well defined. It is commonly assumed that glucose transporters and HK are the key molecules regulating glycolytic flux (36). Interestingly, in the arthritic synovium of mice, we found that GLUT1 mRNA was homogeneously expressed, suggesting that GLUT1 expression is up-regulated as a result of joint hypoxia and inflammation, a mechanism that could be responsible for supporting the glycolytic environment in the joint.
GLUT1 mRNA expression was also correlated with HK2 mRNA expression in FLS samples. Although abundantly expressed in embryonic tissue, HK2 is expressed at high levels only in a limited number of adult tissue, such as adipose, skeletal, and cardiac muscle tissue. HK2 is thus considered the principal inducible HK form. Moreover, HK2 is known not only to localize in the cytosol but also to bind to the mitochondria (37–39). Interestingly, there is increasing evidence that mitochondrial HKs protect mitochondria against tissue insult (37,38). Upregulation of HK2 and increased HK2-associated mitochondrial levels have been suggested to contribute to cell survival (37,38). Given its restricted distribution of expression in normal adult tissue, further studies are needed to address the role of HK2 in FLS functions and in the resistance to programmed cell death, as HK2 constitutes an attractive potential selective target for arthritis therapy and is safer than global glycolysis inhibition.
Multiple cellular pathways might lead to the glycolytic phenotype (4,5). One of these pathways is the PI3K/Akt pathway, which can be activated by hypoxia and several cytokines and growth factors, and is known to be activated in inflamed synovium (40–42). AMP-activated protein kinase (AMPK) is also a crucial sensor of energy status and has an important pleiotropic role in cellular responses to metabolic stress (41). The induction of glycolysis also requires the activation of hypoxia-inducible factor 1α (HIF-1α), which leads to the transcription of genes (such as HKs) required for glucose uptake and glycolysis (43). Although we could not reverse the glycolytic phenotype by inhibiting Akt signaling, other possible targets, such as inhibition of HIF-1α, AMPK, or the p53 pathway, require further attention. Moreover, epigenetic changes in the critical glycolytic genes under conditions of chronic inflammation might also play a role in the RA FLS glycolytic phenotype.
Of interest, other fibroblast types, such as primary human pulmonary fibroblasts, for which the environment differs from that for FLS, may not need this flexibility in metabolism. Cross-talk between the glycolytic and pentose pathways could potentially also help to adapt the cells to the more hypoxic environment present in the joints but not in the lungs. The lack of response to LPS might be a consequence of the FLS being already maximally stimulated because of environmental exposure or because they are epigenetically altered by this environment. Further experiments are needed to understand the differences between both types of cells.
Although a few studies have addressed the metabolic changes in RA or other autoimmune diseases, immune cells, such as lymphocytes and macrophages, show an increase in the rate of glycolysis during activation (44,45). Metabolism and nutrient availability influence T cell activation and function. Activated T cells show induced expression of, among other factors, GLUT1 and several glycolytic enzymes. Moreover, normalization of T cell metabolism through the dual inhibition of glycolysis and mitochondrial metabolism has been shown to be a promising therapeutic venue in patients with systemic lupus erythematosus (11,44). Whether the macrophage obtains its energy primarily through glycolysis or whether it is obtained through oxidative metabolism can give rise to different phenotypes (45). Classically activated (M1) macrophages are known to rely heavily on glycolysis. M2 macrophages, on the other hand, are involved in tissue repair and wound healing and use oxidative metabolism to fuel their longer-term functions (45). The murine model of arthritis used herein is independent of an adaptive response, but macrophages and other cells of the innate immune response play a role in this model. Thus, although glycolytic inhibitors could potentially be used to target other cell types, our study assesses their effect on FLS.
These results could have a major impact relevant to rheumatic diseases. Our findings may shed light on how changes in FLS metabolism could contribute to the pathogenesis of RA. Metabolic changes in FLS can also potentially change the cross-talk between FLS and other cells in the synovium. We believe that targeting metabolic dysfunction offers a novel approach to understanding the mechanisms of susceptibility to RA, and offers potential new treatment options, which could be administered locally as concerns about systemic toxicity will likely be raised. Taken together, findings from our in vitro and in vivo studies suggest that glycolysis is activated in RA FLS under proinflammatory conditions, and that blocking glycolysis might be beneficial in inflammatory arthritis by suppressing disease-associated FLS functions, including FLS migration, cytokine production, and resistance to apoptosis. Therefore, these data provide a rationale for inhibition of glycolysis as a therapeutic approach in RA.
Supplementary Material
Acknowledgments
Supported by the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases grant 1K08-AR-064834 to Dr. Guma). Additional support was provided by the NIH (grant R01-AI-050265 to Dr. Vicente-Suarez and grants DK-054441-15 and CA-188652 to Dr. Murphy) and by Seahorse Bioscience (to Dr. Murphy). Support for the nuclear magnetic resonance facility was provided by the University of Texas Health Science Center at San Antonio and NIH grant NCI P30-CA-54174 (to the Cancer Therapy and Research Center at the University of Texas Health Science Center at San Antonio). Dr. Garcia-Carbonell’s work was supported by a fellowship from the Boehringer-Ingelheim Foundation.
We thank Beth Friedman for assisting with the Maestro in vivo imaging experiments.
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Guma had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Garcia-Carbonell, Divakaruni, Lodi, Vicente-Suarez, Saha, Cheroutre, Boss, Tiziani, Murphy, Guma.
Acquisition of data. Garcia-Carbonell, Divakaruni, Lodi, Vicente-Suarez, Saha, Cheroutre, Boss, Tiziani, Murphy, Guma.
Analysis and interpretation of data. Garcia-Carbonell, Divakaruni, Lodi, Vicente-Suarez, Saha, Cheroutre, Boss, Tiziani, Murphy, Guma.
ROLE OF THE STUDY SPONSOR
Seahorse Bioscience, which provided the XF analyzer, had no role in the study design or in the collection, analysis, or interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by Seahorse Bioscience.
References
- 1.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–61. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 2.Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233:233–55. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9:24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 5.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol. 2012;13:907–15. doi: 10.1038/ni.2386. [DOI] [PubMed] [Google Scholar]
- 8.Ghesquiere B, Wong BW, Kuchnio A, Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature. 2014;511:167–76. doi: 10.1038/nature13312. [DOI] [PubMed] [Google Scholar]
- 9.McDonald G, Deepak S, Miguel L, Hall CJ, Isenberg DA, Magee AI, et al. Normalizing glycosphingolipids restores function in CD4+ T cells from lupus patients. J Clin Invest. 2014;124:712–24. doi: 10.1172/JCI69571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z, Matteson EL, Goronzy JJ, Weyand CM. T-cell metabolism in autoimmune disease. Arthritis Res Ther. 2015;17:29. doi: 10.1186/s13075-015-0542-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7:274ra18. doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guma M, Sanchez-Lopez E, Lodi A, Garcia-Carbonell R, Tiziani S, Karin M, et al. Choline kinase inhibition in rheumatoid arthritis. Ann Rheum Dis. 2015;74:1399–407. doi: 10.1136/annrheumdis-2014-205696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckhart AD, Beebe K, Milburn M. Metabolomics as a key integrator for “omic” advancement of personalized medicine and future therapies. Clin Transl Sci. 2012;5:285–8. doi: 10.1111/j.1752-8062.2011.00388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor invasion: a multidisciplinary study. Cancer Res. 2006;66:5216–23. doi: 10.1158/0008-5472.CAN-05-4193. [DOI] [PubMed] [Google Scholar]
- 15.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 16.Gaber T, Dziurla R, Tripmacher R, Burmester GR, Buttgereit F. Hypoxia inducible factor (HIF) in rheumatology: low O2! See what HIF can do! Ann Rheum Dis. 2005;64:971–80. doi: 10.1136/ard.2004.031641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer. 2002;2:683–93. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 18.Matsui T, Nakata N, Nagai S, Nakatani A, Takahashi M, Momose T, et al. Inflammatory cytokines and hypoxia contribute to 18F-FDG uptake by cells involved in pannus formation in rheumatoid arthritis. J Nucl Med. 2009;50:920–6. doi: 10.2967/jnumed.108.060103. [DOI] [PubMed] [Google Scholar]
- 19.Vijayant V, Sarma M, Aurangabadkar H, Bichile L, Basu S. Potential of 18F-FDG-PET as a valuable adjunct to clinical and response assessment in rheumatoid arthritis and seronegative spondyloarthropathies. World J Radiol. 2012;4:462–8. doi: 10.4329/wjr.v4.i12.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 21.Alvaro-Gracia JM, Zvaifler NJ, Brown CB, Kaushansky K, Firestein GS. Cytokines in chronic inflammatory arthritis. VI. Analysis of the synovial cells involved in granulocyte-macrophage colony-stimulating factor production and gene expression in rheumatoid arthritis and its regulation by IL-1 and tumor necrosis factor-α. J Immunol. 1991;146:3365–71. [PubMed] [Google Scholar]
- 22.Alvaro-Gracia JM, Zvaifler NJ, Firestein GS. Cytokines in chronic inflammatory arthritis. V. Mutual antagonism between interferon-gamma and tumor necrosis factor-α on HLA-DR expression, proliferation, collagenase production, and granulocyte macrophage colony-stimulating factor production by rheumatoid arthritis synoviocytes. J Clin Invest. 1990;86:1790–8. doi: 10.1172/JCI114908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schipper RG, van den Heuvel LP, Verhofstad AA, De Abreu RA. Polyamines and DNA methylation in childhood leukaemia. Biochem Soc Trans. 2007;35:331–5. doi: 10.1042/BST0350331. [DOI] [PubMed] [Google Scholar]
- 24.Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. Caspase 1–independent activation of interleukin-1β in neutrophil-predominant inflammation. Arthritis Rheum. 2009;60:3642–50. doi: 10.1002/art.24959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach Ml, et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci U S A. 2013;110:5422–7. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta. 2001;1504:46–57. doi: 10.1016/s0005-2728(00)00238-3. [DOI] [PubMed] [Google Scholar]
- 27.Cardaci S, Desideri E, Ciriolo MR. Targeting aerobic glycolysis: 3-bromopyruvate as a promising anticancer drug. J Bioenerg Biomembr. 2012;44:17–29. doi: 10.1007/s10863-012-9422-7. [DOI] [PubMed] [Google Scholar]
- 28.Fridman A, Saha A, Chan A, Casteel DE, Pilz RB, Boss GR. Cell cycle regulation of purine synthesis by phosphoribosyl pyrophosphate and inorganic phosphate. Biochem J. 2013;454:91–9. doi: 10.1042/BJ20130153. [DOI] [PubMed] [Google Scholar]
- 29.Saha A, Connelly S, Jiang J, Zhuang S, Amador DT, Phan T, et al. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol Cell. 2014;55:264–76. doi: 10.1016/j.molcel.2014.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315:1006–10. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- 31.Kyburz D, Corr M. The KRN mouse model of inflammatory arthritis. Springer Semin Immunopathol. 2003;25:79–90. doi: 10.1007/s00281-003-0131-5. [DOI] [PubMed] [Google Scholar]
- 32.Ko YH, Verhoeven HA, Lee MJ, Corbin DJ, Vogl TJ, Pedersen PL. A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: from bench side to bedside. J Bioenerg Biomembr. 2012;44:163–70. doi: 10.1007/s10863-012-9417-4. [DOI] [PubMed] [Google Scholar]
- 33.Pedersen PL. 3-Bromopyruvate (3BP) a fast acting, promising, powerful, specific, and effective “small molecule” anti-cancer agent taken from labside to bedside: introduction to a special issue. J Bioenerg Biomembr. 2012;44:1–6. doi: 10.1007/s10863-012-9425-4. [DOI] [PubMed] [Google Scholar]
- 34.Shoshan MC. 3-Bromopyruvate: targets and outcomes. J Bioenerg Biomembr. 2012;44:7–15. doi: 10.1007/s10863-012-9419-2. [DOI] [PubMed] [Google Scholar]
- 35.Ekwall AK, Eisler T, Anderberg C, Jin C, Karlsson N, Brisslert M, et al. The tumour-associated glycoprotein podoplanin is expressed in fibroblast-like synoviocytes of the hyperplastic synovial lining layer in rheumatoid arthritis. Arthritis Res Ther. 2011;13:R40. doi: 10.1186/ar3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–57. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 37.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–18. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–30. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 39.Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25:4683–96. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- 40.Morel J, Audo R, Hahne M, Combe B. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces rheumatoid arthritis synovial fibroblast proliferation through mitogen-activated protein kinases and phosphatidylinositol 3-kinase/Akt. J Biol Chem. 2005;280:15709–18. doi: 10.1074/jbc.M414469200. [DOI] [PubMed] [Google Scholar]
- 41.Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015;22:248–57. doi: 10.1038/cdd.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM, Karagiannis TC. Cancer metabolism and the Warburg effect: the role of HIF-1 and PI3K. Mol Biol Rep. 2015;42:841–51. doi: 10.1007/s11033-015-3858-x. [DOI] [PubMed] [Google Scholar]
- 44.Maciolek JA, Pasternak JA, Wilson HL. Metabolism of activated T lymphocytes. Curr Opin Immunol. 2014;27:60–74. doi: 10.1016/j.coi.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 45.Galvan-Pena S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420. doi: 10.3389/fimmu.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.