

Abstract
The cell of origin for most mesenchymal tumors is unclear. One cell type that contributes to this lineages is the pericyte, a cell expressing Ng2/Cspg4. Using lineage tracing, we demonstrated that bone and soft tissue sarcomas driven by the deletion of the Trp53 tumor suppressor, or desmoid tumors driven by a mutation in Apc can derive from cells expressing Ng2/Cspg4. Deletion of the Trp53 tumor suppressor gene in these cells resulted in the bone and soft tissue sarcomas that closely resemble human sarcomas, while stabilizing β-catenin in this same cell type caused desmoid tumors. Comparing expression between Ng2/Cspg4 expressing pericytes lacking Trp53 and sarcomas that arose from deletion of Trp53, showed inhibition of β-catenin signaling in the sarcomas. Activation of β-catenin inhibited the formation and growth of sarcomas. Thus, pericytes can be a cell of origin for mesenchymal tumors, and β-catenin dysregulation plays an important role in the neoplastic phenotype.
Keywords: sarcoma, Desmoid, pericyte, Ng2, mouse, beta-catenin, lineage tracing, P53
Graphical Abstract

eTOC Blurb
Here we used lineage tracing studies in mice to show that bone and soft tissue sarcomas driven by the deletion of the Trp53 tumor suppressor gene, can derive from Ng2/Cspg4 expressing pericytes. Benign mesenchymal desmoid tumors, driven by a mutation in Apc, can derive from cells expressing Ng2/Cspg4. Driving mutations in Trp53 or β-catenin in Ng2/Cspg4 expressing cells, resulted in sarcoma or desmoid tumor formation. Comparing the expression profiles from RNA sequencing between Ng2/Cspg4 expressing pericytes lacking Trp53 and sarcomas that arose from deletion of Trp53, showed inhibition of β-catenin signaling in the sarcomas. Activation of β-catenin inhibited the formation and growth of sarcomas. Our data shows that pericytes can be a cell of origin for mesenchymal tumors. Furthermore, β-catenin plays a critical role in mesenchymal neoplasia, with malignant sarcomas exhibiting a lower level of lower level of β-catenin activity.
Introduction
Tumors are initiated by mutations in specific cell types. Since progenitor cell populations can survive over longer periods of time, they may be more likely to accumulate mutations that cause neoplasia (Reya et al., 2001). Identifying the cell of origin of a tumor type can be used to identify critical events responsible for tumor formation and driving oncogenesis in the cell of origin can be used to develop animal models that more accurately recapitulate human tumors (Visvader, 2011).
Sarcomas are malignancies found in the connective tissues, composed of cells with mesenchymal characteristics. There is a broad range of sarcoma types, including those that derive in bone, cartilage, fat, muscle, or vascular, tissues. Two of the most common sarcoma types are osteosarcoma and undifferentiated pleomorphic sarcomas, and yet much remains to be established about the critical steps required for tumor formation in these subtypes. Desmoid tumors are locally invasive mesenchymal tumors that do not metastasize. They are composed of fibroblast-like cells with a proliferative advantage, driven by somatic mutations activating β-catenin mediated signaling. Mutations in Apc or in β-catenin itself are identified in almost all cases of this tumor type (Alman et al., 1997a; Cheon et al., 2002). The precise cell of origin for these tumors is unknown. Since they have mesenchymal characteristics, it is likely that they derive from a mesenchymal lineage progenitor cell.
In addition to its role in desmoid tumors, β-catenin protein is also implicated in sarcomas. However, its role in sarcomas has been controversial, with some studies suggesting activated β-catenin signaling is important to drive the neoplastic phenotype, while others found an opposite effect (Cai et al., 2014; Cai et al., 2010; Du et al., 2014; Matushansky et al., 2007; Wan et al., 2014). In mesenchymal cell development, β-catenin is precisely regulated at different stages for normal differentiation, raising the possibility that either high or low β-catenin leads to pathology (Chen et al., 2007; Hoffman and Benoit, 2013; Li et al., 2008; Wan et al., 2013). Understanding the role of β-catenin mediated signaling in neoplasia also has therapeutic implications, as β-catenin modulating therapies are being developed for clinical use.
Pericytes are mesenchymal cells that surround endothelial cells in capillaries, venules, and small arterioles (Diaz-Flores et al., 2009; Hirschi and D'Amore, 1996). These cells express markers such as Chondroitin Sulfate Proteoglycan 4 (CSPG4), also termed Neuron-glial antigen 2 (NG2) and CD146, also known as melanoma cell adhesion molecule (Bergers and Song, 2005; Covas et al., 2008; Crisan et al., 2012; Crisan et al., 2008). This cell type is involved in the stability and contractility of blood vessels, but also can be a progenitor for several mesenchymal cell types (Crisan et al., 2012; Crisan et al., 2008; Dellavalle et al., 2007). Interestingly, human sarcomas are known to express genes that are characteristically expressed in pericytes (Benassi et al., 2009; Schiano et al., 2012). Thus, pericytes could be a cell of origin for some mesenchymal tumors.
Here we addressed the role of Ng2/Cspg4 expressing cells and β-catenin in the origin of mesenchymal tumors. Lineage tracing studies in murine sarcomas driven by the deletion of the Trp53 tumor suppressor, or desmoid tumors driven by a mutation in Apc, were used to investigate Ng2/Cspg4 expressing cells as a cell of origin for mesenchymal. We also determined the ability of Trp53 deletion and/or stabilization of β-catenin in Ng2/Cspg4 expressing cells to result in tumor formation.
Results
Mesenchymal tumors can derive from Ng2/Cspg4 expressing cells
To determine if mesenchymal tumors might derive from Ng2/Cspg4 expressing cells, we undertook lineage-tracing studies in genetically modified mice that are known to develop mesenchymal tumors. We used Trp53 deficient mice to study sarcomas. These mice are a model for Li-Fraumeni syndrome and develop malignancies, including lymphomas and sarcomas (Jacks et al., 1994). To study the origin of a benign tumor, we investigated desmoid tumors, which are benign locally invasive mesenchymal lesions driven by mutations activating β-catenin mediated signaling. The Apc1638N mouse (Smits et al., 1998) harbors a mutation in Apc that results in the development of multiple desmoid tumors.
NG2/CSPG4 is a cell surface proteoglycan expressed by pericytes, neural progenitor cells, chondrocytes, and hair follicles (Feng et al., 2010). To label Ng2/Cspg4 expressing cells, we crossed Ng2/Cspg4-CreER mice (Zhu et al., 2011) with Rosa26RlacZ mice (Soriano, 1999). The transgene was activated by daily tamoxifen injections for one week after weaning (Madisen et al., 2010). β-galactosidase (X-gal) staining was performed to identify the distribution of LacZ-positive cells, and this confirmed that LacZ was expressed in pericytes, neural cells, chondrocytes, and hair follicles (Figs. 1A and S1A). In contrast, osteoblasts did not show expression of LacZ, a finding consistent with other studies using this animal (Feng et al., 2011), in which lacZ staining was only observed in bone during mesenchymal repair processes when the transgene was activated postnatally (Fig. S1B). To verify which cells were expressing lacZ, we dissociated cells and sorted LacZ positive and negative populations as in our previous publications (Amini-Nik et al., 2014; Amini-Nik et al., 2011). There was an increase in RNA expression of Ng2/Cspg4 in the LacZ positive population (Fig. S1C). We next sorted NG2/CSPG4 positive and negative cells using a cell surface antibody, and analyzed the populations for expression of LacZ, finding that the NG2/CSP4 positive population expressed LacZ. We also analyzed the LacZ positive and negative populations for the expression of CD146, a cell surface marker expressed by pericytes (Wei et al., 2015), and found that the LacZ expressing cells also expressed CD146 (Fig. S1E). Taken together, these data show that LacZ effectively labels Ng2/Cspg4 expressing pericytes.
Figure 1. Mesenchymal tumors can derive from Ng2/Cspg4 expressing cells.
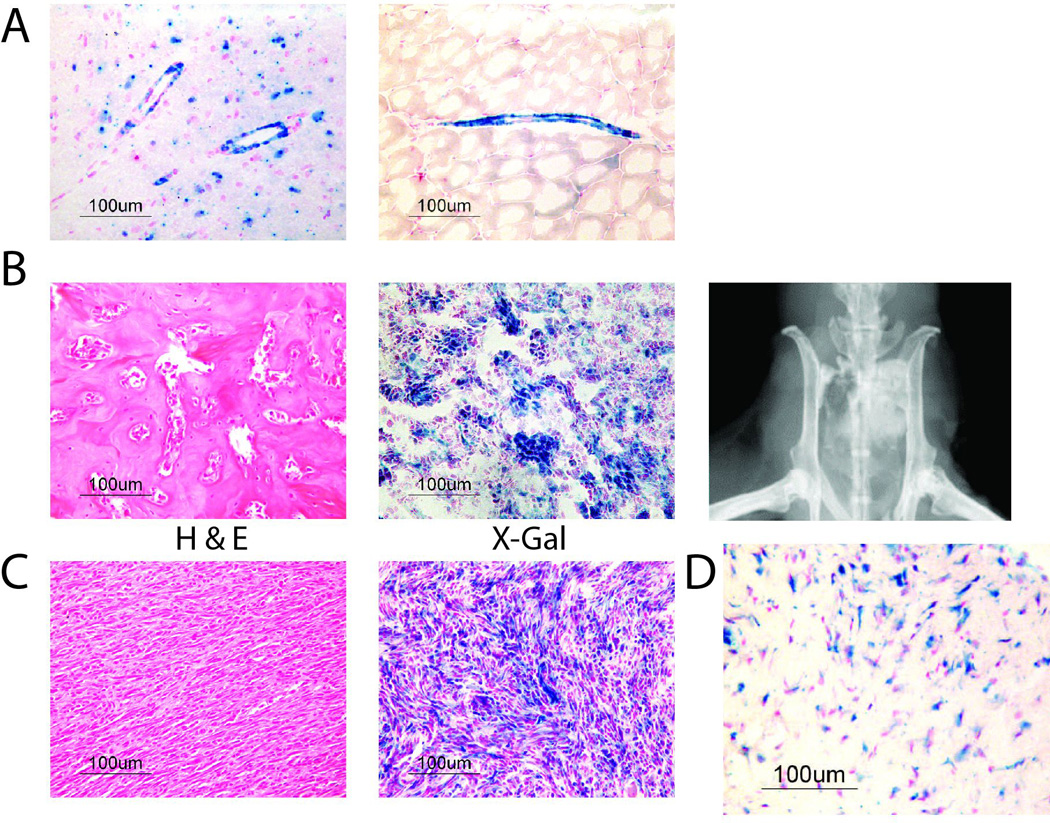
A) X-gal staining in Ng2/Cspg4-CreER;Rosa26RlacZ mice, showing blue staining (LacZ) in the brain (left) and in perivascular tissues in the skeletal muscle (right). B) Representative H & E, X-gal staining, and a radiograph from a mouse osteosarcoma that developed in a Trp53 deficient mouse, showing tumor cells that stained blue, indicating that they are derived from Ng2/Cspg4 expressing cells. C) Representative H & E and X-gal staining from a mouse soft tissue sarcoma that developed in a Trp53 deficient mouse showing histology consistent with undifferentiated pleomorphic sarcoma. Similar to the situation in osteosarcomas, tumor cells stained blue. D) Representative X-gal staining from a mouse desmoid tumor that developing in an Apc1638N mouse showing blue staining in the tumor cells. This shows that these benign mesenchymal tumors also derived from Ng2/Cspg4 expressing cells.
We next crossed Ng2/Cspg4-CreER;Rosa26RlacZ mice with Trp53 deficient mice (Jacks et al., 1994) and injected them with tamoxifen. 12 sarcomas developed in the mice, 5 of which were undifferentiated pleomorphic sarcomas, 6 were osteosarcomas, and one was an angiosarcoma. X-gal staining confirmed that the sarcomas derived from LacZ expressing cells (Figures 1B and 1C). Given the expression pattern of LacZ in normal tissues, this suggests that the tumors derived from pericytes. The Apc1638N mouse (Smits et al., 1998) harbors a mutation in Apc that results in β-catenin activation and the development of multiple desmoid tumors. Ng2/Cspg4-CreER;Rosa26RlacZ mice were crossed with Apc1638N mutant mice and injected them with tamoxifen. X-gal staining showed that the desmoid tumors that developed also express LacZ (Fig. 1D).
Interestingly, not all cells stained blue. Solid tumors contain a subpopulation of non-tumoral cells, including normal stromal cells (Mao et al., 2013), but in mesenchymal tumors, there are no cytologic or cell surface markers to distinguish stromal cells from neoplastic cells, making differentiation between tumoral and stromal cells problematic. We sorted LacZ stained from non-stained cells in these tumors. Between 28 and 49 percent of cells in the tumors did not stain with LacZ. In desmoid tumors, we found a higher level of β-catenin in the LacZ positive cells (Fig. S2). Thus, there is a population of normal cells within bulk mesenchymal tumors that arise from non-Ng2/Cspg4 expressing cells.
Trp53 deletion in Ng2/Cspg4 expressing cells induces bone and soft tissue sarcomas
To determine whether mutations in Ng2/Cspg4 expressing cells could induce sarcomas, we crossed Ng2/Cspg4-Cre mice or Ng2/Cspg4-CreER mice with Trp53flox/flox mice (Marino et al., 2000) to generate Ng2/Cspg4-Cre mediated Trp53 conditional knockout mice. In the case of mice expressing the Ng2/Cspg4-CreER allele, the conditional allele was activated by tamoxifen administration the week following weaning. In Ng2/Cspg4-CreER;Trp53flox/flox mice treated with tamoxifen, 66 % developed bone sarcomas and 20 % developed soft tissue sarcomas. In Ng2/Cspg4-Cre;Trp53flox/flox mice in which Cre is constitutively expressed in Ng2/Cspg4+ cells, 76% developed bone sarcomas and 16.0 % developed soft tissue sarcomas (Table one). The mice succumbed to tumors by 14 months of age, and had a survival that is better than for the Trp53−/− mice that we studied in our lineage tracing analysis (Fig. 2A). This is expected, since these mice only rarely developed tumors that were not sarcomas (Table 1), and as such succumbed from sarcoma-related mortality.
Table 1.
Tumor distribution of Ng2/Cspg4-Cre mediated Trp53 conditional knockout mice
| Genotype | Number of mice |
Bone sarcoma (OS) |
Soft tissue Sarcoma (UPS) |
Lymphoma | Other malignancies |
Average latency of sarcoma development (days ± SD) |
|---|---|---|---|---|---|---|
|
Ng2/Cspg4-Cre; Trp53flox/flox |
50 |
38 (76.0 %) |
8 (16.0 %) |
2 (4.0 %) |
0 | 299 ± 56 |
| Male | 19 | 10 (52.6 %) |
6 (31.6 %) |
0 | 0 | 337 ± 59 |
| Female | 31 | 28 (90.3 %) |
2 (9.7 %) |
2 (6.5 %) |
0 | 280 ± 44 |
|
Ng2/Cspg4-Cre; Trp53flox/− |
16 |
13 (81.2 %) |
3 (18.8 %) |
0 | 0 | 297 ± 61 |
| Male | 4 | 2 (50.0 %) |
2 (50.0 %) |
0 | 0 | 212 ± 49 |
| Female | 12 | 11 (91.7 %) |
1 (8.3 %) |
0 | 0 | 318 ± 43 |
|
Ng2/Cspg4-CreE R; Trp53flox/flox |
30 |
20 (66.0 %) |
6 (20 %) |
0 | 0 | 361± 59 |
| Male | 10 | 6 | 2 | 0 | 0 | 324 ± 64 |
| Female | 20 | 14 | 4 | 0 | 0 | 380 ± 56 |
|
NG2-Cre; Trp53flox/+ |
12 | 0 |
1 (8.3 %) |
0 | 0 | 407 |
Figure 2. Deletion of Trp53 in Ng2/Cspg4 expressing cells causes sarcomas, while expression of a stabilized form of β-catenin in Ng2/Cspg4 expressing cells causes desmoid tumors.
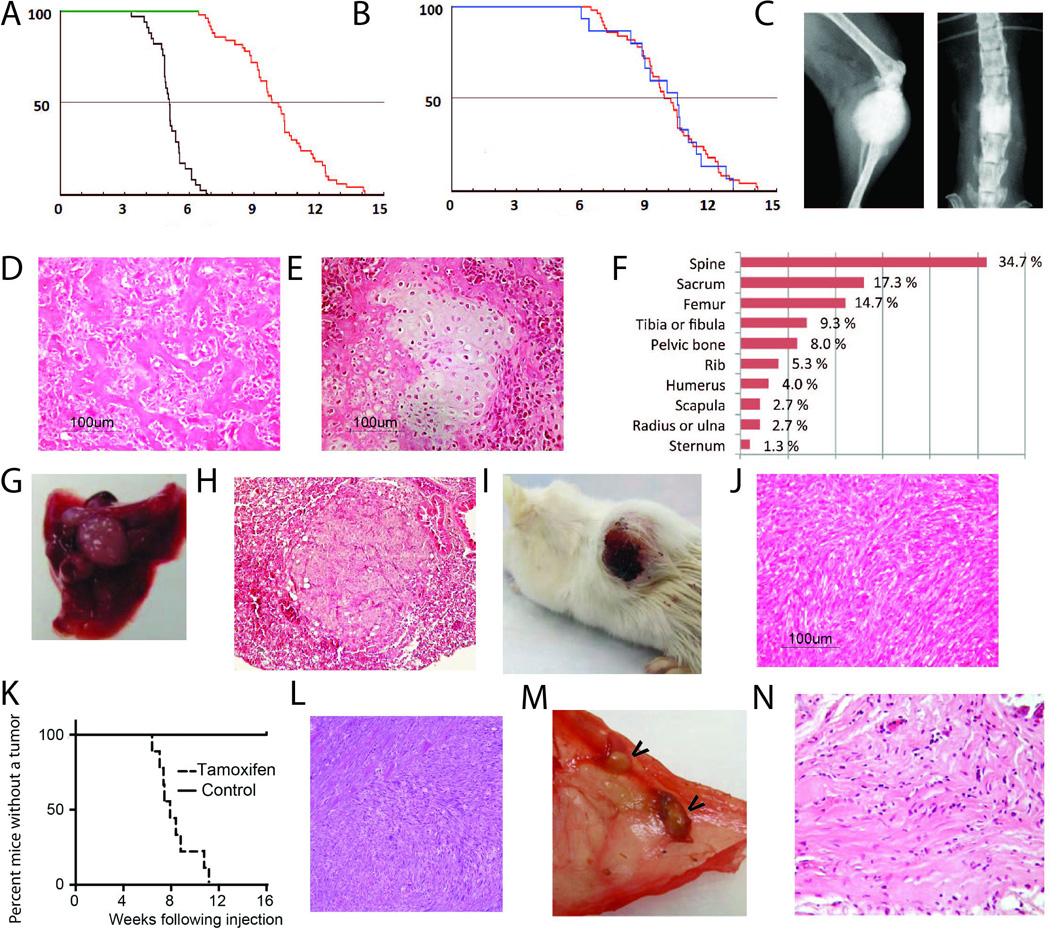
A) Kaplan-Meier survival curves in months of survival for mice expressing conditional Trp53 null alleles driven by Ng2/Cspg4-Cre (red curve), and mice expressing a germline deletion of Trp53 in both alleles (green curve). There is a significantly better survival in mice expressing Trp53 null alleles only in Ng2/Cspg4 expressing cells (p<0.01). B) Kaplan-Meier survival curves in months of survival for Ng2/Cspg4-Cre;Trp53flox/flox (green curve) and Ng2/Cspg4-Cre;Trp53flox/− (red curve) showing little difference in survival. C to H) Osteosarcomas developed in the mice lacking Trp53 in Ng2/Cspg4 expressing cells. C) Radiographs, D) histology showing an osteoblastic or E) a rare chondroblastic phenotype, F) anatomic location of the osteosarcomas, and G and H) lung metastasis that developed. I and J) Soft tissue sarcomas developed in mice lacking Trp53 in Ng2/Cspg4 expressing cells. Gross (I) and histologic (J) view of a tumor, showing typical histology for an undifferentiated pleomorphic sarcoma. K) Kaplan-Meier curves in weeks following localized 4-hydroxy-tamoxifen intramuscular injection in Ng2/Cspg4-CreER;Trp53flox/flox;KrasG12D mice for the development of a palpable tumor. L) Typical histology of the soft tissue tumors that developed, an appearance consistent with undifferentiated pleomorphic sarcoma. M) Gross view of desmid tumors in the peritoneum of a mouse (arrows show tumors). N) Histology showing a typical appearance of a desmoid tumor that developed following tamoxifen regulated activation of the conditional stabilized β-catenin allele.
We then generated Ng2/Cspg4-Cre;Trp53flox/+ and Ng2/Cspg4-Cre;Trp53flox/− mice. The mice expressing only the one conditional allele rarely developed tumors, while the mice also expressing a null allele developed tumors at a frequency equivalent to Ng2/Cspg4-Cre;Trp53flox/flox mice, and had a nearly identical survival curve (Fig. 2B), providing additional support to the concept that loss of Trp53 specifically in an Ng2/Cspg4 expressing cell predisposes to sarcoma formation.
The bone sarcomas displayed poorly-marginated masses with osteoid formation, an appearance characteristic of osteosarcoma (Fig. 2C and D). There was some heterogeneity in the cytology, with two tumors displaying chondroblastic-type osteosarcoma characteristics (Fig. 2E). The osteosarcomas arose from multiple bones (Fig. 2F). 20 % of mice showed lung metastases (Fig. 2G and H). Using X-chromosome inactivation, as previously reported (Tsunashima et al., 1996), we found the same pattern of inactivation in the bone and lung lesions from female mice, suggesting that the lesions derived from the same initial tumor (Fig. S3).
Soft tissue sarcomas were also detected in mice (Fig. 2I). They arose from multiple tissues including the cutaneous tissues, retroperitoneum, muscle, and in one case from the uterus. The soft tissue sarcomas were characterized by spindle shaped cells forming rough bundles and fascicles with hyperchromatic nuclei and abundant atypical mitoses (Fig. 2J). These sarcomas were consistent with undifferentiated pleomorphic sarcomas. Mice developing soft tissue sarcomas did not show distant metastasis.
Localized Trp53 deletion and expression of KrasG12D in Ng2/Cspg4 expressing cells induces soft tissue sarcomas
We then investigated a mouse in which soft tissue sarcomas can be generated using an inducible KrasG12D mutation and Trp53 deletion driven by cre-recombinase (Kirsch et al., 2007). These mice develop localized sarcomas when injected with a virus expressing Cre-recombinase into muscle. To determine if Ng2/Cspg4 expressing cells would drive soft tissue sarcomas in LSL-KrasG12D;Trp53flox/flox mice, we crossed them with Ng2/Cspg4-CreER mice, and drove expression of the conditional alleles using localized tamoxifen injection into muscle (Blum et al., 2013). In this way, recombination would occur in Ng2/Cspg4 expressing cells at the injection site. 12 mice were studied and they developed a palpable soft tissue lesion 12 weeks following injection (Fig. 2K), resulting in tumors with histology identical to that seen in the soft tissue sarcomas generated by Trp53 deletion in the same cell types (Fig. 2L).
β-catenin stabilization in Ng2/Cspg4 expressing cells induces the formation of desmoid tumors
To determine if stabilizing β-catenin mutations could induce desmoid tumors, we crossed Ng2/Cspg4-CreER mice with β-catenin conditionally stabilized Ctnnb1ex3 mice (Harada et al., 1999). The conditional β-catenin Ctnnb1ex3 allele lacks the phosphorylation sites in the amino terminal of β-catenin, preventing its ubiquitin mediated degradation, thus activating β-catenin mediated transcription. Mice were treated with tamoxifen, and developed desmoid tumors, with an identical histology as seen in other murine desmoid tumors, including infiltration into local muscle tissues (Figs. 2M and N)
Sarcomas from Ng2/Cspg4-CreER;Trp53flox/flox mice show expression of genes similar to those seen in human tumors
A similar microarray platform as has been used in human tumors was used to compare mRNA from sarcomas that developed in Ng2/Cspg4-CreER;Trp53flox/flox with human tumors. The mouse sarcoma data were deposited in the GEO database, GSE63631. Gene expression data from a variety of human tumors were downloaded from the Gene Expression Omnibus (GSE2553). Differential expression was compiled as a gene set which was compared to expression data from various human cancer types, and was called for each gene within each cancer type comparing it to the aggregate of all other cancer types using a moderated t-statistic. Gene Set Enrichment Analysis (Mootha et al., 2003) was carried out to identify the significance of enrichment of the mouse genes with the most differentially expressed human genes that differentiate each cancer type. This showed the strongest similarities between the same mouse and human sarcoma subtypes (Fig. 3). The expression pattern for the soft tissue sarcomas was nearly identical to that previously reported (Mito et al., 2009).
Figure 3. Comparative expression between mouse and human tumors showing similarities between mouse and human osteosarcomas.

A) Gene Set Enrichment Analysis demonstrating that genes differentially regulated in mouse osteosarcoma are significantly enriched in human osteosarcoma. B) Table showing Gene Set Enrichment Analysis statistics for comparison of genes differentially regulated in mouse osteosarcoma and various subtypes of human sarcoma.
Mouse sarcomas express genes that are distinct from Trp53 mutant cells from which they derive
Ng2/Cspg4 expressing cells were dissociated from non-cancerous skeletal muscle and sorted using an Ng2/Cspg4 antibody. RNA was extracted and RNA sequencing performed to determine gene expression differences between these cells and sarcomas that developed in the same mice. The data were deposited in the GEO database, GSE63679. Differentially expressed genes were analyzed using Gene Set Enrichment Analysis (Mootha et al., 2003), identifying the differential regulation of multiple genes associated with decreased β-catenin signaling in the tumors compared to the Ng2/Cspg4 expressing cells (Figs. 4A and B). Using RT-PCR we verified differential expression of several β-catenin transcriptional target genes (Figs. 4C and D). While both activation or inactivation of β-catenin transcription in sarcomas have been reported (Dieudonne et al., 2010; Hoang et al., 2004; Iwao et al., 1999; Iwaya et al., 2003; Lin et al., 2013; Matushansky et al., 2007; Sakamoto et al., 2002; Wan et al., 2014), our data showed that β-catenin mediated transcription was inactivated in both the bone and soft tissue sarcomas compared to the Ng2/Cspg4 expressing cells.
Figure 4. β-catenin mediated signaling is downregulated in sarcoma, and its activation inhibits tumor growth.
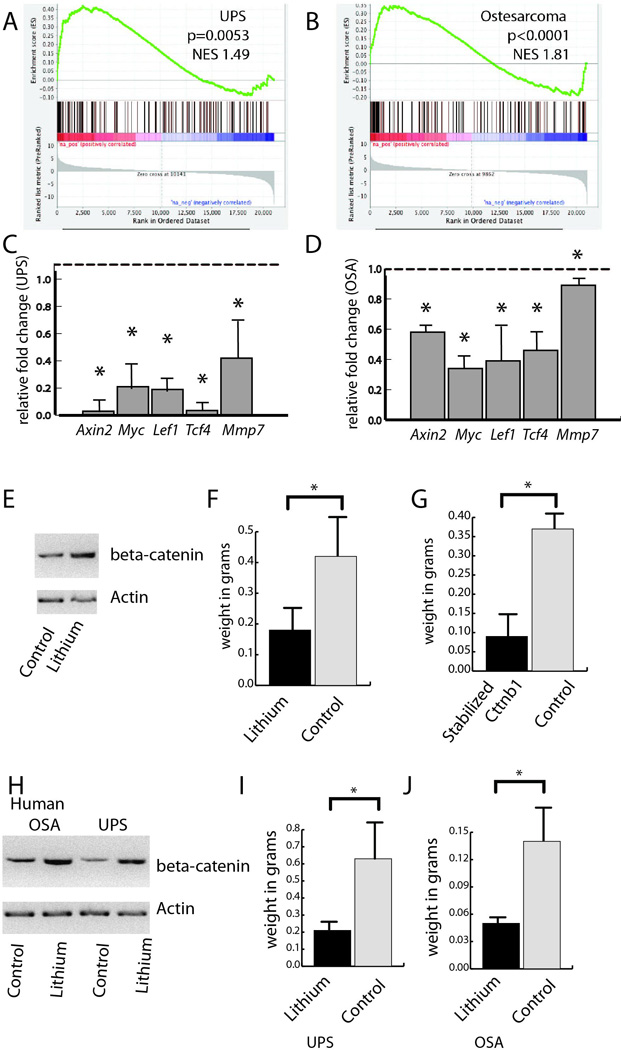
A and B) Gene Set Enrichment Analysis from RNA sequencing data comparing tumors to Ng2/Cspg4 expressing cells lacking Trp53 showing inhibition of β-catenin mediated signaling in both soft tissue sarcomas (A) and osteosarcomas (B). C and D) RT-PCR verifying downregulation of expression of β-catenin transcriptional target genes in murine tumors, soft tissue sarcomas (C) and osteosarcomas (D). Data are given as relative expression compared to Ng2/Cspg4 expressing cells lacking Trp53, which is arbitrarily defined as "1." E and F) Treatment of soft tissue sarcomas from Trp53flox/flox;KrasG12D mice grafted into immunodeficient mice treated with lithium results in a five fold increase in β-catenin protein level (E), and a significantly lower weight in grams (F) as compared to controls. G) Soft tissue sarcomas from Trp53flox/flox;KrasG12D mice expressing a stabilized version of beta-catenin grafted into immunodeficient mice had a significantly lower weight in grams as compared to controls. H to J) Treatment of human undifferentiated pleomorphic sarcomas and osteosarcomas established as xeongrafts in immunodeficient mice with lithium results in a more than five fold increase β-catenin protein level in both tumor types (H) and a lower weight in grams in treated soft tissue sarcomas (I) and osteosarcomas (J). Means and 95 % confidence intervals are shown for all data, with an asterisk indicating a decline with a p < 0.05.
Activation of β-catenin suppresses sarcoma development and growth
To determine the role of β-catenin stabilization in sarcoma formation, we generated Ng2/Cspg4-Cre;Trp53flox/flox;;KrasG12D;Ctnnb1ex3 mice in which Ng2/Cspg4 expressing cells would harbor a mutation causing sarcomas, and also express a stabilized form of β-catenin. When the conditional alleles were activated using tamoxifen mice did not develop tumors. Since mice expressing the Ctnnb1ex3 allele would not form tumors, we analyzed cells from sarcomas induced by a localized injection of an adenovirus expressing cre recombinase into Trp53flox/flox;; KrasG12D mice. Sarcoma cells were dissociated and studied as grafts in immunodeficient NOD-scid IL2rγnull mice (Wang et al., 2012). 10,000 cells were implanted subcutaneously along with matrigel. β-catenin was activated by adding lithium to the drinking water at a known effective dose (Chen et al., 2007). As a second approach, the cell cultures were infected with a lentivirus expressing the ΔN89-β-catenin (a stabilized form of β-catenin that retains its signaling functions) construct, or an empty control, as previously reported (Fuerer and Nusse, 2010; Li et al., 1998). With lithium treatment (Fig. 4E and F) or with expression of the stabilized form of β-catenin (Fig. 4G), there was a significantly smaller weight to the tumors after four weeks.
Individual cells from ten primary human osteosarcomas or undifferentiated pleomorphic sarcomas were used to establish xenografts in immunodeficient NOD-scid IL2rγnull mice as previously reported (Wang et al., 2012). Similar to the work in murine tumors, mice were treated with lithium (Chen et al., 2007). This resulted in a substantial increase in β-catenin protein level in the tumor tissues (Fig. 4H), and a substantial decrease in tumor volume (Figs. 4I and J). These data are consistent with the notion that β-catenin transcriptional activity is lower in sarcomas than in the cells they arise from, and that stabilization of β-catenin in sarcomas can suppress tumor growth.
Discussion
Identifying the cell of origin of tumors is critical to determine the genetic events important in neoplastic progression, and to develop models of cancers in mice that more accurately reflect human disease. However, for common sarcomas, the precise cellular origin is unclear. Since sarcomas have mesenchymal properties, mesenchymal stromal cells (MSCs) have been investigated as the cell of origin. Indeed, driving expression of oncogenes in this cell type can give rise to sarcomas (Mohseny et al., 2009; Rubio et al., 2013; Shimizu et al., 2010; Xiao et al., 2013). However, MSCs are a heterogeneous population of cells. Pericytes are mesenchymal cells that surround endothelial cells, have a multi-differentiation mesenchymal potential, and express genes that can be used as lineage markers in vivo (Covas et al., 2008; Crisan et al., 2008; Dellavalle et al., 2007). Our studies showed that both sarcomas and desmoid tumors can derive from Ng2/Cspg4 expressing cells, most likely from pericytes. By also using tamoxifen-inducible mice, our interpretation of the cell of origin for these mesenchymal tumors is not confounded by unanticipated expression of Cre during development, which has been shown for some constitutive Cre lines, such as Myf6-Cre (Sambasivan et al., 2013).
Ng2/Cspg4 is expressed not only in pericytes but in other cell types. As such, our lineage tracing studies with Ng2/Cspg4-Cre mice cannot rule out the possibility that the mouse tumors are derived from other Ng2/Cspg4 expressing cells. However, our analysis of LacZ labeled cells in the absence of tumors in the limbs show that Ng2/Cspg4 expressing cells express high levels of the pericyte marker CD146. It is also possible that sarcoma cells in this model might activate Ng2/Cspg4 expression during early tumorigenesis and thereby lineage tag the tumor cells. While this possibility cannot be completely eliminated, the generation of sarcomas in Ng2/Cspg4-CreER mice following tamoxifen-inducible Cre suggests that the cell of origin of these mesenchymal tumors expresses Ng2/Cspg4 at tumor initiation. Mice with Trp53 mutations in Ng2/Cspg4 developed both osteogenic and soft tissue sarcomas a finding consistent with the notion that pericytes can differentiate into a variety of mesenchymal cell types. The concept that a mesenchymal progenitor can form both bone and soft tissue sarcomas is in agreement with data from driving oncogenic mutations in MSCs, showing that the same mutation in MSCs can result in either bone or soft tissue sarcomas (Rubio et al., 2010; Rubio et al., 2013). Furthermore, we found that different mutations in the same cell type can cause different mesenchymal tumors. Driving a stabilized form of β-catenin in Ng2/Cspg4 expressing cells results in desmoid tumors, while Trp53 deletion causes sarcomas. Therefore, the same cell of origin can give rise to a variety of benign and malignant tumor types, with the type of mutation determining the tumor type that develops.
Tumors are intimately related to non-neoplastic stromal cells, but since sarcomas have mesenchymal characteristics, the identification of such cells in sarcomas has been problematic. In our lineage tracing studies, we found that sarcomas contain a subpopulation of mesenchymal cells that do not stain for LacZ. These cells likely represent a population of reactive mesenchymal stromal cells within the sarcomas. The intermingling of reactive stromal cells within the neoplastic mesenchymal cells raises complexity in the interpretation of pathologic data in these tumor types, as it is difficult to distinguish these stromal cells from the neoplastic cells. Indeed, some of the controversy regarding roles in cell signaling activation and gene expression in these tumor types may be related to detecting expression or biologic findings from these normal cells. This is a notion supported by the finding of normal mesenchymal progenitor cells in human sarcomas (Morozov et al., 2010).
Other mouse osteosarcoma models have been developed based on the conditional deletion or mutation of Trp53 (Berman et al., 2008; Lin et al., 2009; Walkley et al., 2008). However, driving deletion in a subset of cells and developing a sarcoma does not necessarily identify a specific cell of origin. In our work, a combination of lineage tracing and targeted deletion supports the pericyte as a cell of origin. The anatomic distribution of tumors in our mouse model closely mimics the situation in human sarcomas, the distal femur, proximal tibia, and proximal humerus. In contrast, driving deletion of Trp53 in other cell types such as osteoblasts results in 80 % lesions in axial skeletal sites (Berman et al., 2008; Lin et al., 2009; Walkley et al., 2008).
Desmoid tumors are a clonal proliferation of mesenchymal cells driven by mutations in APC or CTNNB1 driving β-catenin protein stabilization(Alman et al., 1997a; Alman et al., 1997b; Tejpar et al., 1999). Interestingly, while a subset of desmoid tumors were previously thought to be mutation negative when analyzed by traditional Sanger sequencing, deep sequencing(Aitken et al., 2015) found that most of these mutation negative tumors do indeed harbor mutations. Similar to the finding in sarcomas, not all desmoid tumor cells in the mice stained for LacZ in the lineage tracing studies, and these non-staining cells may be non-neoplastic stromal cells in the tumors. A high proportion of normal cells in a tumor mass can mask the detection of a mutation using traditional Sanger sequencing. Previous studies of mice that develop desmoid tumors (Cheon et al., 2002; Smits et al., 1998), are limited for studies comparing the tumors to normal cells, as the most appropriate normal cell control is unknown. Here we found a source of normal precursor cells for such analysis. The finding that these tumors derive from perciytes is consistent with data showing a correlation between numbers of mesenchymal progenitors and numbers of desmoid tumors that from in Apc mutant mice(Wu et al., 2010).
The role of β-catenin in sarcomas has been controversial, with both activation and inhibition reported. In addition, both activation and inhibition are suggested to increase tumor invasiveness (Chen et al., 2007; Hoffman and Benoit, 2013; Li et al., 2008; Wan et al., 2013). We found that β-catenin is inactivated in the sarcomas compared to the Ng2/Cspg4 expressing cell from which they derive. One possibility is that the undifferentiated pericytes maintain a high β-catenin level and this must be downregulated for differentiation into cells that become sarcomas. While our data cannot rule out this possibility, the finding that stabilization of β-catenin in these cells results in desmoid tumors, suggests that high β-catenin alone does not maintain the perciytes in a native undifferentiated state. Furthermore, driving β-catenin stabilization in sarcomas also suppressed tumor growth. Thus, similar to the situation in mesenchymal cell differentiation during development and repair, β-catenin protein level is important in mesenchymal neoplasia, with higher or lower levels contributing to pathology. In this situation, its activation causes a benign locally invasive tumor, but its inhibition is required for sarcoma formation. This is a notion similar to that in colon cancer, where β-catenin needs to be maintained at the right level for cancer development(Albuquerque et al., 2002). The requirement for the precise regulation of β-catenin in mesenchymal neoplasia raises the possibility that modulating its level could be developed into a therapeutic approach for these tumor types.
Methods
Genetically modified mice
We used Trp53 mutant mice (Jacks et al., 1994), Apc1638N (Smits et al., 1998), Trp53flox/flox conditional (Marino et al., 2000), LSL-KrasG12D mice (Tuveson et al., 2004), Catnbex3 mice (Harada et al., 1999), Ng2/Cspg4-Cre and Ng2/Cspg4-CreER transgenic mice (Feng et al., 2010; Zhu et al., 2008; Zhu et al., 2011), and Rosa26RlacZ reporter mice (Soriano, 1999) as previously reported. Ng2/Cspg4-CreER;Rosa26RlacZ mice, Ng2/Cspg4-CrERe;Rosa26RlacZ;Trp53+/− mice, Ng2/Cspg4-CrERe;Rosa26RlacZ;Trp53−/− mice, and Ng2/Cspg4-CreER;Rosa26RlacZ;Apc1638N mice were generated by crossing these mouse lines for lineage tracing studies. Ng2/Cspg4-CreER;Trp53flox/flox mice, Ng2/Cspg-CreER;Trp53flox/− mice, Ng2/Cspg4-CreER;Trp53flox/+ mice, Ng2/Cspg4-CreER;Catnbex3 mice Ng2/Cspg4-Cre;Trp53flox/flox mice, Ng2/Cspg4-Cre;Trp53flox/− mice, and Ng2/Cspg4-Cre;Trp53flox/+ mice were generated by crossing the mice and used to determine if driving mutations in Ng2/Cspg4 expressing cells causes tumors. In addition, Ng2/Cspg4-Cre;Trp53flox/flox;Catnbex3 mice were generated. In the case of inducible Cre strains, the transgene was activated by daily intraperitoneal injection of tamoxifen for one week after weaning. Trp53flox/flox;; LSL-KrasG12D mice (Kirsch et al., 2007), were used in crosses with Ng2/Cspg4-CreER mice to generate soft tissue sarcomas, but tamoxifen was injected locally into the muscle (Blum et al., 2013). All of the comparisons from different genotypes were performed on littermates. An equal number of male and female mice were used in each study. The end point for the Kaplan-Meier survival curve was when a mouse was found dead or was sacrificed due to poor health. Mice that were sacrificed or found dead were investigated using a systematic autopsy to identify the exact tumor type and tumor location. Radiographs of the whole bodies of mice were obtained by using the Faxitron MX20 X-ray system (Faxitron Bioptics, LLC, Tucson, AZ, USA). All mouse protocols were approved by the animal care committee of the Toronto Center for Phenogenomics or the IACUC committee of Duke University.
Quantitative PCR
Total RNAs from mouse sarcomas and non-cancerous tissues were extracted using Trizol reagent (Invitrogen). The RNAs were used to generate single-strand cDNA using SuperScript II reverse transcriptase (Invitrogen). To detect mRNA level, quantitative real-time polymerase chain reaction (RT-PCR) was performed. TaqMan primers were used and the ΔΔCt method was used for the analysis of the data.
Microarray
For microarray, total RNAs were extracted from osteosarcomas (n=4) and soft tissue sarcomas (n=4) which developed in Ng2/Cspg4-Cre;Trp53flox/flox mice. Skeletal muscle tissues (n=2) and bone marrow tissues (n=2) were used as controls. Biotinylated cRNAs were hybridized onto the Mouse WG-6 v2.0 Expression BeadChips (Illumina, San Diego, CA). To identify gene signatures differentially expressed between sarcomas and non-cancerous tissues, linear models for microarray data were used. The false discovery rate was set at 0.01, and evaluated using Benjamini and Hochberg multiple testing procedures. Differential expression was compiled as a gene set which was compared to expression data from various human cancer types, with gene expression called for each gene within each cancer type comparing it to the aggregate of all other cancer types using a moderated t-statistic. Gene Set Enrichment Analysis (Mootha et al., 2003) was carried out to identify the significance of enrichment of the mouse genes with the most differentially expressed human genes that differentiate each cancer type.
Cell sorting
To isolate Ng2/Cspg4 expressing cells, tissues were harvested from mice, and dissociated into individual cells as previously reported (Wang et al., 2012; Wu et al., 2007). Flow cytometry was used to sort Ng2/Cspg4 expressing cells using an NG2 Ab (Abcam). Sorting for LacZ (Amini-Nik et al., 2011) and CD146 (Wei et al., 2015) was performed as we previously reported.
RNA sequencing
For each sample, 10 ng of total RNA was processed using the SMARTTM cDNA synthesis protocol including SMARTScribe Reverse Transcriptase. The amplified cDNA was subject to Illumina paired-end library construction. RNA sequencing analysis was performed by the Genome Sciences Centre at the British Columbia Cancer Agency, Vancouver, Canada, using Illumina HiSeq 2000 sequencing at 75 base PET indexed lane, pooling 2 libraries per lane. Data was processed using the TrimGalore toolkit (Chen et al., 2014) to trim low quality bases and Illumina sequencing adapters from the 3’ end of the reads. Only pairs where both reads were 35 nt or longer were kept for further analysis. Reads were mapped to the GRCm38.73 version of the mouse genome and transcriptome (Kersey et al., 2012) using the STAR RNA-seq alignment tool (Dobin et al., 2013). Gene counts were compiled using the HTSeq tool (http://www-huber.embl.de/users/anders/HTSeq). Normalization and differential expression was carried out using the DESeq2 (Love et al., 2014) Bioconductor (Gentleman et al., 2004) package with the R statistical programming environment (http://www.r-project.org). A negative binomial generalized linear model was employed to identify differentially expressed genes across sample types. Pathway analyses were performed using Gene Set Enrichment Analysis (GSEA) with parameters set to 2,000 gene set per mutations and gene sets size between 8 and 500 (Subramanian et al., 2005). Gene sets were obtained from KEGG, MsigDB-c2, NCI, Biocarta, IOB, Netpath, Human Cyc, Reactome, and the Gene Ontology (GO) databases (Kanehisa and Goto, 2000; Merico et al., 2010). An enrichment map was generated using Cytoscape with parameters set for a nominal P value of < 0.005, FDR < 0.25, and the Jaccard coefficient set to 0.5 (Saito et al., 2012).
Xenograft in immunocompromised mice
Primary sarcomas were dissociated into single cells (Wang et al., 2012). 10,000 dissociated cells were suspended with Matrigel (Becton Dickinson) and were injected subcutaneously into 6-8-week-old NOD-scid IL2rγnull (NSG) mice. After injection, the mice were observed for 3 weeks, and then lithium was added to the drinking water at a dose previously shown to increase β-catenin in mesenchymal tissues (Chen et al., 2007). The ΔN89-β-catenin construct, or empty control, was used as previously reported (Fuerer and Nusse, 2010; Li et al., 1998). Western analysis using an antibody to actin as a loading control(Tejpar et al., 1999) was used to determine β-catenin levels, and the tumors were weighed using an analytical balance, as previously reported (Wang et al., 2012).
Supplementary Material
Highlights.
Pericytes can be a cell of origin for benign and malignant mesenchymal neoplasms.
Malignant sarcomas show a decrease in beta-catenin signaling compared to pericytes.
Benign desmoids show an increase in beta-catenin signaling compared to pericytes.
Acknowledgments
This research is funded by NIH grant R01 CA183811, the Canadian Institutes for health Research (FRN 123493), Restracomp fellowship of the Hospital for Sick Children Research Training Centre, and Grants for foreign study of KANAE Foundation for the Promotion of Medical Science. We thank Pigzhou Ho (The Hospital for Sick Children, Toronto, ON, Canada) for analyzing the microarray data, David Corcoran (Duke University, Durham, NC, USA) for analyzing the RNA sequencing data, and Dr. Hiroaki Kanda (Ariake Cancer Institute Hospital, Tokyo, Japan) for the pathological diagnosis of mouse tumors from our mouse models. We also wish to acknowledge Canada's Michael Smith Genome Sciences Centre (Vancouver, Canada) for performing the RNA sequencing analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
Conceptualization, B.A.A., J.S.W., S.S. and Q.W.; Methodology, B.A.A., D.G.K., S.S., Y.J.T., and S.Y.W.; Validation, A.O. and S.T.; Investigation, S.S., Y.J.T, Q.W, M.H, A.W, I.H., H.W, and P.N.; Writing – Original Draft, S.S., J.S.W., and B.A.A.; Writing – Review & Editing, B.A.A., Y.J.T., and D.G.K.; Funding Acquisition, B.A.A., J.S.W., and D.G.K.; Supervision, J.S.W., D.G.K., and B.A.A.
Accession Numbers: The mouse sarcoma microarray data were deposited in the GEO database, GSE63631. RNA sequencing data from Ng2/Cspg4 expressing cells and tumors were deposited in the GEO database, GSE63679.
Conflict of Interest
No potential conflicts of interest were disclosed.
References
- Aitken SJ, Presneau N, Kalimuthu S, Dileo P, Berisha F, Tirabosco R, Amary MF, Flanagan AM. Next-generation sequencing is highly sensitive for the detection of beta-catenin mutations in desmoid-type fibromatoses. Virchows Archiv : an international journal of pathology. 2015;467:203–210. doi: 10.1007/s00428-015-1765-0. [DOI] [PubMed] [Google Scholar]
- Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJ, Leitao CN, Fodde R, Smits R. The 'just-right' signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Human molecular genetics. 2002;11:1549–1560. doi: 10.1093/hmg/11.13.1549. [DOI] [PubMed] [Google Scholar]
- Alman BA, Li C, Pajerski ME, Diaz-Cano S, Wolfe HJ. Increased beta-catenin protein and somatic APC mutations in sporadic aggressive fibromatoses (desmoid tumors) The American journal of pathology. 1997a;151:329–334. [PMC free article] [PubMed] [Google Scholar]
- Alman BA, Pajerski ME, Diaz-Cano S, Corboy K, Wolfe HJ. Aggressive fibromatosis (desmoid tumor) is a monoclonal disorder. Diagnostic molecular pathology : the American journal of surgical pathology, part B. 1997b;6:98–101. doi: 10.1097/00019606-199704000-00005. [DOI] [PubMed] [Google Scholar]
- Amini-Nik S, Cambridge E, Yu W, Guo A, Whetstone H, Nadesan P, Poon R, Hinz B, Alman BA. beta-Catenin-regulated myeloid cell adhesion and migration determine wound healing. J Clin Invest. 2014;124:2599–2610. doi: 10.1172/JCI62059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amini-Nik S, Glancy D, Boimer C, Whetstone H, Keller C, Alman BA. Pax7 expressing cells contribute to dermal wound repair, regulating scar size through a beta-catenin mediated process. Stem cells. 2011;29:1371–1379. doi: 10.1002/stem.688. [DOI] [PubMed] [Google Scholar]
- Benassi MS, Pazzaglia L, Chiechi A, Alberghini M, Conti A, Cattaruzza S, Wassermann B, Picci P, Perris R. NG2 expression predicts the metastasis formation in soft-tissue sarcoma patients. J Orthop Res. 2009;27:135–140. doi: 10.1002/jor.20694. [DOI] [PubMed] [Google Scholar]
- Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro-oncology. 2005;7:452–464. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, Fonhoue BD, Caron A, Bronson R, Bouxsein ML, et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11851–11856. doi: 10.1073/pnas.0805462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Cai T, Chen Y. Wnt pathway in osteosarcoma, from oncogenic to therapeutic. Journal of cellular biochemistry. 2014;115:625–631. doi: 10.1002/jcb.24708. [DOI] [PubMed] [Google Scholar]
- Cai Y, Mohseny AB, Karperien M, Hogendoorn PC, Zhou G, Cleton-Jansen AM. Inactive Wnt/beta-catenin pathway in conventional high-grade osteosarcoma. The Journal of pathology. 2010;220:24–33. doi: 10.1002/path.2628. [DOI] [PubMed] [Google Scholar]
- Chen C, Khaleel SS, Huang H, Wu CH. Software for pre-processing Illumina next-generation sequencing short read sequences. Source code for biology and medicine. 2014;9:8. doi: 10.1186/1751-0473-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Whetstone HC, Lin AC, Nadesan P, Wei Q, Poon R, Alman BA. Beta-catenin signaling plays a disparate role in different phases of fracture repair: implications for therapy to improve bone healing. PLoS Med. 2007;4:e249. doi: 10.1371/journal.pmed.0040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon SS, Cheah AY, Turley S, Nadesan P, Poon R, Clevers H, Alman BA. beta-Catenin stabilization dysregulates mesenchymal cell proliferation, motility, and invasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:6973–6978. doi: 10.1073/pnas.102657399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covas DT, Panepucci RA, Fontes AM, Silva WA, Jr, Orellana MD, Freitas MC, Neder L, Santos AR, Peres LC, Jamur MC, Zago MA. Multipotent mesenchymal stromal cells obtained from diverse human tissues share functional properties and gene-expression profile with CD146+ perivascular cells and fibroblasts. Experimental hematology. 2008;36:642–654. doi: 10.1016/j.exphem.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Crisan M, Corselli M, Chen WC, Peault B. Perivascular cells for regenerative medicine. Journal of cellular and molecular medicine. 2012;16:2851–2860. doi: 10.1111/j.1582-4934.2012.01617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell stem cell. 2008;3:301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, Innocenzi A, Galvez BG, Messina G, Morosetti R, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nature cell biology. 2007;9:255–267. doi: 10.1038/ncb1542. [DOI] [PubMed] [Google Scholar]
- Diaz-Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E, Martin-Vasallo P, Diaz-Flores L., Jr Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histology and histopathology. 2009;24:909–969. doi: 10.14670/HH-24.909. [DOI] [PubMed] [Google Scholar]
- Dieudonne FX, Marion A, Hay E, Marie PJ, Modrowski D. High Wnt signaling represses the proapoptotic proteoglycan syndecan-2 in osteosarcoma cells. Cancer research. 2010;70:5399–5408. doi: 10.1158/0008-5472.CAN-10-0090. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Yang J, Yang D, Tian W, Zhu Z. The genetic basis for inactivation of Wnt pathway in human osteosarcoma. BMC cancer. 2014;14:450. doi: 10.1186/1471-2407-14-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Mantesso A, De Bari C, Nishiyama A, Sharpe PT. Dual origin of mesenchymal stem cells contributing to organ growth and repair. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:6503–6508. doi: 10.1073/pnas.1015449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Mantesso A, Sharpe PT. Perivascular cells as mesenchymal stem cells. Expert opinion on biological therapy. 2010;10:1441–1451. doi: 10.1517/14712598.2010.517191. [DOI] [PubMed] [Google Scholar]
- Fuerer C, Nusse R. Lentiviral vectors to probe and manipulate the Wnt signaling pathway. PloS one. 2010;5:e9370. doi: 10.1371/journal.pone.0009370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome biology. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. The EMBO journal. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschi KK, D'Amore PA. Pericytes in the microvasculature. Cardiovascular research. 1996;32:687–698. [PubMed] [Google Scholar]
- Hoang BH, Kubo T, Healey JH, Yang R, Nathan SS, Kolb EA, Mazza B, Meyers PA, Gorlick R. Dickkopf 3 inhibits invasion and motility of Saos-2 osteosarcoma cells by modulating the Wnt-beta-catenin pathway. Cancer research. 2004;64:2734–2739. doi: 10.1158/0008-5472.can-03-1952. [DOI] [PubMed] [Google Scholar]
- Hoffman MD, Benoit DS. Agonism of Wnt-beta-catenin signalling promotes mesenchymal stem cell (MSC) expansion. Journal of tissue engineering and regenerative medicine. 2013 doi: 10.1002/term.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwao K, Miyoshi Y, Nawa G, Yoshikawa H, Ochi T, Nakamura Y. Frequent beta-catenin abnormalities in bone and soft-tissue tumors. Japanese journal of cancer research : Gann. 1999;90:205–209. doi: 10.1111/j.1349-7006.1999.tb00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaya K, Ogawa H, Kuroda M, Izumi M, Ishida T, Mukai K. Cytoplasmic and/or nuclear staining of beta-catenin is associated with lung metastasis. Clinical & experimental metastasis. 2003;20:525–529. doi: 10.1023/a:1025821229013. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Current biology : CB. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey PJ, Staines DM, Lawson D, Kulesha E, Derwent P, Humphrey JC, Hughes DS, Keenan S, Kerhornou A, Koscielny G, et al. Ensembl Genomes: an integrative resource for genome-scale data from non-vertebrate species. Nucleic acids research. 2012;40:D91–D97. doi: 10.1093/nar/gkr895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, Nielsen GP, Quade BJ, Chaber CJ, Schultz CP, et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nature medicine. 2007;13:992–997. doi: 10.1038/nm1602. [DOI] [PubMed] [Google Scholar]
- Li C, Bapat B, Alman BA. Adenomatous polyposis coli gene mutation alters proliferation through its beta-catenin-regulatory function in aggressive fibromatosis (desmoid tumor) The American journal of pathology. 1998;153:709–714. doi: 10.1016/s0002-9440(10)65614-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HX, Luo X, Liu RX, Yang YJ, Yang GS. Roles of Wnt/beta-catenin signaling in adipogenic differentiation potential of adipose-derived mesenchymal stem cells. Molecular and cellular endocrinology. 2008;291:116–124. doi: 10.1016/j.mce.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Lin CH, Guo Y, Ghaffar S, McQueen P, Pourmorady J, Christ A, Rooney K, Ji T, Eskander R, Zi X, Hoang BH. Dkk-3, a secreted wnt antagonist, suppresses tumorigenic potential and pulmonary metastasis in osteosarcoma. Sarcoma. 2013;2013:147541. doi: 10.1155/2013/147541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PP, Pandey MK, Jin F, Raymond AK, Akiyama H, Lozano G. Targeted mutation of p53 and Rb in mesenchymal cells of the limb bud produces sarcomas in mice. Carcinogenesis. 2009;30:1789–1795. doi: 10.1093/carcin/bgp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer metastasis reviews. 2013;32:303–315. doi: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes & development. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- Matushansky I, Hernando E, Socci ND, Mills JE, Matos TA, Edgar MA, Singer S, Maki RG, Cordon-Cardo C. Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. J Clin Invest. 2007;117:3248–3257. doi: 10.1172/JCI31377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PloS one. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito JK, Riedel RF, Dodd L, Lahat G, Lazar AJ, Dodd RD, Stangenberg L, Eward WC, Hornicek FJ, Yoon SS, et al. Cross species genomic analysis identifies a mouse model as undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma. PloS one. 2009;4:e8075. doi: 10.1371/journal.pone.0008075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohseny AB, Szuhai K, Romeo S, Buddingh EP, Briaire-de Bruijn I, de Jong D, van Pel M, Cleton-Jansen AM, Hogendoorn PC. Osteosarcoma originates from mesenchymal stem cells in consequence of aneuploidization and genomic loss of Cdkn2. The Journal of pathology. 2009;219:294–305. doi: 10.1002/path.2603. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Morozov A, Downey RJ, Healey J, Moreira AL, Lou E, Franceschino A, Dogan Y, Leung R, Edgar M, LaQuaglia M, et al. Benign mesenchymal stromal cells in human sarcomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:5630–5640. doi: 10.1158/1078-0432.CCR-09-2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Rubio R, Garcia-Castro J, Gutierrez-Aranda I, Paramio J, Santos M, Catalina P, Leone PE, Menendez P, Rodriguez R. Deficiency in p53 but not retinoblastoma induces the transformation of mesenchymal stem cells in vitro and initiates leiomyosarcoma in vivo. Cancer research. 2010;70:4185–4194. doi: 10.1158/0008-5472.CAN-09-4640. [DOI] [PubMed] [Google Scholar]
- Rubio R, Gutierrez-Aranda I, Saez-Castillo AI, Labarga A, Rosu-Myles M, Gonzalez-Garcia S, Toribio ML, Menendez P, Rodriguez R. The differentiation stage of p53-Rb-deficient bone marrow mesenchymal stem cells imposes the phenotype of in vivo sarcoma development. Oncogene. 2013;32:4970–4980. doi: 10.1038/onc.2012.507. [DOI] [PubMed] [Google Scholar]
- Saito R, Smoot ME, Ono K, Ruscheinski J, Wang PL, Lotia S, Pico AR, Bader GD, Ideker T. A travel guide to Cytoscape plugins. Nature methods. 2012;9:1069–1076. doi: 10.1038/nmeth.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto A, Oda Y, Adachi T, Saito T, Tamiya S, Iwamoto Y, Tsuneyoshi M. Beta-catenin accumulation and gene mutation in exon 3 in dedifferentiated liposarcoma and malignant fibrous histiocytoma. Archives of pathology & laboratory medicine. 2002;126:1071–1078. doi: 10.5858/2002-126-1071-CAAGMI. [DOI] [PubMed] [Google Scholar]
- Schiano C, Grimaldi V, Casamassimi A, Infante T, Esposito A, Giovane A, Napoli C. Different expression of CD146 in human normal and osteosarcoma cell lines. Medical oncology. 2012;29:2998–3002. doi: 10.1007/s12032-012-0158-3. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Ishikawa T, Sugihara E, Kuninaka S, Miyamoto T, Mabuchi Y, Matsuzaki Y, Tsunoda T, Miya F, Morioka H, et al. c-MYC overexpression with loss of Ink4a/Arf transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene. 2010;29:5687–5699. doi: 10.1038/onc.2010.312. [DOI] [PubMed] [Google Scholar]
- Smits R, van der Houven van Oordt W, Luz A, Zurcher C, Jagmohan-Changur S, Breukel C, Khan PM, Fodde R. Apc1638N: a mouse model for familial adenomatous polyposis-associated desmoid tumors and cutaneous cysts. Gastroenterology. 1998;114:275–283. doi: 10.1016/s0016-5085(98)70478-0. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejpar S, Nollet F, Li C, Wunder JS, Michils G, dal Cin P, Van Cutsem E, Bapat B, van Roy F, Cassiman JJ, Alman BA. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor) Oncogene. 1999;18:6615–6620. doi: 10.1038/sj.onc.1203041. [DOI] [PubMed] [Google Scholar]
- Tsunashima K, Endo Y, Asakura H, Kanda H, Nomura K, Kitagawa T, Kominami R. A novel clonality assay for the mouse: application to hepatocellular carcinomas induced with diethylnitrosamine. Molecular carcinogenesis. 1996;15:33–37. doi: 10.1002/(SICI)1098-2744(199601)15:1<33::AID-MC5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, Rodda SJ, Snay E, Dunning P, Fahey FH, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes & development. 2008;22:1662–1676. doi: 10.1101/gad.1656808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Lu C, Cao J, Zhou R, Yao Y, Yu J, Zhang L, Zhao H, Li H, Zhao J, et al. Osteoblastic Wnts differentially regulate bone remodeling and the maintenance of bone marrow mesenchymal stem cells. Bone. 2013;55:258–267. doi: 10.1016/j.bone.2012.12.052. [DOI] [PubMed] [Google Scholar]
- Wan Y, Zhao W, Jiang Y, Liu D, Meng G, Cai Y. beta-catenin is a valuable marker for differential diagnosis of osteoblastoma and osteosarcoma. Human pathology. 2014;45:1459–1465. doi: 10.1016/j.humpath.2014.02.022. [DOI] [PubMed] [Google Scholar]
- Wang CY, Wei Q, Han I, Sato S, Ghanbari-Azarnier R, Whetstone H, Poon R, Hu J, Zheng F, Zhang P, et al. Hedgehog and Notch signaling regulate self-renewal of undifferentiated pleomorphic sarcomas. Cancer research. 2012;72:1013–1022. doi: 10.1158/0008-5472.CAN-11-2531. [DOI] [PubMed] [Google Scholar]
- Wei Q, Tang YJ, Voisin V, Sato S, Hirata M, Whetstone H, Han I, Ailles L, Bader GD, Wunder J, Alman BA. Identification of CD146 as a marker enriched for tumor-propagating capacity reveals targetable pathways in primary human sarcoma. Oncotarget. 2015;6:40283–40294. doi: 10.18632/oncotarget.5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Amini-Nik S, Nadesan P, Stanford WL, Alman BA. Aggressive fibromatosis (desmoid tumor) is derived from mesenchymal progenitor cells. Cancer research. 2010;70:7690–7698. doi: 10.1158/0008-5472.CAN-10-1656. [DOI] [PubMed] [Google Scholar]
- Wu C, Wei Q, Utomo V, Nadesan P, Whetstone H, Kandel R, Wunder JS, Alman BA. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer research. 2007;67:8216–8222. doi: 10.1158/0008-5472.CAN-07-0999. [DOI] [PubMed] [Google Scholar]
- Xiao W, Mohseny AB, Hogendoorn PC, Cleton-Jansen AM. Mesenchymal stem cell transformation and sarcoma genesis. Clinical sarcoma research. 2013;3:10. doi: 10.1186/2045-3329-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]
- Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A. Age-dependent fate and lineage restriction of single NG2 cells. Development. 2011;138:745–753. doi: 10.1242/dev.047951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.