

Abstract
Polymer nanofiber based materials have been widely investigated for use as tissue engineering scaffolds. While promising, these materials are typically fabricated through techniques that require significant time or cost. Here we report a rapid and cost effective air-brushing method for fabricating nanofiber scaffolds using a simple handheld apparatus, compressed air, and a polymer solution. Air-brushing also facilities control over the scaffold degradation rate without adversely impacting architecture. This was accomplished through a one step blending process of high (Mw ≈ 100 000 g mol−1) and low (Mw ≈ 25 000 g mol−1) molecular weight poly(DL-lactide) (PDLLA) polymers at various ratios (100:0, 70:30 and 50:50). Through this approach, we were able to control fiber scaffold degradation rate while maintaining similar fiber morphology, scaffold porosity, and bulk mechanical properties across all of the tested compositions. The impact of altered degradation rates was biologically evaluated in human bone marrow stromal cell (hBMSC) cultures for up to 16 days and demonstrated degradation rate dependence of both total DNA concentration and gene regulation.
Keywords: nanofiber, degradation, tissue engineering scaffold, air-brush, solution blow spinning
1. Introduction
Polymeric nanofiber scaffolds have been extensively studied for use in tissue engineering applications. Nanofiber scaffolds are inherently high in porosity and have a high surface to volume ratio, allowing for cell attachment and in-growth (Pham et al 2006). Nanofibers promote cell migration and integration by mimicking endogenous extracellular matrix (ECM) structure and function (Xu et al 2004). Electrospinning is the most widely investigated technique for generating nanofiber-based scaffolds (Sill et al 2008). While useful, the electrospinning process suffers from low deposition rates, requires high voltages, conductive targets, and is typically only used for deposition onto planar substrates. These limitations, as well as concerns with scalability, have led to the investigation of other fabrication strategies.
More scalable approaches including rotary jet spinning (Badrossamay et al 2010), melt spinning (Zuo et al 2013), nanofiber production by gas jet (Benavides et al 2012), and melt extrusion (Kim et al 2014), have all been investigated to varying success. These fabrication methods are well suited for producing scaffolds at scale but often require specialized laboratory equipment. Furthermore, the ability to construct on-demand scaffolds has not been demonstrated or explored. Airbrushing (also known as solution blow spinning) is an alternative technique that allows for rapid nanofiber deposition using a compressed gas, a concentric nozzle, and concentrated polymer solution (Medeiros et al 2009). Fabrication of polymer fiber scaffolds with this technique has been demonstrated in medical (Tutak et al 2013, Behrens et al 2014, 2015, Hoffman et al 2015) and non-medical (Zhuang et al 2013, Vural et al 2015) applications using a commercial air-brush or a simple apparatus. A low equipment cost coupled with a rapid deposition rate makes solution blow spinning a promising fabrication method for developing clinically translatable tissue engineering scaffolds.
Polymer solution concentration, solvent, and viscosity are the major determinants for generating a nanofiber scaffold through this technique. A polymer solution concentration above the chain overlap condition is required for fiber scaffold generation (Zhuang et al 2013). By changing polymer type, molecular weight, or through the use of polymer blends, properties such as polymer crystallization kinetics, bulk scaffold Young’s modulus, and degradation rate can be tailored for specific applications (Kim et al 2003, Dong et al 2009, Andjelic et al 2015).
Control over degradation is required for successful implementation of any tissue engineering scaffold. Effective scaffolds must retain the desired physical and chemical properties over their functional lifetime, thereby allowing for tissue ingrowth and regeneration while providing structural support. Degradation must be finely tuned to promote early stage cell migration and/or differentiation while matching the rate of tissue ingrowth as to not impede later stage reconstruction (Zhang et al 2014). Through the use of synthetic polymers, degradation rate can be controlled through a variety of routes including end group functionalization, or varying copolymer composition or molecular weight (Langer et al 1983, Siepmann et al 2001). These modifications regularly affect other design parameters such as mechanical strength and may introduce variability in the fabrication process, thereby sacrificing the ability to accurately control scaffold architecture without significant effort and optimization.
Here we report a method for rapidly generating degradable poly(DL-lactide) (PDLLA) nanofiber scaffolds by solution blow spinning with a simple custom built co-axial air-brush (figures 1 and S1) (stacks.iop.org/BMM/11/035001/mmedia). Through a simple one step blending process using polymers of varying molecular weights, we were able to control the rate of in vitro degradation while maintaining comparable fiber diameter, Young’s modulus, and percent porosity across all compositions. The effect of degradation rate on cellular response was investigated by performing transcriptional analysis and measuring total deoxyribonucleic acid (DNA) concentration using human bone marrow stromal cells (hBMSCs). This combined material and fabrication approach provides a powerful and attainable tool for studying and developing tissue engineering scaffolds.
Figure 1.

(A) Custom made air-brush apparatus used for nanofiber deposition. White arrow indicates the nozzle of the apparatus. (B)–(D) SEM of air-brushed 12% (w/w) PDLLA blends in acetone. (B) 100:0 high molecular weight: low molecular weight, (C) 70:30, (D) 50:50. Scale bars represent 1 μm.
2. Materials and methods
2.1. Materials
Solutions of 12% (w/w) PDLLA Resomer R 207 S (1.7–2.6 dl g−1]) and R 203 H (0.25–0.35 dl g−1) (PDLLA-COOH) in acetone (Sigma Aldrich) were prepared at 100:0, 70:30, and 50:50 ratios. The fibers were synthesized at ~15 psi air at 30 standard cubic feet per hour (SCFH) or 103.4 kPa at 14.2 l min−1. Gas pressure was measured using an USG pressure gauge (pressure range: 0–1379 kPa (0–200 PSI)) mounted on a ‘Victor’ gas reducer. Air flow was controlled using a Cole-Parmer air flow meter (flow range: 14.16–141.58 l min−1 (30–300 SCFH)).
A design of the custom airbrush was inspired by a layout outlined in a publication by Medeiros et al (Medeiros et al 2009). Our design had slightly different ratio between inner/outer nozzle diameters (0.55) and featured a stainless steel needle (G 22, McMaster Carr) to deliver the polymer solution (figures 1 and S1) (Tutak et al 2015). The nozzle was made out of a commercially available 1/8 inch brass water plug and the brush housing was machined from an aluminum block. The needle was centered in the middle of the device, threaded through the nozzle (protruding 1–2 mm) and sealed in the back to prevent gas escape. Fibers were deposited by supplying a polymer solution using a syringe pump (NE 1000 X, New Era System Inc., Farmingdale, NY) at a feed rate of 10 ml h−1 for ≈8 min.
2.2. Fiber collection and material analysis
All of the polymer fiber specimens were collected on a fiberglass or teflon mesh 15–20 cm away from the tip of the nozzle. After deposition, the scaffolds, were removed and sectioned into samples used for the degradation study, morphological analysis, and mechanical testing.
For cell culture studies, the nanofiber scaffolds were deposited directly onto 12 mm tissue culture polystyrene (TCPS) discs. The discs were hot-punched from the bottom of TCPS petri dishes (100 mm dia.) and placed on the supporting metal mesh. After the fiber deposition was completed, the scaffolds were heat sealed around the edges and stored in a desiccator under vacuum for later use.
2.3. Scanning electron microscopy (SEM) imaging
PDLLA nanofiber scaffolds were cut into 2 × 5 mm pieces, were mounted onto SEM stubs using carbon tape (Ted Pella, Inc.), placed under house vacuum overnight with desiccant, and gold sputter-coated (120 s at 75 mA, Desk V HP, Denton Vacuum) with approximately 10 nm of gold. Six scanning electron micrographs, three high mag (13 000 × or 15 000×) and three low mag (500×), were captured of each sample (Hitachi S4700 SEM, 3 kV, 7 mA, ≈13 mm working distance). Images were taken at 13 000 × and 15 000 × magnification.
2.4. Degradation
The fabricated fibrous scaffolds were cut into 1.5 × 1.5 cm squares and weighed to select samples with comparable size and weight. Samples were enclosed into single plastic cassettes (with large perfusion holes and a stainless steel washer to weigh each cassettes down), hung on stainless steel wire, and placed in separate glass vials. The cassettes were submerged in 50 ml of 1× phosphate buffered saline (PBS, 10010–023) placed on a stirring plate (Variomag-Poly) at 250 rpm and kept in large incubator at 37 °C. The PBS was not changed throughout the duration of the study.
Experimental groups were removed from the stirring plate at time points of 0, 2, 4, 6, 8, and 12 weeks. PBS was removed, samples were rinsed with DI water, and then samples were dried and stored in a vacuum desiccator. For each time point, molecular weight distribution was determined by gel permeation chromatography (GPC) using a Waters Alliance Separations Module e2695, Waters 2414 refractive index (RI) Detector, and Waters HSPgel columns in series (HR MB-L and HR 3.0 columns, 6.0 mm I.D. × 15 cm). Both of the columns and the RI detector were heated to 30 °C during all data acquisition. Samples were dissolved at 3 mg ml−1 in tetrahydrofuran (THF) and filtered with Whatman Mini-Uniprep 0.2 μm polytetrafluoroethylene (PTFE) membrane filters. THF was also used as the eluent at a flow rate of 0.6 ml min−1. Molecular weights are reported as polystyrene relative molecular weights, as calculated from a 10-point calibration curve generated using Agilent EasiCal polystyrene standards. Each sample was injected 3 separate times with a 25 μl injection volume, data is presented as the average of these injections. All data analysis was performed using Waters Empower 3 Chromatography Data software. Samples were imaged at each time point using scanning electron microscopy (SEM, S-4700-II FE-SEM, Hitachi). Prior to imaging, the samples were sputter-coated with thin layer of gold (Denton Vacuum Desk II, 15 kV, 70 s). pH was also monitored over 3 weeks of degradation in either PBS that was not changed and in cell culture medium that was changed weekly. These samples were also imaged by SEM.
2.5. Nanofiber diameter and scaffold morphology assessment
SEM micrographs of PLGA nanofibers were assessed using an ImageJ (Schneider et al 2012) plugin called DiameterJ and a previous published method (Hotaling et al 2015). Briefly, nanofiber diameter histograms were obtained using DiameterJ. All histograms from replicate images of treatments were combined and the peaks of the histogram were then fit using a Gaussian peak fitting in Fityk (Wojdyr 2010). The means (centers) and standard deviations of the peaks were then calculated and recorded. Fiber diameter for each of the polymer blends was calculated based on n ≥ 20 000 measurements. Pore size histograms were constructed by analyzing all images at the same magnification, working distance, brightness/contrast settings on the SEM, using the same segmentation algorithms, and finding the area of each pore not touching the image edge. Pore areas were then combined from replicate images of treatments and histograms were constructed by taking the pore size range and dividing it into 250 bins, or 150 nm2 bins, and finding the frequency of pore area occurrence within each bin. Pore size distribution data was based on n ≥ 100 measurements. A logarithmic binning of pore sizes was also performed to examine pore size distributions in four distinct area ranges. Additionally, percent porosity, intersection density, and characteristic length of fibers were all reported in the software and averaged across all replicate images per treatment. Percent porosity was defined as the total pore area segmented at the given focal plane divided by the total image resolution. Intersection density was defined as the number of fiber intersections divided by the total area of the image. Characteristic length was defined as the mean fiber length between intersections.
2.6. Mechanical testing
Tensile tests were performed using a TA Instruments RSA3 with fiber/film tools installed. Samples (n ≥ 3) were 2 cm long, 1 cm wide, and 200–300 μm thick. A constant elongation of 0.2 mm min−1 was used and the normal force transducer was set in the low range. Young’s Modulus was reported by taking the slope in the linear region at low strains.
2.7. Cell culture
Prior to the cell culture experiments, the scaffold/TCPS discs were sterilized by exposure to UV (~3 min) on each side (top/bottom), rinsed one time in 70% ethanol (v/v) and three times with PBS. The discs were dried and mounted over vacuum grease (Corning) in 48-well polystyrene plates (Corning, NY). Primary human bone marrow stromal cells (hBMSCs (donor 7083, 24 years old, male, P4)) were counted using a hemocytometer and concentration was adjusted to seed 10 000 cells per scaffold (n = 3). The cells were cultured using 5% (v/v) CO2 at 37 °C in α-minimum essential medium (Invitrogen) containing 16.5% (v/v) fetal bovine serum (Atlanta Biologicals) and 4 mmol L-glutamine (Kumar et al 2011) changed weekly. The cells were cultured up to 21 d without using osteogenic supplements.
2.8. Cell DNA content
Picogreen® DNA assay kit (Invitrogen) was utilized to quantify cellular DNA on experimental scaffolds. Specimens were rinsed with PBS and then treated with lysis buffer (PBS with 0.175 U ml−1 Papain and 14.5 mmol L-cysteine) for 17 h at 60 °C to produce cellular lysate. The lysate was then transferred to a 96-well plate (0.2 ml/well), mixed with of Picogreen® reagent (0.2 ml/well) and measured using a plate reader to assess DNA concentration (excitation 485 nm, emission 538 nm). Cell-scaffold constructs were collected at day 1, 7, 12, and 16 d to determine total amount of cell DNA for each scaffold. Total DNA concentration was determined by constructing a standard curve from known DNA concentrations.
2.9. Cell fluorescent staining
Cells were stained by fixing cell cultures with 3.7% (v/v) formaldehyde in PBS for 15 min then washed with PBS. Cells were then treated with 0.2% (v/v) Triton X-100 in PBS for 5 min and then washed twice with PBS. The scaffold-seeded cells, were fluorescently stained with F-actin stain (AlexaFluor 546-phalloidin, Invitrogen) for 30 min, washed with PBS and air-dried. Samples were then imaged with a Nikon TE200 inverted fluorescent microscope.
2.10. Quantitative RT-PCR
Total RNA was purified from hBMSCs using the RNeasy Kit (Qiagen, Valencia, CA) followed by DNase treatment (Promega, San Luis Obispo, CA) to eliminate contaminating genomic DNA. Complementary DNA (cDNA) was synthesized using iScript cDNA Synthesis kit (Bio Rad, Hercules, CA) according to the manufacturer’s protocol. For the polymerase chain reaction (PCR) 2X iQ SYBR Green Supermix (Bio Rad, Hercules, CA) was used with the CFX96 Real-Time PCR Detection System (Bio Rad, Hercules, CA). Reactions were done in technical triplicates using 1 μl of cDNA as a template in a 20 μl reaction volume. The amount of starting cDNA was normalized to Gapdh. Each experiment was repeated three times with biological triplicates. Real-Time PCR primers were designed using Primer3 software. Heat maps were generated by using PlotLy software. Fold change for hBMSCs grown on PDLLA nanofiber scaffolds was calculated for days 12, 16 and 21 using day 7 as a baseline. Degree of change is represented using a heat map, red being the most change and blue being the least. The y-axis represents the names of genes in the row.
2.11. Data analysis and statistics
A 1-way ANOVA with Tukey’s Posttest (R v3.2.1) was used to analyze data for each deposition type (table S2). The skewness, kurtosis, and data residuals were used to assess data normality and found to indicate all had a normal data distribution (skewness/kurtosis < |x| where −1 ≤ x ≤ 1 and no trend in plotted residuals). Statistical significance level was p = 0.05.
3. Results and discussion
An air-brush was constructed with concentric nozzles in which compressed air at 30 SCFH (≈100 kPa) flows through the outer nozzle and a 12% (w/w) PDLLA solution in acetone was fed through the center nozzle at a rate of 10 ml h−1 (figure 1(A)). A simple one step approach to controlling degradation rate was carried out by preparing a series of high and low molecular weight polymer blends. Relatively higher molecular weight PDLLA (Mw ≈ 100 000 g mol−1) and lower molecular weight carboxylic acid terminated PDLLA (PDLLA-COOH, Mw ≈ 25 000 g mol−1) were blended in ratios of 100:0, 70:30, and 50:50 (PDLLA:PDLLA-COOH). These ratios were confirmed by GPC and are listed in table S1. All of the compositions tested with these conditions were able to generate polymer nanofibers (figures 1(B)–(D)). Polymer blending can be utilized to adjust polymer fiber degradation rates while improving material processability. The Low MW polymers presumably act as plasticizers and improve scaffold quality by forming longer fibers (Andjelic et al 2015).
PDLLA blend composition drastically influenced scaffold degradation rate. Degradation was evaluated over 12 weeks for morphologic and molecular weight change in vitro (figure 2). Incorporation of the relatively lower molecular weight PDLLA resulted in higher degradation rate for both the 50:50 blend (−3000 g mol−1 week−1) and the 70:30 blend (−3600 g mol−1 week−1) when compared to the 100:0 composition (−1300 g mol−1 week−1). The 50:50 and 70:30 polymer blends lost approximately 60% of their initial molecular weight in comparison with the 100:0 composition that only lost 12% of its initial molecular weight at week 12 (figures 2(A)–(C)). During this time frame, scaffold morphology noticeably changed. The 100:0 composition was the least drastically changed over 12 weeks, maintaining fibrous morphology (figure 2(D)). Both the 70:30 and 50:50 blends lost a majority or all of their fiber morphology by 12 weeks due to faster degradation rates (figures 2(E) and (F)). The collected SEM Images and GPC data suggests that the fibers degraded throughout the scaffolds uniformly and therefore likely underwent bulk degradation. PDLLA fiber scaffolds typically experience bulk material degradation due to the high surface to volume ratio of the fibers (Kim et al 2003, Zong et al 2003, Bini et al 2004, Andjelic et al 2015).
Figure 2.
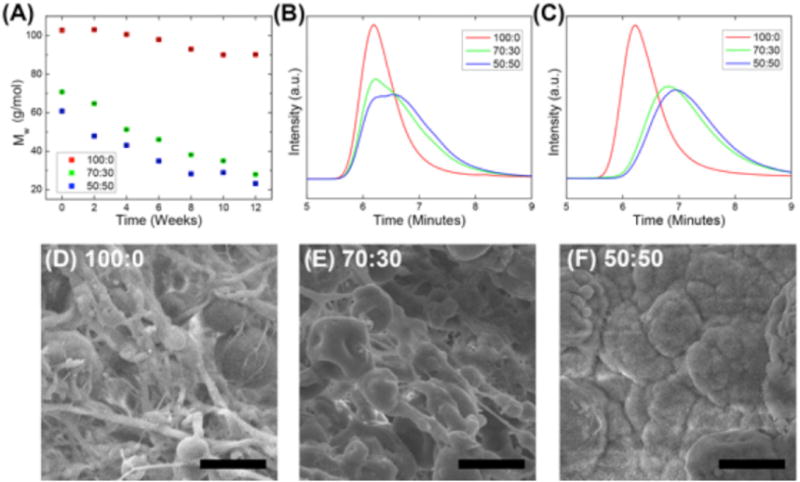
(A) Weight average molecular weight decrease overtime for varying polymer blends in vitro. (B) and (C) GPC of polymer blends at week 0 (B) and week 12 (C). (D)–(F) SEMs of PDLLA nanofiber degradation after 12 weeks for each blend. Scale bars represent 20 μm.
Initial nanofiber diameter was not significantly altered with changing polymer composition. The nanofiber scaffolds had average fiber diameters and standard deviations of 165 ± 50, 256 ± 90, and 129 ± 40 nm, for 100:0, 70:30, and 50:50, respectively. Distribution of fiber diameters was found to be bimodal (figures 3(A) and (B)). Figure 3(A) shows the raw histogram of occurrence of fiber diameter for each ratio of PDLLA and figure 3(B) shows the fitted peaks of these histograms. When comparing the first peak or second peak of each treatment, no statistical difference in diameter was found between any of the groups (supplementary table S2 provides a statistical summary and P-value comparisons). In general, each polymer solution composition (100:0, 70:30, 50:50) had the highest frequency of fiber diameter between 130 and 260 nm and then a secondary smaller peak that occurred between 250 and 550 nm. The second smaller peak was due to the relatively smaller homogeneous fibers frequently banding together to form segments of larger diameter fibers that then disperse again, as shown in supplementary figure S2.
Figure 3.
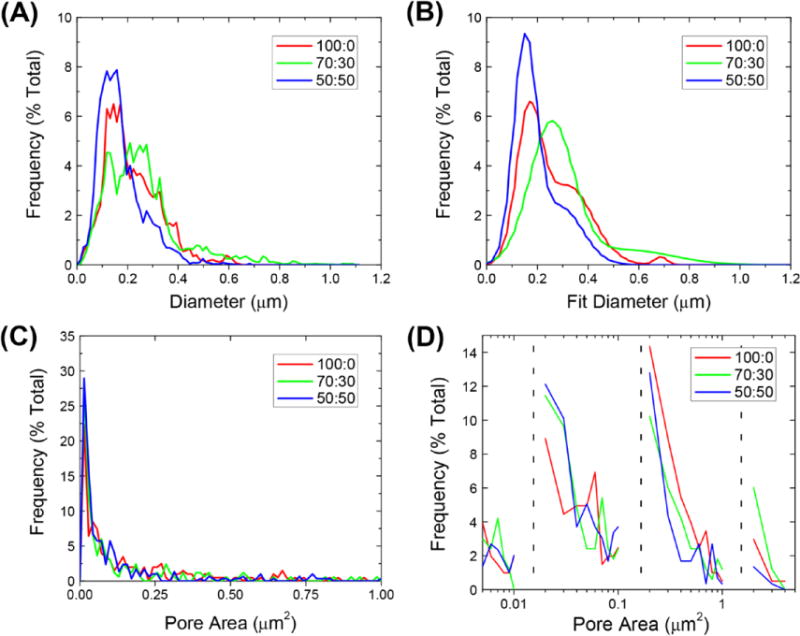
(A) Raw nanofiber diameter distributions. (B) Fitted nanofiber distributions. (C) Raw pore area distributions. (D) Binned pore areas.
The micro-porosity (pore areas <100 μm2) of the nanofiber scaffolds was also found to be statistically similar across the different polymer compositions (table S2). Pores displayed an exponential decrease in frequency of pore area (figure 3(C)). While large pores can lead to significantly altered bulk mechanical properties, pores in this size range do not provide insight into a cells local environment when interacting with the scaffold (Karageorgiou et al 2005). Thus, the pore areas were logarithmically binned to identify trends in four distinct cell relevant size regimes. The first bin shows increments of 0.001 μm2 increments, the second bin size has increments of 0.010 μm2, the third bin has increments of 0.100 μm2, and the fourth bin has increments of 1.0 μm2 (figure 3(D)). In the smallest regime pore sizes were relatively normally distributed, while in larger regimes an exponential decrease in pore size was seen. This indicates that at each scale the smallest pore size was the most prevalent. The logarithmic binning of the pore areas was chosen to represent four different size scales that have relevance to a cell: 10 s of nanometers (Park et al 2007), 100 s of nanometers (You et al 2010), submicron, and micrometer scale for cellular migration and patterning (Markert et al 2009, Olivares-Navarrete et al 2010). The trends within each one of these regimes follows expected trends for area fractions of inclusions in hierarchically structured solids (Coppens et al 2001). Complete scaffold morphology metrics are contained in table 1. The mechanical properties did not significantly vary with polymer composition (figure 4). Young’s modulus was calculated by the slope of the linear region of each stress–strain curve. The nanofiber scaffolds had average Young’s moduli of 19.8 ± 3.2, 20.1 ± 2.7, and 24.2 ± 5.6 kPa, for 100:0, 70:30, and 50:50, respectively.
Table 1.
Scaffold morphology metrics.
| 100:0 | 70:30 | 50:50 | |
|---|---|---|---|
| P1 diameter (μm) | 0.165 ± 0.050 | 0.256 ± 0.088 | 0.129 ± 0.040 |
| P2 diameter (μm) | 0.322 ± 0.098 | 0.552 ± 0.18 | 0.252 ± 0.075 |
| Mean pore size (μm2) | 0.26 ± 0.08 | 0.29 ± 0.13 | 0.15 ± 0.05 |
| Percent porosity (%) | 46.8 ± 6.2 | 38.0 ± 1.2 | 48.2 ± 10.0 |
| Intersection density (Int./μm2) | 2.4 ± 1.5 | 2.0 ± 1.1 | 4.8 ± 2.0 |
| Characteristic length (μm) | 1040 ± 290 | 1150 ± 280 | 680 ± 100 |
n = 3 for each polymer ratio.
Table lists the average and standard deviation for tested polymer blends.
Figure 4.
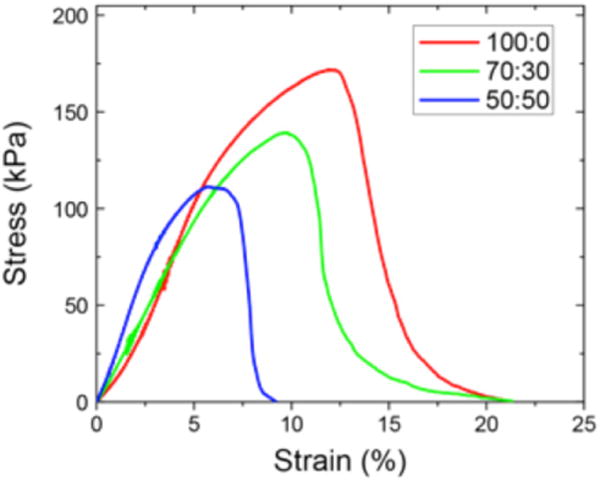
Representative stress/strain curves of nanofiber scaffolds composed of the various polymer blends.
With initial nanofiber diameter, scaffold porosity, and mechanical properties remaining largely unchanged with alteration in polymer composition, the impact of the degradation rate was evaluated in vitro. 100:0 and 70:30 nanofiber scaffolds were selected for cell culture experiments because the scaffolds had similar average fiber diameter, pore area, and Young’s modulus, but greatly differed in degradation rate. hBMSCs were cultured on either 100:0 or 70:30 nanofiber scaffolds for up to 21 d without using osteogenic supplements. In order to ensure that nanofiber scaffold degradation did not alter in the cell culture conditions when compared to PBS, both change in pH and scaffold morphology were monitored over the same 21 d period. Neither pH nor morphology changed drastically over this timescale (supplementary figures S4–S6). Total DNA concentration indicated that at day 1 and 5 hBMSC cell number was comparable (figure 5(A)). At day 16, cells seeded on the 100:0 nanofiber scaffolds appeared to have higher overall number as indicated by a greater total DNA concentration. However, the results were not statistically significant and the high standard deviation values could have been caused by differences in scaffold morphology after degradation and non-uniform cell penetration (supplementary figure S3).
Figure 5.
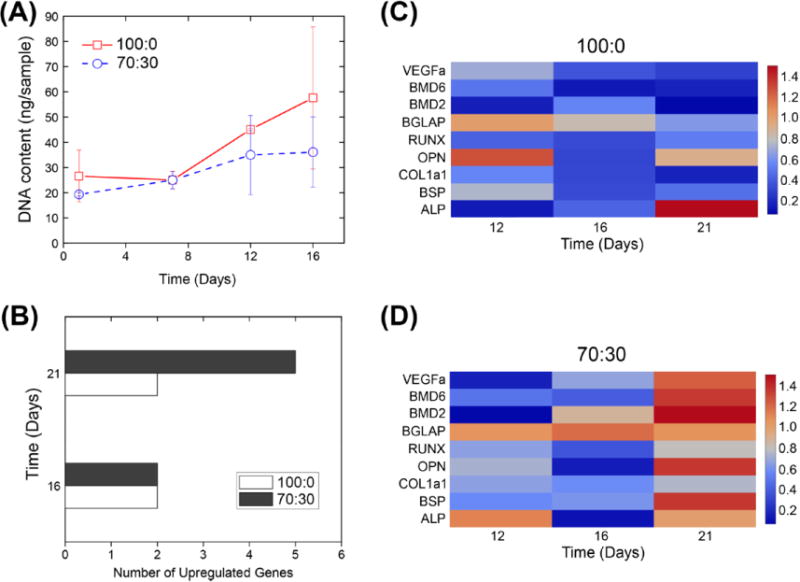
(A) Total DNA content over time. Error bars represent standard deviation (n = 3). (B) Total number of upregulated genes (n = 3). (C) and (D) Heat maps of gene upregulation in hBMSCs in response to the 100:0 (C) and 70:30 (D) polymer blend nanofiber scaffolds at 12, 16, and 21 d.
At time points of 12, 16, and 21 d, real-time PCR was used to evaluate the expression of 9 genes that have implications in bone osteogenesis and vascular formation (figures 5(B)–(D)). Gene expression fold change as calculated from average cycle threshold values was used to generate heat maps (figures 5(C)–(D)) The gene expression patterns of hBMSCs grown on 100:0 nanofiber scaffolds remained stable and largely unchanged over the 21 d period. However, 5 out of 9 genes of hBMSCs grown on 70:30 nanofiber scaffolds began to be upregulated by day 21. Differences in gene regulation and total DNA concentration showed the nanofiber scaffold degradation rate has biological effects on cell growth and bone formation in hBMSCs. The use of a polymer blend consisting of PDLLA and PDLLA-COOH introduces the possibility for differences in surface chemistry leading to the described changes in cell response. Degradation is still likely the dominant mechanism of influence as carboxylic acid end groups are generally associated with faster degradation rates (Lee et al 2003). Additionally, upon hydrolytic degradation carboxylic acid groups are generated and should have a similar or greater effect than the original end groups (Gentile et al 2014). Overall, these results demonstrate the applicability of the polymer blending approach to easily control scaffold degradation through the rapid fabrication polymer nanofiber based scaffolds by air-brushing. Other important relationships such as the effect of polymer chemistry or fiber/scaffold structural degradation on cellular response will be explored in more detail in future research.
4. Conclusions
Polymer nanofiber constructs have been widely investigated for use as tissue engineering scaffolds. However, fabrication of polymer fiber based scaffolds with tuned degradation rates is often resource and time intensive. Here we demonstrated a rapid fabrication method through the use of a simple custom-made apparatus and compressed air. We have also shown the ability to tune the degradation rate of these scaffolds with a one step process using polymer blends. The use of blends did not lead to significant differences in scaffold micro-porosity, nanofiber diameter, or Young’s modulus. The biological effects of degradation rate were assessed through investigating hBMSC gene expression in vitro, showing upregulation of a variety of genes in cells grown on faster degrading scaffolds. This facile and versatile approach allows for inexpensive, tunable, and potentially scalable fabrication of tissue engineering scaffolds.
Supplementary Material
Acknowledgments
We would like to acknowledge the American Dental Association Foundation (ADAF) and the Warren Citrin Graduate Fellowship for supporting this work. Research reported in this publication was also supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award No. R01EB019963. AMB was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award No. F31EB019289. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplementary material for this article is available online
Notes
The authors declare no competing financial interest. Official Contribution of the National Institute of Standards and Technology; not subject to copyright in the United States.
Disclaimer
Certain commercial equipment, instruments, or materials are identified in this paper in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by the National Institute of Standards and Technology, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
References
- Andjelic S, Scogna RC. Polymer crystallization rate challenges: the art of chemistry and processing. J Appl Polym Sci. 2015;132:42066. [Google Scholar]
- Badrossamay MR, McIlwee HA, Goss JA, Parker KK. Nanofiber Assembly by rotary jet-spinning. Nano Lett. 2010;10:2257–61. doi: 10.1021/nl101355x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens AM, Casey BJ, Sikorski MJ, Wu KL, Tuta KW, Sandler AD, Kofinas P. In situ deposition of PLGA nanofibers via solution blow spinning. ACS Macro Lett. 2014;3:249–54. doi: 10.1021/mz500049x. [DOI] [PubMed] [Google Scholar]
- Behrens AM, Lee NG, Casey BJ, Srinivasan P, Sikorski MJ, Daristotle JL, Sandler AD, Kofinas P. Biodegradable-polymer-blend-based surgical sealant with body-temperature-mediated adhesion. Adv Mater. 2015;27:8056–61. doi: 10.1002/adma.201503691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides RE, Jana SC, Reneker DH. Nanofibers from scalable gas jet process. ACS Macro Lett. 2012;1:1032–6. doi: 10.1021/mz300297g. [DOI] [PubMed] [Google Scholar]
- Bini TB, Gao SJ, Xu XY, Wang S, Ramakrishna S, Leong KW. Peripheral nerve regeneration by microbraided poly(L-lactide-co-glycolide) biodegradable polymer fibers. J Biomed Mater Res A. 2004;68A:286–95. doi: 10.1002/jbm.a.20050. [DOI] [PubMed] [Google Scholar]
- Coppens MO, Sun JH, Maschmeyer T. Synthesis of hierarchical porous silicas with a controlled pore size distribution at various length scales. Catal Today. 2001;69:331–5. [Google Scholar]
- Dong YX, Liao S, Ngiam M, Chan CK, Ramakrishna S. Degradation behaviors of electrospun resorbable polyester nanofibers. Tissue Eng B: Rev. 2009;15:333–51. doi: 10.1089/ten.TEB.2008.0619. [DOI] [PubMed] [Google Scholar]
- Gentile P, Chiono V, Carmagnola I, Hatton P. An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int J Mol Sci. 2014;15:3640–59. doi: 10.3390/ijms15033640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman K, Skrtic D, Sun JR, Tutak W. Airbrushed composite polymer Zr-ACP nanofiber scaffolds with improved cell penetration for bone tissue regeneration. Tissue Eng C: Methods. 2015;21:284–91. doi: 10.1089/ten.tec.2014.0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotaling NA, Bharti K, Kriel H, Simon CG. Diameter J: a validated open source nanofiber diameter measurement tool. Biomaterials. 2015;61:327–38. doi: 10.1016/j.biomaterials.2015.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karageorgiou V, Kaplan D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials. 2005;26:5474–91. doi: 10.1016/j.biomaterials.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kim SE, Wang J, Jordan AM, Korley LTJ, Baer E, Pokorski JK. Surface modification of melt extruded poly(epsilon-caprolactone) nanofibers: toward a new scalable biomaterial scaffold. ACS Macro Lett. 2014;3:585–9. doi: 10.1021/mz500112d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Yu M, Zong XH, Chiu J, Fang DF, Seo YS, Hsiao BS, Chu B, Hadjiargyrou M. Control of degradation rate and hydrophilicity in electrospun non-woven poly(D,L-lactide) nanofiber scaffolds for biomedical applications. Biomaterials. 2003;24:4977–85. doi: 10.1016/s0142-9612(03)00407-1. [DOI] [PubMed] [Google Scholar]
- Kumar G, Tison CK, Chatterjee K, Pine PS, McDaniel JH, Salit ML, Young MF, Simon CG. The determination of stem cell fate by 3D scaffold structures through the control of cell shape. Biomaterials. 2011;32:9188–96. doi: 10.1016/j.biomaterials.2011.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer R, Peppas Chemical and physical structure of polymers as carriers for controlled release of bioactive agents—a review. J Macromol Sci, Rev Macromol Chem Phys. 1983;C23:61–126. [Google Scholar]
- Lee JW, Gardella JA. Simultaneous time-of-flight secondary ion MS quantitative analysis of drug surface concentration and polymer degradation kinetics in biodegradable poly(L-lactic acid) blends. Anal Chem. 2003;75:2950–8. doi: 10.1021/ac034305i. [DOI] [PubMed] [Google Scholar]
- Markert LD, et al. Identification of distinct topographical surface microstructures favoring either undifferentiated expansion or differentiation of murine embryonic stem cells. Stem Cells Dev. 2009;18:1331–42. doi: 10.1089/scd.2009.0114. [DOI] [PubMed] [Google Scholar]
- Medeiros ES, Glenn GM, Klamczynski AP, Orts WJ, Mattoso LH. Solution blow spinning: a new method to produce micro-and nanofibers from polymer solutions. J Appl Polym Sci. 2009;113:2322–30. [Google Scholar]
- Olivares-Navarrete R, Hyzy SL, Hutton DL, Erdman CP, Wieland M, Boyan BD, Schwartz Z. Direct and indirect effects of microstructured titanium substrates on the induction of mesenchymal stem cell differentiation towards the osteoblast lineage. Biomaterials. 2010;31:2728–35. doi: 10.1016/j.biomaterials.2009.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Bauer S, von der Mark K, Schmuki P. Nanosize and vitality: TiO2 nanotube diameter directs cell fate. Nano Lett. 2007;7:1686–91. doi: 10.1021/nl070678d. [DOI] [PubMed] [Google Scholar]
- Pham QP, Sharma U, Mikos AG. Electrospinning of polymeric nanofibers for tissue engineering applications: a review. Tissue Eng. 2006;12:1197–211. doi: 10.1089/ten.2006.12.1197. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH image to imageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepmann J, Gopferich A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv Drug Deliv Rev. 2001;48:229–47. doi: 10.1016/s0169-409x(01)00116-8. [DOI] [PubMed] [Google Scholar]
- Sill TJ, von Recum HA. Electrospinning: applications in drug delivery and tissue engineering. Biomaterials. 2008;29:1989–2006. doi: 10.1016/j.biomaterials.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Tutak W, Gelven G, Marckle C, Palmer X. Rapid polymer fiber airbrushing: Impact of a device design on the fiber fabrication and matrix quality. J Appl Polym Sci. 2015;132:42813. [Google Scholar]
- Tutak W, Sarkar S, Lin-Gibson S, Farooque TM, Jyotsnendu G, Wang DB, Kohn J, Bolikal D, Simon CG. The support of bone marrow stromal cell differentiation by airbrushed nanofiber scaffolds. Biomaterials. 2013;34:2389–98. doi: 10.1016/j.biomaterials.2012.12.020. [DOI] [PubMed] [Google Scholar]
- Vural M, Behrens AM, Ayyub OB, Ayoub JJ, Kofinas P. Sprayable elastic conductors based on block copolymer silver nanoparticle composites. ACS Nano. 2015;9:336–44. doi: 10.1021/nn505306h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojdyr M. Fityk: a general-purpose peak fitting program. J Appl Crystallogr. 2010;43:1126–8. [Google Scholar]
- Xu CY, Inai R, Kotaki M, Ramakrishna S. Electrospun nanofiber fabrication as synthetic extracellular matrix and its potential for vascular tissue engineering. Tissue Eng. 2004;10:1160–8. doi: 10.1089/ten.2004.10.1160. [DOI] [PubMed] [Google Scholar]
- You MH, Kwak MK, Kim DH, Kim K, Levchenko A, Kim DY, Suh KY. Synergistically enhanced osteogenic differentiation of human mesenchymal stem cells by culture on nanostructured surfaces with induction media. Biomacromolecules. 2010;11:1856–62. doi: 10.1021/bm100374n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HB, Zhou L, Zhang WJ. Control of scaffold degradation in tissue engineering: a review. Tissue Eng B: Rev. 2014;20:492–502. doi: 10.1089/ten.TEB.2013.0452. [DOI] [PubMed] [Google Scholar]
- Zhuang XP, Shi L, Jia KF, Cheng BW, Kang WM. Solution blown nanofibrous membrane for microfiltration. J Membr Sci. 2013;429:66–70. [Google Scholar]
- Zong XH, Ran SF, Kim KS, Fang DF, Hsiao BS, Chu B. Structure and morphology changes during in vitro degradation of electrospun poly(glycolide-co-lactide) nanofiber membrane. Biomacromolecules. 2003;4:416–23. doi: 10.1021/bm025717o. [DOI] [PubMed] [Google Scholar]
- Zuo F, Tan DH, Wang ZF, Jeung S, Macosko CW, Bates FS. Nanofibers from melt blown fiber-in-fiber polymer blends. ACS Macro Lett. 2013;2:301–5. doi: 10.1021/mz400053n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.