

Abstract
Ketoreductases (KRs) are the most widespread tailoring domains found in individual modules of assembly line polyketide synthases (PKSs), and are responsible for controlling the configurations of both the α-methyl and β-hydroxyl stereogenic centers in the growing polyketide chain. Because they recognize substrates that are covalently bound to acyl carrier proteins (ACPs) within the same PKS module, we sought to quantify the extent to which protein-protein recognition contributes to the turnover of these oxidoreductive enzymes using stand-alone domains from the 6-deoxyerythronolide B synthase (DEBS). Reduced 2-methyl-3-hydroxyacyl-ACP substrates derived from two enantiomeric acyl chains and four distinct ACP domains were synthesized and presented to four distinct KR domains. Two KRs, from DEBS modules 2 and 5, displayed little preference for oxidation of substrates tethered to their cognate ACP domains over those attached to the other ACP domains tested. In contrast, the KR from DEBS module 1 showed a ca. 10-50-fold preference for substrate attached to its native ACP domain, whereas the KR from DEBS module 6 actually displayed a ca. 10-fold preference for the ACP from DEBS module 5. Our findings suggest that recognition of the ACP by a KR domain is unlikely to affect the rate of native assembly line polyketide biosynthesis. In some cases, however, unfavorable KR-ACP interactions may suppress the rate of substrate processing when KR domains are swapped to construct hybrid PKS modules.
Introduction
Polyketide synthases (PKS) are large multidomain enzyme complexes that synthesize a variety of bioactive natural products1,2,3. One family of PKS systems, most vividly illustrated by the 6-deoxyerythronolide B synthase (DEBS; Figure 1A), has an assembly line architecture, with individual catalytic domains organized into successive modules4,5. Each catalytic domain performs a unique reaction in the overall catalytic cycle of the PKS. A typical module of an assembly line PKS consists of a ketosynthase (KS), acyltransferase (AT), and acyl carrier protein (ACP), which together yield a β-ketoacyl-ACP intermediate. This intermediate is then modified by other tailoring domains, such as the ketoreductase (KR), dehydratase (DH) and enoyl reductase (ER) domains, to introduce functional group diversity along the carbon chain backbone of the polyketide.
Figure 1.
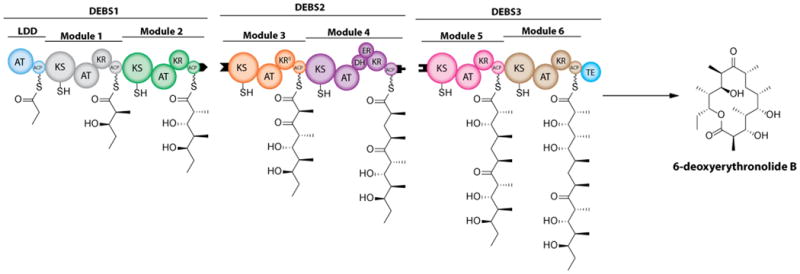
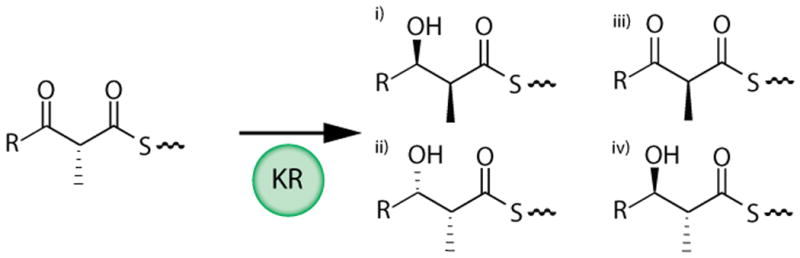
(A) DEBS schematic (B) Reactions carried out by KR domains of DEBS catalyzed by (i) KR1; (ii) KR2, KR5, KR6; (iii) KR3; and (iv) KR4. Following the condensation reaction, the substrate for each KR is a (2R)-2-methyl-3-ketoacyl-ACP which is then processed to yield a (i) (2S, 3R)-2-methyl-3-hydroxy; (ii) (2R, 3S)-2-methyl-3-hydroxy; (iii) (2S)-2-methyl-3-keto; (iv) or (2R, 3R)-2-methyl-3-hydroxy, acyl product.
The KR domains of assembly line PKSs are the most prevalent tailoring domains, and are responsible for establishing the stereo-configuration of both the α and β carbon atoms of their (2R)-2-methyl-3-ketoacyl-ACP substrates (Figure 1B). In addition to catalyzing NADPH-dependent ketone reduction, some KR domains also harbor epimerase activity, allowing them to catalyze epimerization of the methyl groups of their acyl-ACP substrates. Earlier studies have primarily focused on elucidating the stereocontrol principles of individual KR domains6,7,8, including their ability to catalyze α-methyl epimerization9. Here we have utilized a previously developed assay system to interrogate the contribution of protein-protein interactions to the overall substrate specificity of KR domains10.
Materials and Methods
Instrumentation
UV-VIS spectrophotometric assays were performed on a BioTek Synergy HT Microplate Reader. LC-ESI-MS analysis was carried out using a Waters single quadrupole mass spectrometer and Acquity H Class UPLC with photodiode array detector. LC-ESI-MS samples were run on a 10 min gradient, eluting with increasing acetonitrile containing 0.1% formic acid over a C8 column (Zorbax 300SB-C8).
Plasmids
Plasmids expressing the Sfp phosphopantetheinyl transferase, KRs, and ACPs were constructed using standard molecular cloning procedures. Detailed descriptions of these plasmids can be found in SI Tables 1-2.
Protein purification
Expression plasmids were introduced into E. coli BL21(DE3) cells via electroporation, then plated onto Luria-Bertani (LB) media plates with the appropriate antibiotic. Colonies were selected and grown in 5 mL seed cultures for 10-15 h at 37 °C, then used to inoculate 1 L autoclaved LB with the appropriate antibiotic. Cells were grown to an A600 of 0.6-0.8 then induced with 250 μL of 1 M IPTG, cooled to 18 °C, and incubated at this temperature for 12-15 h.
Cells were centrifuged at 6 370 g, then resuspended in 30 mL lysis buffer (50 mM sodium phosphate, 450 mM sodium chloride, 10 mM imidazole, 20% glycerol, adjusted to pH 7.6) per liter of original cell culture. The resulting cell slurry was lysed on ice, using a Branson Sonifier 450. The lysate was centrifuged at 29 000 g for 1 h, and the supernatant was mixed with Ni-NTA agarose resin (2.5 mL resin per L culture) and incubated with gentle agitation for at least 1 h. After incubation, the loaded resin was applied to a Kimble-Kontes Flex column and washed with 20 column volumes of lysis buffer, 10 column volumes of wash buffer (50 mM sodium phosphate, 300 mM sodium chloride, 25 mM imidazole, 10% glycerol, adjusted to pH 7.6), and eluted with 2-6 column volumes of elution buffer (150 mM sodium phosphate, 40 mM sodium chloride, 500 mM imidazole, 10% glycerol, adjusted to pH 7.6). For ACPs, the eluate was further processed with GE PD-10 desalting columns and Buffer A (50 mM sodium phosphate, 10% glycerol, adjusted to pH 7.6) in order to improve the efficacy of downstream purification steps. The eluate was further purified using anion exchange chromatography over a GE HiTrap Q column with Buffer A (as above) and a linear gradient of Buffer B (50 mM sodium phosphate, 500 mM sodium chloride, 10% glycerol, adjusted to pH 7.6). FPLC fractions were tested using SDS polyacrylamide gel electrophoresis, and fractions containing the protein of interest were pooled, concentrated, and buffer exchanged into Buffer A using Amicon Ultra-15 centrifugal filter devices. Purified proteins were aliquoted and flash frozen in liquid nitrogen, and stored at -80 °C. Protein concentrations were determined using the BCA Protein Assay Kit (Pierce) with BSA as a reference.
Synthesis of (2S, 3R)-2-methyl-3-hydroxypentanoyl-CoA (NDK-CoA) and (2S, 3R)-2-methyl-3-hydroxypentanoyl-CoA (EDK-CoA)
NDK-CoA and EDK-CoA were synthesized, as described previously11,12,13. Ellman's reagent was used to precisely quantify the concentration of each stock solution. Specifically, 20 μL of 2 M NaOH was added to a 40 μL aliquot of each purified acyl-CoA and incubated at RT for 15 min to hydrolyze the thioester. The reaction was quenched with 20 μL 2 M HCl. The amount of acyl-CoA was determined by measuring the thiol content using Ellman's reagent, and comparison to a standard curve generated from different concentrations of L-cysteine.
Synthesis of NDK-ACP and EDK-ACP substrates
A typical acyl-ACP substrate was synthesized by co-incubating the corresponding apo-ACP (100 μM) and acyl-CoA (100-600 μM) with 1 μM Sfp phosphopantetheinyl transferase in a buffer containing 10 mM magnesium chloride and 100 mM sodium phosphate, pH 7.214. After reaction at RT for ca. 2 h, the mixtures were applied serially to five Zeba spin desalting columns with a 7 kDa molecular weight cutoff to remove unreacted acyl-CoA (using 100 mM sodium phosphate, pH 7.2, as equilibration buffer). Thereafter, a portion of the sample was concentrated using Amicon Ultra-0.5 mL centrifugal filter devices.
Ketoreductase activity assays
Each batch of a purified KR was tested for activity as well as consistency with previous preparations of the same enzyme by measuring its ability to reduce the model substrate, trans-decalone15. Assays were performed in 100 μL volumes with 2 mM trans-decalone, 1 mM NADPH, and 5 μM KR in 100 mM sodium phosphate buffer, pH 7.2. After initiation with NADPH, reaction curves were monitored at 340 nm corresponding to oxidation of the cofactor NADPH to NADP+ (SI Figure 1).
KR activity against acyl-CoA substrates was quantified in 100 mM sodium phosphate, pH 7.2, with ∼25 ug/mL BSA, 1 uM KR, and 2 mM NADP+. After initiation with NADP+, the entire reaction volume was immediately transferred into a 96 well plate (Corning 3993), and readings were taken using a 360/460 nm filter to monitor the production of NADPH. Reaction progress curves in relative fluorescence units were converted to μM NADPH by comparison to a linear standard curve of NADPH in 100 mM sodium phosphate, pH 7.2.
KR activity against acyl-ACP substrates was quantified analogously in 50 μL reaction volumes containing 100 mM sodium phosphate, pH 7.2, with 0.5-1 μM KR and ∼25 μg/mL BSA to stabilize protein components. Reactions were initiated by adding the protein mixture directly to 2.5 μL of 40 mM NADP+, resulting in a final concentration of 2 mM NADP+. Negative controls contained the cofactor NADP+ and included the acyl-ACP without KR, the KR only, and the KR with desalted ACP that had been mixed with acyl-CoA in the absence of Sfp (to confirm retention of acyl-CoA by the desalting column). Assays were performed in a 96 well plate (Corning 3993) using a 360/460 nm filter to monitor the production of NADPH. Reaction progress curves in relative fluorescence units were converted to μM NADPH by comparison to a linear standard curve of NADPH in 100 mM sodium phosphate, pH 7.2. A portion of the desalted acyl-ACP was set aside. Part of this aliquot was flash frozen and later processed with the BCA assay to determine the amount of acyl-ACP recovered after the desalting procedure. Additionally, the chemical composition of each acyl-ACP was verified by LC-ESI-MS. Most ACPs had minor post-translational modifications such as acetylation, gluconoylation, and phosphogluconoylation16. When modified with the acyl-CoA and Sfp, the apo-ACP peaks all increased by the mass corresponding to the added acyl-phosphopantetheine group. In all cases, only small amounts (<10%) of unloaded apo-ACP remained (SI Figures 2-3).
Results
The catalytic activities of stand-alone DEBS KR1 and KR6 were initially quantified by oxidation of 2-methyl-3-hydroxyacyl-CoA substrates that correspond to their respective natively produced ACP-bound (2S, 3R)- and (2R, 3S) enantiomers. As seen in Figure 2A, both enzymes oxidize their substrates with comparable catalytic efficiency; the kcat/KM of DEBS KR1 for NDK-CoA was 4 mM-1.h-1, whereas the corresponding value of KR6 for EDK-CoA was 0.9 mM-1.h-1. Neither enzyme had a particularly high affinity for its CoA substrate; only KR1 showed evidence of saturation under our assay conditions, with an estimated KM of 0.2 mM.
Figure 2.

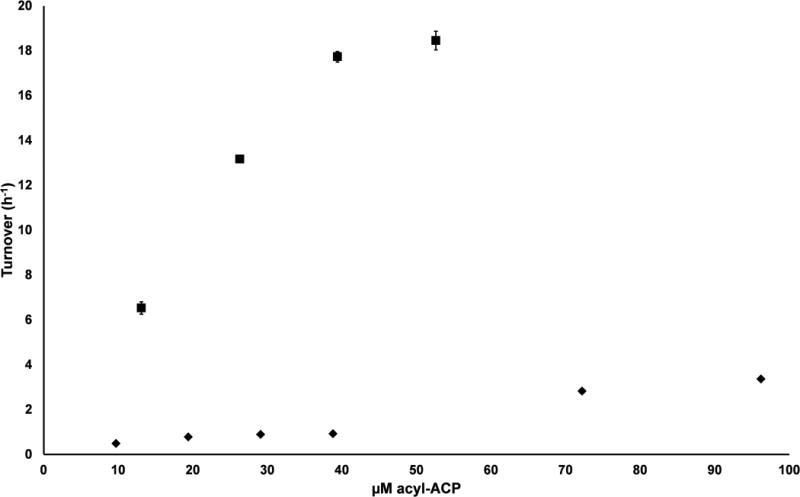
Activity of DEBS KR1 (squares) and KR6 (diamonds) with (A) their preferred acyl-CoA substrates and (B) the corresponding acyl-ACP substrates ACP1 and ACP6, respectively. The preferred substrates for KR1 were NDK ((2S, 3R)-2-methyl-3-hydroxypentanoyl-) thioesters, whereas the preferred substrates for KR6 were EDK ((2R, 3S)-2-methyl-3-hydroxypentanoyl-) thioesters. All reactions were performed in triplicate with error bars showing one standard deviation. The lower limit of quantification of turnover in this assay is ca. 0.01 h-1.
In order to evaluate the specificity of the same KR domains for their acyl-ACP substrates, we first needed to attach the acyl substrate onto the pantetheine arms of these small helical proteins. To do so, a phosphopantetheinyl transferase, Sfp, with broad substrate specificity for preformed acyl-CoA substrates was used17. In the presence of NDK-CoA or EDK-CoA, Sfp was used to prepare acyl-ACP derivatives of DEBS ACP1, ACP2, ACP3, ACP5, and ACP6, which were then individually incubated with DEBS KR1, KR2, KR5, or KR6 in the presence of saturating amounts of NADP+. As seen in the cases of KR1 and KR6 (Figure 2B), acyl-ACP substrates are substantially better (kcat/KM 10-100 fold higher) than their CoA thioester counterparts, suggesting that protein-protein recognition between the KR and the ACP contributes significantly to catalysis of this critical reaction in polyketide biosynthesis.
To assess the ability of representative KR domains to discriminate between alternative ACP carriers, the kcat/KM values of each of the above four DEBS KR domains was measured against a panel of acyl-ACP substrates harboring the identical acyl group. As summarized in Table 1, KR1 shows approximately 10-50 fold kinetic preference for its cognate (2S, 3R) attached to its native ACP1 over the other four ACPs tested. In contrast, the specificities of KR2 and KR5 for their cognate (2R, 3S) ACP were more modest, showing a smaller 4-5-fold advantage in measured kcat/Km. Unexpectedly, KR6 was relatively non-discriminatory between its cognate (2R, 3S) ACP substrate and either ACP1, ACP2, or ACP3, but showed an approximately 10-fold preference for substrate attached to ACP5. It should be noted that the absolute turnover rates of KR5 and especially KR2 are low, and therefore present lower confidence limits to the reported specificity analysis of these two domains.
Table 1.
Specificity constants (kcat/KM; mM-1.h-1) for KR catalyzed oxidations of their preferred acyl-ACP substrates (as before, KR1 preferred NDK thioesters whereas KR 6 preferred EDK thioesters) loaded onto a panel of ACPs. Reaction progress was monitored by measuring appearance of the co-product NADPH; the linear portion of the curve was used to calculate initial rates (tabulated in Supplemental File 2) which were fit to the standard Michaelis-Menten equation. Abbreviation NT, not tested.
| KR | ACP 1 | ACP 2 | ACP 3 | ACP 5 | ACP 6 |
|---|---|---|---|---|---|
| KR 1 | 500 | 33 | 11 | 43 | 31 |
| KR 2 | 1.6 | 9.9 | NT | 2.6 | 2.7 |
| KR 5 | 2.5 | 12 | NT | 50 | 11 |
| KR 6 | 35 | 38 | 16 | 310 | 35 |
Given the enhanced reactivity of acyl-ACP substrates relative to acyl-CoA substrates, we attempted to determine if a given KR could oxidize the enantiomer of its natural substrate. To do so, KR1 was presented with 43 μM EDK-ACP1. With NDK-ACP1 at a comparable concentration, KR1 displayed turnover of ∼20 h-1, however, with the enantiomeric counterpart, no reduction of NADP+ above background levels could be detected. Similar results were obtained with KR 6 and NDK-ACP6. Thus, KRs appear to have at least a 1000-fold specificity for acyl chains with the natural α- and β-carbon stereochemistry as compared to enantiomeric forms.
Discussion
The importance of specific protein-protein interactions in the catalytic cycles of assembly line PKSs has been well established18. For example, the KS domains of DEBS have significant preference (∼10-1000 fold) for their cognate ACP domains when catalyzing polyketide chain elongation19 as well as intermodular chain translocation20. Similarly, AT domains show preference for their cognate ACP domains in the context of extender unit transacylation21. Our data presented here suggests that the specificity of KR domains for cognate ACP domains is relatively low. Moreover, given the unexpected preference of KR6 for a neighboring ACP domain (ACP5), there appears to be little evolutionary pressure on KR-ACP recognition.
Computational analysis of the recognition of ACP1 by KR1 of DEBS identified possible interactions between the two domains, including Arg 1857 (KR): Asp 45 (ACP), Phe 1856 (KR): Leu 47 (ACP), and 4 other residues on the KR that were predicted to make contact with the ACP22. Both the Asp and Leu residues of ACP1 are conserved across the five ACPs tested in our study, suggesting that conserved interactions contribute to the bulk of KR-ACP recognition in assembly line PKSs. Our findings have encouraging implications for the engineering of PKSs through KR domain substitution, as they suggest that protein-protein interactions are unlikely to be important considerations in the design of a chimeric assembly line.
Supplementary Material
Acknowledgments
This research was supported by grants from the NIH (GM 087934 to C.K., and GM 022172 to D.E.C.). M.P.O. is a recipient of an NSF Graduate Research Fellowship and a Stanford Graduate Fellowship. LC-ESI-MS analysis was carried out at the Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University Mass Spectrometry (http://mass-spec.stanford.edu).
References
- 1.Bruegger J, Caldara G, Beld J, Burkart MD, Tsai S. In: Natural Products: Discourse, Diversity, and Design. Osbourn A, Goss RJ, Carter GT, editors. John Wiley & Sons, Inc; Hoboken, USA: 2014. pp. 219–243. [Google Scholar]
- 2.Wang H, Fewer DP, Holm L, Rouhiainen L, Sivonen K. Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc Natl Acad Sci USA. 2014;111:9259–64. doi: 10.1073/pnas.1401734111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balskus EP. Colibactin: understanding an elusive gut bacterial genotoxin. Nat Prod Rep. 2015;32:1534–1540. doi: 10.1039/c5np00091b. [DOI] [PubMed] [Google Scholar]
- 4.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu Rev Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 5.Khosla C, Herschlag D, Cane DE, Walsh CT. Assembly line polyketide synthases: mechanistic insights and unsolved problems. Biochemistry. 2014;53:2875–83. doi: 10.1021/bi500290t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young-Ok Y, Khosla C, Cane DE. Stereochemistry of Reductions Catalyzed by Methyl-Epimerizing Ketoreductase Domains of Polyketide Synthases. J Am Chem Soc. 2013;135:7406–7409. doi: 10.1021/ja4014776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, et al. Polyketide Intermediate Mimics as Probes for Revealing Cryptic Stereochemistry of Ketoreductase Domains. ACS Chem Biol. 2014;9:2914–2922. doi: 10.1021/cb5006883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mugnai ML, Shi Y, Keatinge-Clay AT, Elber R. Molecular Dynamics Studies of Modular Polyketide Synthase Ketoreductase Stereospecificity. Biochemistry. 2015;54:2346–2359. doi: 10.1021/bi501401g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garg A, Xie X, Keatinge-Clay A, Khosla C, Cane DE. Elucidation of the Cryptic Epimerase Activity of Redox-Inactive Ketoreductase Domains from Modular Polyketide Synthases by Tandem Equilibrium Isotope Exchange. J Am Chem Soc. 2014;136:10190–10193. doi: 10.1021/ja5056998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen AY, Cane DE, Khosla C. Structure-Based Dissociation of a Type I Polyketide Synthase Module. Chem Biol. 2007;14:784–792. doi: 10.1016/j.chembiol.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu T, Cane DE, Khosla C. Assessing the Balance between Protein–Protein Interactions and Enzyme–Substrate Interactions in the Channeling of Intermediates between Polyketide Synthase Modules. J Am Chem Soc. 2001;123:6465–6474. doi: 10.1021/ja010219t. [DOI] [PubMed] [Google Scholar]
- 12.Cane DE, Lambalot RH, Prabhakaran PC, Ott WR. Macrolide biosynthesis. 7 Incorporation of polyketide chain elongation intermediates into methymycin J Am Chem Soc. 1993;115:522–526. [Google Scholar]
- 13.Harris RC, et al. Enantiospecific Synthesis of Analogues of the Diketide Intermediate of the Erythromycin Polyketide Synthase (PKS). J Chem Res. 1998;6:283. [Google Scholar]
- 14.Garg A, Khosla C, Cane DE. Coupled Methyl Group Epimerization and Reduction by Polyketide Synthase Ketoreductase Domains. Ketoreductase-Catalyzed Equilibrium Isotope Exchange J Am Chem Soc. 2013;135:16324–16327. doi: 10.1021/ja408944s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siskos AP, et al. Molecular Basis of Celmer's Rules: Stereochemistry of Catalysis by Isolated Ketoreductase Domains from Modular Polyketide Synthases. Chem Biol. 2005;12:1145–1153. doi: 10.1016/j.chembiol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 16.Geoghegan D, et al. Spontaneous alpha-N-6-phosphogluconoylation of a “His tag” in Escherichia coli: the cause of extra mass of 258 or 178 Da in fusion protein. Anal Biochem. 1999;267:169–184. doi: 10.1006/abio.1998.2990. [DOI] [PubMed] [Google Scholar]
- 17.Beld J, Sonnenschein EC, Vickery CR, Noel JP, Burkart MD. The phosphopantetheinyl transferases: catalysis of a post-translational modification crucial for life. Nat Prod Rep. 2014;31:61–108. doi: 10.1039/c3np70054b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu W, Qiao K, Tang Y. Structural analysis of protein–protein interactions in type I polyketide synthases. Crit Rev Biochem Mol Biol. 2013;48:98–122. doi: 10.3109/10409238.2012.745476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. Extender unit and acyl carrier protein specificity of ketosynthase domains of the 6-deoxyerythronolide B synthase. J Am Chem Soc. 2006;128:3067–3074. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu N, Cane DE, Khosla C. Quantitative Analysis of the Relative Contributions of Donor Acyl Carrier Proteins, Acceptor Ketosynthases, and Linker Regions to Intermodular Transfer of Intermediates in Hybrid Polyketide Synthases. Biochemistry. 2002;41:5056–5066. doi: 10.1021/bi012086u. [DOI] [PubMed] [Google Scholar]
- 21.Dunn BJ, Cane DE, Khosla C. Mechanism and specificity of an acyltransferase domain from a modular polyketide synthase. Biochemistry. 2013;52:1839–1841. doi: 10.1021/bi400185v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anand S, Mohanty D. Modeling holo-ACP:DH and holo-ACP:KR complexes of modular polyketide synthases: a docking and molecular dynamics study. BMC Struct Biol. 2012;12:10. doi: 10.1186/1472-6807-12-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.