

Abstract
Microbial fermentation provides as an attractive alternative to chemical synthesis for the production of structurally complex natural products. In most cases, however, production titers are low and need to be improved for compound characterization and/or commercial production. Owing to advances in functional genomics and genetic engineering technologies, microbial hosts can be engineered to overproduce a desired natural product, greatly accelerating the traditionally time-consuming strain improvement process. This review covers recent developments and challenges in the engineering of native and heterologous microbial hosts for production of bacterial natural products, focusing on the genetic tools and strategies for strain improvement. Special emphasis is placed on bioactive secondary metabolites from actinomycetes. The considerations for the choice of host systems will also be discussed in this review.
Graphical Abstract
1. Introduction
The importance of natural products in medicine and health is indisputable and documented by civilizations throughout human history.1 Today, natural products from plants and microorganisms remain a fertile source of pharmaceuticals and bioactive scaffolds.2 Beside their enormous structural complexity and diversity, natural products are evolutionarily selected over millions of years for interactions with biomolecules using core chemical scaffolds that are optimized for bioactivity. Indeed, the subnanomolar potency and striking target specificity of these privileged chemical scaffolds are used to probe the functional contributions of individual proteins and signaling pathways in key biological processes, understand biological phenomena, discover new therapeutic drug targets and inspire new drug designs.2, 3
With breakthroughs in whole genome sequencing and functional genomics, it is increasingly apparent that genetic engineering of natural product-producing microorganisms can address several major challenges associated with natural product discovery and development. Large-scale microbial fermentation provides an attractive alternative to the otherwise challenging and often impractical chemical syntheses of the structurally diverse and complex natural products or their derivatives. With biosynthesis, enantiopure compounds can be attained without generation of diastereomeric byproducts, purification of intermediates and tedious work-ups after each step of chemical synthesis. Additionally, microbial production allows reliable natural product supply from cheap renewable starting materials. These cellular factories can be naturally occurring microorganisms with the innate biosynthetic capability to make the target natural product (native hosts) or surrogate hosts into which the specific genetically encoded biosynthetic capability for making the target secondary metabolite is introduced (heterologous hosts). For both native and heterologous production systems, the microbial host producing the target secondary metabolite can be rationally engineered to improve production titers and reduce unwanted byproducts. During natural product discovery, high production titers can facilitate compound validation, isolation and identification from complex microbial extracts, which can be extremely challenging if the target compound is produced at low levels. During the downstream production phase, high production titers will be needed for an economically viable industrial bioprocess.
This review covers recent advances in the engineering of native and heterologous microbial hosts for the overproduction of natural products from bacteria. Special emphasis will be placed on biosynthetic gene clusters (BGCs) and secondary metabolites from actinomycetes, which produce a large proportion of the bioactive compounds from bacteria. The advantages and limitations of native and various heterologous host production systems are discussed in sections 2 and 5 respectively. In section 3, we focus on the tools for genetic engineering of actinomycetes, including the development of genetic parts and genome editing technologies. Section 4 covers the application of these technologies to improve the production of target secondary metabolites in native actinomycete producers by directing metabolic flux towards desired products, manipulating regulatory pathways, and engineering cellular translational and transcriptional machineries. In section 6, we highlight the conceptual and technological advances for the engineering of heterologous host systems, including dynamic metabolic regulation, genome-scale engineering and genome minimization. Other related topics of host engineering for cryptic BGC activation or combinatorial protein, pathway and precursor engineering to generate novel natural product analogs were recently reviewed and will not be discussed in detail.4–6 We expect that many of the host engineering strategies described here can be adopted for the production of natural products from other bacteria such as Cyanobacteria and Bacillus spp. with the development of advanced genetic manipulation techniques, which are currently lacking in these systems.
2. Advantages and limitations of native host production systems
There are advantages to engineering native hosts to improve secondary metabolite production. While much progress has been made to identify BGCs and predict the functions of the genes within,7–10 identification, refactoring and reconstitution of all the necessary genes for heterologous production still involve substantial pathway engineering, especially for uncharacterized BGCs (Figure 1). In comparison, native hosts are likely to be equipped with all the necessary cellular factors to produce the natural product of interest, including those needed for precursor and product biosynthesis, pathway regulation, self-resistance and transport. For this reason, relatively smaller-scale and fewer genetic manipulations are needed to significantly improve titers and productivity in native host (Section 4). With better understanding of the regulatory pathways that control secondary metabolite production and new technologies that allow efficient genetic manipulation of native hosts such as actinomycetes, rational host engineering can greatly accelerate the strain improvement process.
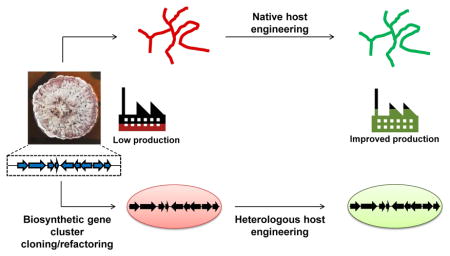
Fig. 1. Production of bacterial natural products using native and heterologous host systems.

Native producers encode all the necessary genes in a biosynthetic gene cluster (BGC) for the biosynthesis of a target natural product. The BGC can be cloned from the native host and refactored for expression in a heterologous production host. In the case of heterologous production, biosynthetic genes can also be obtained through de novo gene synthesis or cloned from environmental DNA. Additionally, the choice of hosts and pathway engineering strategies are critical in ensuring production of the desired metabolite in heterologous systems. Both native and heterologous production hosts usually produce very low amounts of the natural compound and host engineering will be needed to improve production titers and productivity to obtain sufficient quantities for product purification/identification or for commercial production. Engineered hosts with improved production properties can be used as heterologous production hosts. Further bioprocess optimization can be performed after host engineering.
On the other hand, introducing exogenous DNA can be a major hurdle for genetic manipulation of non-model strains with unknown restriction-modification (RM) systems and for which no transformation protocol exists. There are many methods of introducing recombinant DNA into actinomycetes, including variations of electroporation, chemical transformation, and interspecies conjugal transfer with highly variable efficiencies depending on the host strain.11 In general, however, transformation efficiencies in actinomycetes are still orders of magnitude lower than that in E. coli or yeast. Furthermore, RM systems that recognize and selectively degrade exogenous DNA greatly hamper genetic engineering of non-model actinomycetes. Mimicking host DNA-methylation significantly improved transformation efficiencies for multiple Streptomyces strains but determining the methylation motifs of a target host can be a lengthy process.12 Other solutions include avoidance of RM recognition sites and disruption of native RM systems.13, 14 Technologies that improve transformation or conjugation efficiencies in actinomycetes will increase the scope of native producers that can be engineered and the size of DNA libraries that can be screened for desired phenotypes. Sections 3 and 4 focus on the technologies and strategies for engineering native actinomycete hosts granted that introduction of exogenous DNA is possible.
3 Advances in genetic engineering of actinomycetes
3.1 Genetic parts for engineering of actinomycete hosts
Engineering an actinomycete host for improved natural product biosynthesis involves the disruption and rewiring of native regulatory networks, redirection of metabolic flux and controlled expression of native and heterologous genes. To do so, one needs a collection of characterized regulatory elements including promoters and ribosomal binding sites (RBSs) that cover a wide dynamic range in various actinomycetes. The GC–rich genome content (>70% GC) and unusual metabolism of actinomycetes preclude facile transfer of existing genetic elements and tools developed in more GC-balanced organisms such as Escherichia coli.15 For example, stronger promoters are more G-rich in the −10 and spacer motifs in Streptomyces lividans,16 which are in stark contrast to the AT-rich E. coli promoters. In another example, the common lacZ reporter cannot be used due to the high endogenous beta-galactosidase activity in Streptomyces. It is evident that even established genetic parts from other organisms need to be tested and characterized in actinomycetes before they can be used for strain engineering.
With bacterial gene expression predominantly regulated at the level of transcriptional activation at promoters, well-characterized promoters that function reliably in actinomycetes are crucial in efforts to rewire regulatory networks and tune gene expression for the optimization of natural product biosynthesis. Distinct but complementary strategies have greatly expanded the promoter repertoire and dynamic range in Streptomyces. The first strategy involves the cloning and characterization of native promoters. From an RNA-seq screen, Luo and coworkers identified ten constitutive housekeeping and heat/cold shock gene promoters from Streptomyces albus that are 2 to 13-fold stronger than the widely used ermE*p derived from Saccharopolyspora erythrea.17, 18 In the second strategy, synthetic promoters are generated by randomization of promoter sequences. Synthetic promoter libraries of Streptomyces vegetative promoters and the constitutive ermEp1 yielded promoters with dynamic ranges of more than 10 and 100-fold, respectively.16, 19 The third strategy involves rational promoter engineering. Starting from a native promoter with core motifs similar to the consensus sequence recognized by the S. coelicolor housekeeping sigma factor σHrdB, removal of repressor binding sites yielded the 97 bp kasO*p that is one to two orders of magnitude stronger than ermE*p in different Streptomyces strains.20 kasO*p remains one of the strongest promoters in Streptomyces. Recently, randomization of the −10 and spacer sequences of kasO*p generated 180 variants with activities spanning two orders of magnitudes, including 6 variants that are stronger than kasO*p in Streptomyces venezuelae.21 Most constitutive promoters identified using these strategies retain their function when transplanted to other Streptomyces strains,18, 20–22 but it remains to be determined if they will function similarly in rare actinomycetes (non-Streptomycetes).19 Notably, many of the strong constitutive promoters, including kasO*p, are recognized by σHrdB and exhibit growth-dependent activity, with high activity confined to vegetative growth and significantly lower activity upon exit from exponential growth.18, 20 Identification of native and synthetic promoters recognized by alternative sigma factors that are dominant during later growth stages, when majority of secondary metabolites are produced, will be useful for host engineering applications.
Inducible promoters allow the temporal coordination of gene expression to maximize the yield of a natural product of interest. Besides initiating biosynthesis when precursors are available, coordinated expression of late and early biosynthetic genes may be needed to reduce unwanted and/or toxic side products during titer improvement. Furthermore, inducible promoters can be used to temporally control gene expression for multiplex genome and transcriptome engineering (Section 3.2). Of the few available inducible promoters, the thiostrepton-inducible tipA promoter is most widely used and has been employed to modulate the expression of the T7 RNA polymerase in a T7 expression system adapted for Streptomyces.23 Though a strong promoter, tipAp exhibits significant background expression and requires the presence of the TipAL activator and thiostrepton resistance gene tsr.15 In contrast, resistance genes are not needed for the nitAp/nitR ε-caprolactam24- and anhydrotetracycline25-inducible systems.
Complementing inducible promoters are post-transcriptional inducible switches that can be paired with different promoters to tune induction strength. The theophylline riboswitch E*, tunable by varying inducer dosage and promoter combinations, can achieve 30 to 260-fold induction with low basal expression in S. coelicolor.26 Aminoglycosides-27 and neomycin-28 sensing riboswitches have been developed but their portability and utility in different actinomycetes have not been determined. Likewise, the use of short (50–155 bp) antisense sequences or synthetic peptide nucleic acids to modulate gene expression post-transcriptionally has been demonstrated but its general utility is unknown.29, 30 Employing a heterologous system of amber suppression by pyrrolysyl-tRNA synthetase and suppressor tRNAPyl in the presence of unnatural amino acid pyrrolysine,31 Myronovski and co-workers established an induction system that functions at the translational level.32 Despite the negligible background activity and high induction factor, the absolute reporter activity upon induction was relatively low,32 making this inducible platform more suitable for applications where leaky basal expression is untolerated.
A greater selection of well-characterized genetic parts enables the creation of more sophisticated genetic constructs to better engineer actinomycete hosts for natural product biosynthesis. In fact, some of the more advanced strategies for strain improvement in E. coli and yeast, such as dynamic metabolic regulation (Section 6.1) and feedback-regulated evolution (Section 6.2), can be applied to actinomycetes with assembly of the right genetic parts. Therefore, tools that facilitate quantitative characterization of individual and/or combinations of genetic regulatory elements are critical. When fused to regulatory elements, reporter genes provide quantifiable signals that can be used to monitor the performance of genetic parts and entire genetic circuits. Quantitative reporter genes shown to work in actinomycetes include gfp encoding the green fluorescent protein (GFP),21 xylE encoding for catechol 2,3-dioxygenase that converts colorless catechol to yellow hydroxymuconic semialdehyde,33 luxAB and luxCDABE operons encoding luciferases,25, 34 and gusA encoding for a beta-glucuronidase that converts different substrates for chromogenic, fluorescent, spectroscopic or chemiluminescent readouts.32 When selecting the appropriate reporter gene, important considerations include the scalability, sensitivity (signal-to-noise) and dynamic range of the assay in the host of interest, as well as the option of a real-time or end-point assay. Using superfolder GFP as a reporter, Bai and coworkers developed a flow cytometry-based single-cell quantitation method for Streptomyces and used it to characterize 200 promoters and 200 RBSs, as well as to improve the modularity of promoter-RBS pairings.21 The single-cell resolution achieved with this method enables the evaluation of population heterogeneity and eliminates the need for normalization to dry cell weight, a necessary step for enzyme-based reporter assays. More importantly, this scalable method allows for high-throughput characterization and optimization of new genetic parts, and possibly genetic circuits to enable more complex engineering of native actinomycete hosts.
3.2 Technologies for genetic engineering of actinomycetes
Genetic manipulation of actinomycetes has traditionally been challenging due to their GC-rich genomes and our incomplete understanding of their native DNA repair mechanisms. The development and application of integrases, homing and RNA-guided endonuclease technologies have significantly improved the genetic tractability of these prolific natural product producers (Figure 2). Complemented by the expanding collection of genetic parts for actinomycetes (Section 3.1), these technologies will be critical for rational strain engineering and accelerating strain improvement programs for secondary metabolite production.
Fig. 2. Tools for site-specific genetic engineering of actinomycetes.

(A) Genomic integrations and deletions mediated by site-specific phage recombinases. Serine integrases mediate the integration reactions between cognate attP sites with endogenous attB sites in genome (left scheme). The integrated fragment is flanked by new attachment sites (attR, attL). Cre (loxP) and FLP (FRT) tyrosine recombinases recognize and favor the excision of recombination sites-flanked regions (right scheme). (B) Stable tandem amplification of RsA and RsB flanked genomic regions is achieved in the presence of the ZouA relaxase. Tandem copies of 3–12 BGCs have been achieved using this system. (C) Multiplex CRISPR/Cas9 genome editing technology. The Cas9 nuclease can be directed to any site on the genome by a synthetic guide RNA (sgRNA), requiring only a PAM sequence at the target site. Non-homologous end-joining introduces indel mutations for gene inactivation whereas homologous recombination in the presence of donor DNA as the repair fragment can be used to generate large deletions as well as gene replacements or insertions.
Conventional methods of gene disruption, deletion and replacement involve the use of suicide plasmids or plasmids with temperature-sensitive origins in conjunction with two antibiotic cassettes to select and screen for single- and double-crossover recombination events respectively. Traditionally, this process is inefficient and time consuming due to low homologous recombination (HR) efficiency and the multiple replica plating steps needed to distinguish single-crossover (antibiotic-resistant) from the much less abundant double-crossover (antibiotic-sensitive) mutants. Chromogenic reporter genes such as xylE,33 gusA,32 and idgS35 facilitate the workflow by allowing visual identification of the second crossover event without colony replication. The counterselectable marker cytosine deaminase CodA, which converts 5-fluorocytosine (5FC) to the highly toxic 5-fluorouracil (5FU), has been used to select for double-crossover mutants36 and plasmid curing37 without the need for further genetic modifications.38, 39 An alanine mutant (D314A) enables counterselection to be performed at lower 5FC concentrations that can be tolerated by many strains.36 Alternatively, exploiting the inefficient non-homologous end-joining (NHEJ) in vegetative cells and the lethality of double stranded chromosomal breaks in the absence of HR repair, the meganuclease I-SceI from Saccharomyces cerevisiae has been adapted to select for double-crossover recombinants in actinomycetes.40, 41 In the first of this two-step method, an 18 bp I-SceI recognition site is co-introduced with a selection marker into the genome by single-crossover recombination. When I-SceI is introduced in a second step, only double-crossover recombinants (revertants or desired mutants) that have repaired the targeted chromosomal lesion created by I-SceI will survive. This strategy has been successfully used to generate large markerless gene deletions in S. coelicolor and S. venezuelae.40, 41
Site-specific recombination systems based on bacteriophage integrases have been a cornerstone of Streptomyces genetics (Figure 2A). Well-characterized phiC3142 and phiBT143, 44 attachment/integration (att/int) systems are found in many integrative Streptomyces plasmids, allowing the efficient integration of large inserts into highly conserved and relatively neutral attB sites in Streptomyces genomes.45 Innate attB sites are less conserved in rare actinomycetes but can be introduced prior to integration.45 In addition, there are similar TG1,46 R4,47 and SV148 systems involving large serine recombinases that catalyze recombination between attP with bacterial attB sites. Tyrosine integrases such as the cre/loxP and Flp/FRT systems have also been reconstituted in actinomycetes.49–52 Readers are referred to reviews comparing the integrases, their recombination requirements and associated accessory proteins controlling recombination directionality.45, 53 Head-to-tail orientation of recombination sites have been used to generate unmarked mutations via marker excision49, 51, 52, 54 or deletion of flanked genes or entire clusters,52, 55 leaving behind short scar sequences. Du and coworkers exploited phiBT1-mediated excision to clone entire BGCs from the genome into a multi-copy plasmid that can be propagated and isolated, while at the same time generating a deletion strain. An interesting aspect of this strategy is that it also increases the copy number of the target cluster, which can significantly increase natural product titers.55 The development of multiple non-compatible attB/attP pairs,56, 57 as well as non-overlapping attachment sites for different integrase systems are a harbinger of their utility in multiplex genome engineering of actinomycetes.45, 53
The ability to amplify specified genomic regions can have important applications in increasing gene dosage and redirect metabolic flux towards secondary metabolite production. Murakami and co-workers discovered that ZouA relaxase and two 16 nucleotide recombination sites, RsA and RsB, are required for the tandem duplication of the 145 kb kanamycin BGC in the native producer Streptomyces kanamyceticus.58 Reconstituting the three components in S. coelicolor is sufficient to direct gene amplification, generating 4–12 tandem copies of the RsA-RsB-flanked 23 kb actinorhodin BGC within 5 generations.58 Recombination between RsA and RsB was also demonstrated for longer fragments of 110 kb in S. coelicolor and on a plasmid in E. coli, suggesting the utility of this ZouA-mediated targeted amplification system in the engineering of diverse hosts (Figure 2B).59
CRISPR (clustered regularly interspaced palindromic repeat) technology has revolutionized genome engineering, enabling the genetic manipulation of genetically recalcitrant organisms, including mammals, plants and Streptomyces (Figure 2C).37, 60–62 Compared to other site-specific genome engineering technologies in actinomycetes, the S. pyogenes Cas9 nuclease can be directed to any site on the genome simply by transcribing a synthetic guide RNA (sgRNA), requiring only a protospacer adjacent motif (PAM) sequence at the target site. Notably, S. pyogenes Cas9 PAMs (NGG) are particularly abundant in the GC-rich actinomycete genomes, greatly increasing the number of potential target sites and coverage of CRISPR-Cas9 genome editing in these natural product producers. CRISPR-Cas9 has been reconstituted in multiple Streptomyces strains to perform precise deletions of individual genes37, 61 and entire BGCs of up to 82.2 kb60, 62 at high efficiencies of 60–100% with minimal off-target activity.37 This efficient recovery of desired mutants at 50–66% less time compared to conventional methods can be attributed to the fact that CRISPR-Cas9 selects against the wild type sequence while selecting for double-crossover recombinants in the presence of repair templates.37, 60–62 In the absence of a repair template, gene inactivation by NHEJ was achieved by co-introducing a LigD homolog from Streptomyces carneaus.61 Showcasing the multiplex nature of the CRISPR technology, co-transcribing two sgRNAs allows simultaneous deletion of two unlinked ORFs from distinct BGCs in single step at 45–100% efficiency.60, 62 It is conceivable that this technology can be adapted to perform gene replacement, knock-in and site-directed mutagenesis. In addition, the CRISPR platform can also be used for transcriptional engineering to control the expression of single and multiple genes. CRISPRi (CRISPR interference) involves a catalytically inactive Cas9 (dCas9) which serves as a versatile RNA-guided transcriptional repressor that can be targeted to specified genetic locus or loci by transcribed sgRNA to sterically hinder cellular transcriptional machinery. By strategically targeting dCas9 to the promoter region or non-template strand of actIORFI in S. coelicolor, actinorhodin production can be reversibly inhibited, with production restored by curing the dCas9-sgRNA expressing plasmid.61 In other organisms, dCas9 has been fused with a variety of proteins or protein domains while the sgRNA scaffold have been engineered to recruit proteins for genome-scale regulation of gene expression and marking of genomic loci.63, 64 Both CRISPR and CRISPRi technologies in Streptomyces are still in their infancy but their potential in native host engineering is huge, offering multiplex genome editing and control over transcriptional programs that regulate natural product biosynthesis.
Collectively, these complementary technologies greatly facilitate the genetic manipulation of actinomycetes. The CRISPR-Cas9 technology is the more efficient and rapid method to make large deletions and introduce indels, site-directed mutations, and possibly domain or gene replacements compared to conventional methods (Figure 2C). Nonetheless, CRISPR-Cas9 has not been demonstrated to function in rare actinomycetes. Site-specific recombination systems allow efficient integration or excision of large DNA fragments that can contain entire BGCs (Figure 2A), with the latter function being able to be coopted for direct BGC capture into a plasmid (Figure 5D). The ZouA system allows stable amplification of a target genomic locus (Figure 2B). A possible application integrating these technologies is the use of CRISPR-Cas9 for multiplex insertion, deletion or modification of recombination sites for large-scale genome engineering or genome rearrangement.65
Fig. 5. Methods for direct capture of large DNA fragments.

(A) RecET-mediated linear-linear homologous recombination (LLHR) or transformation-associated recombination (TAR).233, 234 Fragmented/digested genomic DNA with target of interest is assembled with linearized capture vector with homology sequences via HR in either E. coli (LLHR) or yeast (TAR). (B) CRISPR/Cas9-mediated TAR.236 RNA-guided Cas9 is used to cleave chromosomes at designated target sites in vitro. The resultant fragment is assembled with linearized TAR vector in yeast. (C) In CATCH,237 RNA-guided Cas9 is employed to cleave the isolated chromosome at the designated target sites in vitro and the target fragment is captured via Gibson assembly. (D) phiBT1 integrase-mediated site-specific recombination. Suicide plasmids are used to introduce attB and attP recombination flanking the gene locus of interest by HR. Expression of phiBT1 integrase leads to excision of target fragment into a plasmid backbone that is propagated in vivo.55
4 Strategies for strain improvement
Traditional strain improvement involves multiple rounds of chemical or UV mutagenesis followed by screens for high-producing mutants. Though laborious, this strategy has proven effective over the past decades in generating several high-producing industrial strains.66–68 Whole-genome shuffling 69–72 and targeted gene shuffling73 are additional strategies to rapidly introduce genetic diversity that can be screened for desired phenotypes, requiring significantly less time and labor to obtain the same improvement as iterative mutagenesis methods.74–76 As we try to better understand and navigate the intricate regulatory networks governing secondary metabolite production, such “brute force” random strain improvement approaches will continue to be important for improving natural product titers in actinomycetes and as a source of new leads for rational host engineering.
4.1 Engineering of transcriptional and translational machineries
Coined by Ochi and coworkers, “ribosome engineering” is a well-established method to increase secondary metabolite production titer and productivity.77–81 This economical and scalable strategy stemmed from observations that a high frequency of streptomycin-, rifampin- and gentamicin-resistant mutants also exhibited substantial improvement in secondary metabolite production.82, 83 By selecting for strains that are resistant to drugs targeting the transcriptional and translational machinery, laboratory and industrial actinomycetes strains exhibiting up to an order of magnitude improvement in the production of structurally diverse natural products have been isolated.77, 84, 85 Stepwise increases in production are achieved by accumulating mutations in multi-drug resistant mutants with concurrent and/or sequential drug selections,83, 86–88 suggesting additive or synergistic effects of the different resistance-conferring mutations.89, 90 Isolating S. coelicolor strains that are resistant to eight drugs, Wang and coworkers dramatically improved actinorhodin production 180-fold to a final 1.63 g/L titer (Figure 3B).86 Global derepression of secondary metabolite production through “ribosome engineering” has also enabled the discovery of new molecule classes from transcriptionally silent BGCs.84, 90,89
Fig. 3. Native host engineering strategies.

(A) Comparison of the main native host engineering strategies. (B) Graphical summary of native host engineering studies covered in this review. Data points are plotted as fold change in titer relative to the wild type parent strain (x-axis) against final production titers of the engineered strain (y-axis) and grouped according to the engineering strategy used. Commercial strains for the production of penicillin, erythromycin and clavulanic acid obtained via classical strain improvement methods are shown for comparison. Studies involving the engineering of high producing strains are boxed. Studies involving actinorhodin production in S. coelicolor are circled with the red arrow highlighting the stepwise improvement in titer with additional drug selections during ribosome engineering. Information on the data points and their associated references is documented in Supplementary Table S1.
Almost all secondary metabolite-overproducing mutations identified to date are mapped to translational and transcriptional machineries, majority of which occur in genes encoding the ribosomal protein S12 (rpsL) and the RNA polymerase β subunit (rpoB).77 To a lesser extent, mutations in genes encoding the 16S rRNA methyltransferase (rsmG)91 and ribosomal recycling factor (rrf) have also been identified.92, 93 There are likely mutations in other genes that are yet to be determined.89, 90, 94 Together, these mutations enhance protein translation during late growth phases and stimulate secondary metabolite production by modulating primary metabolism and global gene expression programs78, 89, 95, 96. Clusters of rpsL and rpoB mutations routinely identified in secondary metabolite-overproducing Streptomyces and rare actinomycetes suggested opportunities for rational strain improvement via the introduction of precise mutations.77, 84 One of the most frequently detected rpoB mutations,77, 84 substitutions in histidine 437, have been postulated to trigger secondary metabolite production by mimicking ppGpp-bound RNAP.78 Introduction of these mutations is sufficient to alter the promoter affinity of RNAPs and induce metabolite production.90, 97, 98 Recently, a functionally-divergent rpoB(R) gene is preferentially found in rare actinomycetes.99–101 Consistent with the presence of a mutation that confers rifampicin-resistance in the duplicated rpoB allele, rpoB(R) expression confers rifampicin resistance to otherwise sensitive actinomycetes.99, 101 Interestingly, overexpression of the Nonomueraea sp. rpoB(R) allele in S. lividans induced secondary metabolite production, raising the possibility of general rpoB(R)-based technology for the purpose of improving natural product titers.100 In rpsL, commonly detected K88 missense mutations allow for efficient protein translation by ribosomes in stationary phase, but only in specific genetic backgrounds.90, 102 In addition, strains with rpsL and rsmG mutations showed higher protein synthesis activity during late growth phases and increased metabolite production.92, 96 Recent development of single subunit RNA-stapled ribosomes allows the introduction of otherwise dominant lethal mutations and significantly expands the functional space that can be sampled for novel ribosome functions.103, 104 Introduction of such RNA ribosome tethers into strains prior to “ribosome engineering” may unveil new mutations with desirable overproduction phenotypes. It is conceivable that introduction of engineered orthogonal ribosomes for robust stationary phase protein translation could be a broadly applicable strain improvement strategy.
4.2 Overexpression of biosynthetic genes
Overexpression of key structural genes, either by promoter engineering or increasing gene dosage, is one of the most straightforward strategies to improve titers in laboratory and industrial strains for a specific metabolite. In fact, improvement in production in some high-producing industrial strains has been attributed to tandem amplifications of BGCs.105–107 BGC duplication by integrating an additional copy of the target cluster at innate attB sites of the original host has been demonstrated to increase nikkomycin and gougerotin production in Streptomyces ansochromogenes and Streptomyces graminearus respectively to achieve g/L titers.108, 109 Gene dosage can be further increased via in situ tandem amplification of the target BGC using the ZouA recombination system.58 A 20-fold increase in actinorhodin production was observed in S. coelicolor strains harboring 4–12 tandem copies of the ACT cluster.58 A Streptomyces hygroscopicus strain with 3–5 tandem copies of the 40 kb validomycin A cluster showed a 34% increase in production and a maximum titer of ~20 g/L.59 While stable over multiple generations, these tandemly amplified DNA units can be gradually lost without selection59 and disappear upon sporulation,59, 107 with the latter attributed to limited chromosome packaging space within the spore. Strategic insertion of promoters in front of key structural genes involved in rate-limiting steps within the target cluster prior to integration or duplication can further improve production.110, 111 The oxytetracycline titer of an industrial Streptomyces rimosus strain was increased by 33% when an additional copy of the minimal PKS genes under ermE*p was introduced.111 A dose-dependent increase of 10–73% in chlortetracycline for a maximum titer of 25.9 g/L was achieved by increasing the copy number of the ctcP halogenase gene driven by ermE*p in Streptomyces aureofaciens.112 Interestingly, no titer improvement was observed when more than three copies of ermE*p-ctcP was introduced,112 suggesting that negative feedback mechanisms may be involved in modulating the final halogenation step of chlortetracycline biosynthesis.
Overexpression of non-biosynthetic genes, in particular self-resistance determinants, can also improve secondary metabolite production. Increasing the expression of export-related genes improved avermectin and doxorubicin production in S. avermitilis and Streptomyces peucetius respectively.113, 114 In another example, the upregulation of cluster-associated resistance genes actAB in S. coelicolor enhanced actinorhodin production by up to 5-fold.115 While the exact mechanisms remain to be determined, it is hypothesized that expression of resistance genes relieves potential cellular toxicity and feedback inhibition that limit production titers.
In general, overexpression of key biosynthetic genes, especially those involved in rate-limiting steps of the biosynthetic pathway, improves secondary metabolite production but success of this approach may be limited by negative feedback control mechanisms. Upregulation of self-resistance genes may help relieve feedback inhibition of natural production biosynthesis.
4.3 Manipulation of pathway-specific regulators
Secondary metabolite production is a highly regulated process that involves capturing and integrating a plethora of environmental and developmental signals, relaying the information to pleiotropic and pathway-specific regulators, which in turn modulate expression of their respective BGCs.116, 117 This regulatory complexity is supported by the large number of regulatory genes (~12.5% of genes in S. coelicolor) found in Streptomyces genomes.118 With the wealth of information from characterization of mutations affecting natural product biosynthesis and comparative genomic, transcriptomic, proteomic and metabolomic studies, we are beginning to understand the families of regulators governing secondary metabolism.116, 117 In many cases, severing the connections to native regulatory networks by overexpression of activators or deletion of repressors is sufficient to significantly boost secondary metabolite production.119, 120
To improve production of target secondary metabolites, it is evident to manipulate the lowest level pathway-specific regulators that are situated within their respective BGCs. Of these pathway-level regulators, members of the SARP (Streptomyces antibiotic regulatory protein) family specific to actinomycetes are heavily represented.118 Containing N-terminal winged helix-turn-helix (HTH) motifs, SARPs typically recognize and bind 5–7 bp repeats near promoters and activate the expression of their target biosynthetic genes.121, 122 Overexpression of SARPs and/or by increasing SARP gene dosage using multi-copy plasmids have been demonstrated to increase production titers, including a 4-fold increase in nikkomycin production in S. ansochromogenes,123 a 5.6-fold improvement in fredericamycin production in Streptomyces griseus,124 and doubling of tylosin production in Streptomyces fradiae.125, 126 In addition, members of the LAL (large ATP-binding regulators of the LuxR family) family, characterized by an N-terminal ATP/GTP-binding motif and a C-terminal HTH motif, also generally function as transcriptional activators. Overexpression of LAL regulators is sufficient to induce secondary metabolite production from cryptic BGCs in S. albus, Burkholderia thailandensi and S. ambofaciens.127–129 Constitutive overexpression of LAL-type activators increased FK506 and rapamycin production in Streptomyces tsukubaensis and S. hygroscopicus, respectively.130, 131 Members of the PAS-LuxR family of transcriptional activators, which possess an N-terminal PAS sensory domain and C-terminal LuxR-type HTH motif, are key activators of many polyene polyketide BGCs.132 The archetype of PAS-LuxR regulators and the final regulator of pimaricin biosynthesis in Streptomyces natalensis, PimM binds to operator sequences near the −35 region of promoters and triggers expression of polyene biosynthetic and transport genes.132 Overexpression of PimM and its homologs enhanced production in native pimaricin producers, S. natalensis, Streptomyces chattanoogensis and Streptomyces lydicus, by 2.4-, 4.6- and 3-fold, respectively.133–135 Supporting its role as a general transcriptional activator, heterologous pimM overexpression was sufficient to significantly boost polyene synthesis, including filipin and oligomycin in S. avermitilis, rimocidin in S. rimosus, amphotericin in Streptomyces nodosus, and antimycins in S. albus.128, 132, 136 The apparent functional conservation of PAS-LuxR regulators makes them attractive targets for engineering polyene overproducing strains and for the discovery of novel polyenes.132 Titer improvement can also be achieved by the functional disruption of transcriptional repressors. Cluster-situated regulators from the GntR (N-terminal HTH and C-terminal effector-binding domains) and TetR families tend to be transcriptional repressors. For example, deletion of ptmR1 encoding a GntR-like repressor in Streptomyces platensis dramatically upregulated platensimycin and platencin production.137 While strongly preferred, the activator or repressor roles of these highlighted regulator families are not absolute and exceptions exist. Other transcriptional regulator families contain both activators and repressors, making it challenging to determine their context-dependent function from sequence analysis.138 In these cases, comparing production titers of knockout and overexpression strains of the pathway-specific regulator in question will shed light on their regulatory function.
Many aspects of pathway-specific regulators are yet to be understood. Feedback control or unknown regulatory mechanisms can confound strain improvement efforts. Moreover, there is evidence of crosstalk between pathway-specific regulators and that they may be involved in the “global” control of cellular processes beyond secondary metabolite production.136, 139 In S. avermitilis, overexpression of positive LAL regulator aveR counterintuitively reduced avermectin production and cell growth.140 For BGCs containing multiple regulatory genes, improvement in secondary metabolite production by combinatorial genetic manipulation of transcriptional activators and repressors has been less than predictable. Co-overexpression of two or three pathway-specific activators, which increased titers when overexpressed individually, yielded only wild type level production of the enediyne antitumor antibiotic C-1027 in Streptomyces globisporus.141 On the other hand, overexpression of an activator sgcR1 in conjunction with deletion of a repressor increased C-1027 titers 8-fold.142 These examples highlight the next set of challenges in engineering pathway regulation for strain improvement. Until the intricacies of regulatory networks and functional interactions between multiple pathway-specific regulators are better understood to allow synergistic manipulation, a more systematic empirical approach can be employed to evaluate the synergy and antagonism between regulators.143
4.4 Manipulation of pleiotropic regulators
γ-Butyrolactones (GBL) are small signaling molecules produced by most Streptomyces and some rare actinomycetes. Akin to autoinducers in bacterial quorum sensing, GBLs function as microbial hormones to coordinate secondary metabolism and/or cellular differentiation.117 At the core of the GBL signalling paradigm is the GBL receptor, which directly regulates its own expression and GBL biosynthesis. In general, GBL receptors function as repressors of secondary metabolite production and GBL binding relieves this repression.144–147 Deletion of GBL receptors have by large resulted in improved titers148 and led to the discovery of novel natural products from cryptic BGCs.149, 150 Tandem deletion of two GBL receptors in S. hygroscopicus increased validomycin titers and productivity by up to 26% (24 g/L) and 45% (9.7 g/L/day), respectively, which are the highest reported values to date.151 A few studies have suggested that disruption of GBL receptors may negatively affect antibiotic production, though the lower titers observed may be a result of increased production of other secondary metabolites competing for the same biosynthetic precursors.148 Although GBL identification is hindered by the fact that they are produced at minute levels and our incomplete knowledge of their biosynthesis, GBL receptors and their homologs can be easily identified by sequence analysis.152 Given their key roles in GBL signalling and secondary metabolite production, GBL receptors are attractive candidates for strain improvement.
Sigma factors are needed for the specific recruitment of RNA polymerase to recognized promoters and transcription initiation. The housekeeping σHrdB in Streptomyces is responsible for the expression of most genes during vegetative growth, including genes involved in secondary metabolite biosynthesis. By screening for σHrdB variants that increase the expression of a positive regulator aveR, Zhuo and coworkers achieved more than 50% improvement in avermectin B1a production (6.38 g/L) in S. avermitilis at a 180,000 L scale.153 The strong sequence similarity of hrdB across Streptomyces species and actinomycete genera suggests that sigma factor engineering is a viable universal strategy to improve natural product titers in laboratory and industrial strains.154 In contrast to housekeeping sigma factor, alternative sigma factors function in different cellular contexts, binding to different sets of promoters and driving expression of relevant genes. Overexpression of whiGch encoding the sporulation-specific σwhiGch increased pimaricin production by 26% in S. chattanoogensis.155 A group of alternative sigma factors particularly enriched in actinomycetes is the extracytoplasmic (ECF) sigma factor family, which mediates global transcriptome changes in response to environmental and physiological stimuli. ECF sigma factors have been shown to be required for the expression of key biosynthetic genes and the production of lantibiotics in rare actinomycetes156, 157 and antimycins in S. albus.158 In Planomonospora alba, introducing a second copy of ECF sigma factor pspX increased planosporicin production, while disruption of its cognate anti-sigma factor significantly increased the expression of the target biosynthetic genes and led to constitutive production of the lantibiotic.157 Interestingly, ECF σ25 in S. avermitilis differentially regulates production of two macrolides oligomycin and avermectin, where overexpression of sig25 increases oligomycin production but decreases avermectin titer.159 Given that σ25 directly binds to promoter regions of oligomycin biosynthetic genes, it is likely that the associated reduction in avermectin production is due to competition for common precursors.159 From a host engineering perspective, sigma factors function as resource allocators to distribute the transcriptional resources (e.g. RNAP) within the cell.160 Even though the roles and targets of many sigma factors in actinomycetes are unclear, promoter recognition sequences of close homologs are similar and target prediction is possible for those with characterized counterparts. The conservation of sigma factor binding sites near promoters of antimycin biosynthetic genes in different Streptomyces strains158 and the retention of sigma factor function when transplanted into a heterologous host157 indicate the opportunities of sigma factor engineering in the strain improvement process. Quantitative and/or temporal control of naturally occurring or engineered sigma factors and their cognate anti-sigma factors can be useful strategies to channel transcriptional resources towards the production of a target natural product.160
4.5 Precursor engineering
Primary metabolism provides the energy and biosynthetic precursors for secondary metabolism, which normally starts at later stages of growth. With precursor availability being a key determinant of secondary metabolite production, genetic manipulations that modulate carbon flux, increase precursor pool and/or remove competitive sinks constitute one of the main strategies for strain improvement.
Biosynthetic precursor pools can be boosted by manipulating key enzymes that direct carbon flux through core biochemical pathways involved in glucose, fatty acid and amino acid metabolism.119 Modulation of carbon flux between the pentose-phosphate pathway and Embden-Meyerhof pathway increased production of actinorhodin and undecylprodigiosin in S. lividans and S. coelicolor,161–163 as well as production of oxytetracycline in S. rimosus.164 Disruption of glyceraldehyde-3-phosphate dehydrogenase function in S. clavuligerus increased levels of glyceraldehyde-3-phosphate, a precursor of clavulanic acid, and improved production of the antibiotic 3-fold with exogenous supplementation of arginine, a second biosynthetic building block.165 Increasing the overall levels of common polyketide building blocks such as acetyl-CoA, malonyl-CoA and methylmalonyl-CoA by upregulating enzymes responsible for their biosynthesis enhanced the production of polyketides in different producing strains. Duplication of the methylmalonyl-CoA mutase operon channeled carbon flux towards methylmalonyl-CoA from succinyl-CoA in an oil-based medium and improved erythromycin production in S. erythraea.166, 167 Overexpression of acc genes encoding for acetyl-CoA carboxylase increased both acetyl-CoA and malonyl-CoA biosynthesis, leading to enhanced actinorhodin production in S. coelicolor.161 Mithramycin production was elevated 374% in Streptomyces argillaceus when intracellular pools of malonyl-CoA and glucose-1-phosphate were increased by upregulating their synthesis and disrupting pathways that use these precursors.168 With better understanding of primary metabolism in actinomycetes, it is conceivable that for a given natural product with known biosynthetic pathway, there will be a prescribed set of tools and genetic manipulations (e.g. deletion, overexpression, introduction of heterologous genes) to enhance the flux towards the relevant building blocks.169
4.6 Emerging strategies
Other general strategies that have been demonstrated to be feasible in improving natural product titers include the overexpression of genes coding for bacterial hemoproteins and phosphopantetheinyl transferases (PPTases). Expression of the Vitreoscilla hemoglobin gene (vgb) has been shown to significantly improve growth, stress resistance and the production of recombinant proteins and valuable metabolites in many prokaryotic systems, primarily by increasing oxygen uptake and delivery.170, 171 Consistent with studies demonstrating a positive correlation between antibiotic production levels and oxygen uptake,172 heterologous vgb expression improved production of pimaricin in Streptomyces gilvosporeus and S. lydicus,173, 174 actinorhodin in S. coelicolor,175 and avermectin in S. avermitilis.176 Expressing a different hemoprotein from Shinorhizobium meliloti has similar effects on metabolite production.177 With oxygen expected to be limited in high-density production cultures, heterologous expression of bacterial hemoproteins may be a general strategy to increase titers and productivities in actinomycetes. Recent work also highlights the possibility of using broad-specificity PPTases to improve the production of polyketides, non-ribosomal peptides and hybrid peptide-polyketides. Classified into AcpS-type or Sfp-type, PPTases in actinomycetes are required for post-translational modification and activation of carrier proteins for the biosynthesis of fatty acids, polyketides and non-ribosomal peptides. Gene disruption studies in S. coelicolor, Streptomyces verticillus, S. ambofaciens and S. chattanoogenesis reveal that a continuum of selectivity exists for PPTase homologs of both types. For example, AcpS-type PPTases preferentially modifying fatty acid synthases and/or type II PKS ACPs whereas Sfp-type PPTases tend to be associated with secondary metabolism and preferentially modifying type I and II PKS ACPs as well as NRPS PCPs.178–181 By overexpressing a type I PKS-specific Sfp-type PPTase, Jiang and colleagues improved pimaricin production titer by 40% in an industrial S. chattanoogenesis strain.180
4.7 An integrated approach for strain improvement
The major native host engineering strategies presented in this section have their advantages and limitations (Figure 3A). Each strategy has been demonstrated to improve natural product titers in wild type and high-producing strains, albeit with varying degrees of success depending on the identities of the natural product and host strain (Figure 3B). With a few exceptions, each engineering strategy generally yields modest (<10-fold) improvement in production titers. This underscores the need to combine different strategies to achieve the same improvement observed for production strains that are optimized by iterative mutagenesis. Rational host engineering to achieve industrial production titers is not impossible, especially when functional transcriptional changes in an industrial strain can be largely attributed to changes in a few key regulatory nodes that are targeted by the rational engineering strategies described (Section 4.2–2.5).182
Given the differences in host physiology and the unique metabolic requirements for different secondary metabolites, it is unlikely that is a one-size-fits-all strain improvement program. For example, it is likely that BGC expression, not precursor availability, is the major limiting factor for actinorhodin production in S. coelicolor. Hence, strategies that increase BGC expression (increasing gene dosage, SARP overexpression) yielded more dramatic improvement in actinorhodin production than increasing precursor availability (Figure 3B). Global transcriptional and translational changes also have a positive impact on actinorhodin titers, suggesting additional determinants of actinorhodin production besides BGC expression. Determining the biosynthetic bottlenecks for a target metabolite in a specific host can aid the prioritization of host engineering strategy/strategies for the greatest titer increase and accelerate the strain improvement process.
Identification of biosynthetic bottlenecks can be guided by proteomic, transcriptomic, metabolomic studies,183, 184 or computational metabolic models,185–187 if available. Empirically, one can conceive an integrated platform to rapidly test and identify the main strategy/strategies that would yield significant titer improvement for a target natural product in a given host. This is now within reach with technological advances that enable efficient genome editing, including gene deletion, insertion, replacement, cluster amplification, and integration of large inserts in actinomycetes (Section 3.2). The additive or synergistic effects of different strategies can be interrogated, a process that will be facilitated by multiplex genome editing. Subsequent efforts can be directed towards “debugging” the bottlenecks and optimizing production. Notably, this will be an iterative process until the desired titer is reached since new bottlenecks or rate-limiting steps are expected to surface in the engineered strains. An important caveat to the various rational engineering approaches is the need to introduce recombinant DNA into host, which may not be possible for some strains. For these cases, and in cases where no significant bottlenecks can be identified and the main strategies have been exhausted, random mutagenesis will be the most effective approach.
5. Heterologous production of bacterial natural products
With the advent of recombinant DNA and synthetic biology technologies that allow efficient capture and refactoring of BGCs, heterologous hosts are increasingly employed for microbial natural product production. In this section, we present the motivations for heterologous biosynthesis of natural products, considerations and typical workflow for heterologous BGC expression as well as recently developed tools and strategies that facilitate engineering of heterologous hosts for the production of natural products.
5.1. Advantages and limitations of heterologous production systems
There are several advantages to heterologous production systems. First, the use of surrogate hosts allows access to natural products encoded by BGCs from difficult-to-culture or uncultivated microorganisms, including rare terrestrial and marine actinomycetes.188–191 Next, commonly used hosts for heterologous production are more genetically amenable than most native hosts, enabling efficient optimization of production by rational or semi-rational host engineering strategies. Lastly, heterologous hosts can be engineered to be more stable genetically, helping to ensure steady production performance.
On the other hand, it is unlikely that a “super host” capable of expressing and producing metabolites from all biosynthetic pathways exists or can be engineered. One of the main challenges with heterologous production systems is the lack of critical biosynthetic precursors or enzyme function, which differ for different classes of BGCs.192, 193 Other challenges include incompatible regulatory systems, requiring the introduction of regulators or replacement of native promoters for expression of transplanted BGCs and metabolite production.120 Uncoordinated expression of biosynthetic genes can result in higher metabolic burden and/or production of toxic compounds, which limits production titers. For example, buildup of mevalonate-based isoprenoid biosynthetic intermediates such as HMG-CoA downregulates fatty acid biosynthesis via feedback regulation and negatively impacts host growth.194 While these limitations impact our choice of heterologous production hosts for a given natural product (Section 5.2), understanding these BGC-dependent limitations will help guide various host engineering strategies to improve production titers of a target metabolite (Sections 4 and 6).
5.2. Heterologous hosts for biosynthesis of bacterial natural products
The typical workflow for heterologous expression of a BGC involves (Figure 4): 1) choosing a suitable heterologous host, 2) cloning and/or refactoring target BGC, 3) transferring native or refactored BGC into the heterologous host, and 4) optimizing production in the heterologous host. Host compatibility and pathway cloning/refactoring efficiency are two critical determinants of success for heterologous production of natural products.
Fig. 4. The typical workflow for heterologous expression of a target BGC.

1) choose a suitable heterologous host, 2) clone and/or refactor a target BGC, 3) transfer the native or refactored BGC into the heterologous host, and 4) optimize metabolite production.
When choosing heterologous hosts, target BGC-host compatibility is one of the top considerations (Table 1). BGCs of eukaryotic origins are most likely to be expressed in a eukaryotic host such S. cerevisiae whereas polycistronic BGCs from prokaryotes are more easily reconstituted in bacterial hosts such as E. coli, Pseudomonas putida, Bacillus subtilis and Streptomyces spp.. Additionally, the availability of genetic tools associated with a heterologous host should be considered. Last but not least, knowledge of the metabolism of heterologous hosts is also important, since precursor availability and endogenous biosynthetic pathways can affect the biosynthesis of the target natural product.
Table 1.
Comparison between different heterologous host systems for bacterial natural product biosynthesis.
| Heterologous host | Advantages | Disadvantages |
|---|---|---|
| E. coli (Gram-negative) |
|
|
| P. putida (Gram-negative) |
|
|
| B. subtilis (Gram-positive) |
|
|
| Streptomyces spp.- (Gram-positive) |
|
|
E. coli and P. putida are the main Gram-negative hosts used for heterologous production of bacterial natural products. Owing to its fast growth and facile genetic manipulation, E. coli is one of the most widely used hosts for heterologous production.5, 195–197 A major concern when using E. coli as a production host is the lack of necessary biosynthetic machinery and/or precursors. For example, a PPTase must be introduced into E. coli in order to activate the acyl carrier protein (ACP) and peptidyl carrier protein (PCP) domains of PKS and NRPS respectively. Recently, erythromycin A and its analogs were successfully produced in the engineered E. coli BAP1 strain and its derivatives.198, 199 Due to the extensive genetic manipulations needed, E. coli may not be the ideal production host. An alternative host with established culture and genetic manipulation techniques, P. putida is known for its versatile metabolism and diverse enzymatic capabilities that are absent in E. coli. For example, the P. putida KT2440 strain possesses a broad substrate range PPTase that can activate ACP and PCP domains,200, 201 as well as a wealth of cofactors that are required for a variety of heterologous biosynthetic enzyme functions including oxidoreductases.202 P. putida offers additional unique advantages as a production host such as its adaptability to different physicochemical and nutritional niches, its remarkable xenobiotic tolerance203, 204 and its high NADPH regeneration rate.205 Thus, P. putida has been employed to produce a broad portfolio of natural products including amino acid derived compounds, non-ribosomal peptides, polyketides, rhamnolipids and terpenoids.206
B. subtilis is a Gram-positive, non-pathogenic host for heterologous production of natural products. The Bacillus genus is known to produce a wide assortment of biologically active small molecules, including non-ribosomal cyclic lipopeptides of the surfactin and gageotetrin families,207 polyketides such as macrolactin and bacillaene, polyketide-peptide hybrids like amicoumacin and ieodoglucomides,208 and discoipyrrole alkaloids.209–212 Therefore, B. subtilis serves as an ideal host for heterologous production of natural products from the Bacillus genus. Its fast growth rate, thorough genetic characterization and natural competence facilitate strain engineering efforts. However, the lack of autonomous plasmids to facilitate cloning, transfer and heterologous expression of large biosynthetic gene clusters in B. subtilis limits its utility as a production host.213 Only a few examples of small PKS and NRPS pathways introduced by chromosomal transfer or cosmid library expression have been reported to date.214–219 Recently, the Moore group developed the first direct cloning TAR-based capture-expression platform for B. subtilis.220
Another important group of Gram-positive heterologous hosts are the Streptomyces spp.221 In addition to matching genomic GC-content, Streptomyces spp. are the preferred hosts for expressing BGCs of actinomycete origins, one of the richest sources of natural products, because they possess the necessary metabolic precursors, posttranslational modification machinery and enzymes to support the expression and production of transplanted biosynthetic pathways. Various Streptomyces hosts have been used to heterologously produce diverse natural products including polyketides and non-ribosomal peptides,222 lantibiotics, uridylpeptides, thiopeptides, aminocoumarins, liponucleosides, peptidyl nucleosides and phenazines.221, 223, 224 While S. lividans 66/TK24 and S. albus J1074 are traditionally preferred hosts due to their relatively low secondary metabolite backgrounds and weak restriction barriers, engineered strains of S. coelicolor and S. avermitilis have recently proven to be robust hosts for heterologous production.225–230
Recently, there is an obvious industrial drive towards the development of eukaryotic cell factories for the production of bioactive molecules, as epitomized by the yeast production of artemisinic acid, the precursor of antimalarial drug artemisinin. In particular, with its efficient homologous recombination system coupled with the availability of advanced genetic tools, S. cerevisiae is increasingly used for the overproduction of plant and fungal natural products, such as isoprenoids, terpenoids, flavonoids and alkaloids.231 Due to the lack of biosynthetic precursors, the range of bacterial natural products that can be produced from yeast is relatively limited. Since this review focuses on the production of bacterial natural products that are preferably produced in prokaryotic host systems, we will not elaborate on eukaryotic host systems. Readers are referred to reviews on the use of yeast and other fungi (e.g. Asperigillus genus) as hosts for the biosynthesis of natural products of eukaryotic origins,5, 222, 232 as well as a review by Li and Smolke that focuses on the heterologous production of fungal natural products. Here, we present S. cerevisiae as a platform host for the cloning, assembly and refactoring of bacterial BGCs for heterologous expression (Section 5.3).
5.3. Cloning and refactoring of target BGCs for heterologous expression
For heterologous production, target BGCs, which tend to be relatively large, must be cloned into suitable vectors and engineered for expression in the chosen host(s). Several synthetic biology tools have been developed to directly capture entire BGCs from complex genomic DNA sources (Figure 5), circumventing the laborious construction and screening of genomic libraries. Efficient linear-linear homologous recombination (LLHR) mediated by full length RecE and its partner RecT in E. coli was used to clone ten megasynthetase clusters of up to 52 kb into specified expression vectors.233 A conceptually similar method, transformation associated recombination (TAR), which uses in vivo yeast HR to capture genomic fragments,234 has been successfully used to directly clone the 67 kb taromycin A BGC from Saccharomonospora sp. for heterologous expression in S. coelicolor.235 Yet a major limitation to these powerful methods is the requirement for unique restriction sites flanking the target genomic locus, which may not be readily available. Use of the RNA-guided Cas9 nuclease to cleave both ends of the target DNA in CRISPR/Cas9-mediated TAR and Cas9-associated targeting of chromosome segments (CATCH) methods relieves these constraints and these approaches can be used to clone almost any genomic sequences up to 100 kb in length.236, 237 Direct cloning of very large BGCs (>100 kb) is likely to be hindered by physical shearing of DNA prior to capture. In an alternative approach, recombination sites are first introduced on both ends of the target genomic locus in the native host by homologous recombination. Target genomic fragments up to 157 kb are subsequently captured into a multi-copy plasmid in vivo via site-specific recombination mediated by the phiBT1 integrase with more than 80% efficiency.55 Given that native BGCs may not be expressed even when transferred to another host,238 it is noteworthy that these direct capture methods are compatible with downstream cluster refactoring involving λ RED recombineering in E. coli or HR in yeast. In yeast, TAR has been further adapted for simultaneous capture and refactoring of BGCs by insertion of multiple promoter cassettes for expression in an S. albus host, demonstrating streamlined and scalable workflow for heterologous production of microbial natural products.239
BGCs can also be assembled from smaller de novo synthesized or PCR-amplified DNA fragments employing scarless assembly approaches such as isothermal assembly,240 endonuclease-induced homologous recombination assembly,241 sequence- and ligation-independent cloning (SLIC),242 Golden Gate technology-based methods involving type IIs restriction enzymes that cut outside their recognition sites243–245 and yeast HR-based DNA assembler (Table 2).246, 247 Notably, for HR-based methods, unwanted recombination events can occur for BGCs with repeating sequences, such as PKS and NRPS clusters. The fact that target BGCs can be combinatorially refactored for improved heterologous expression during assembly is one of the main strengths of these approaches. During cluster refactoring, regulatory networks can be rewired, cluster architecture can be reorganized, proteins can be reengineered, and native genetic elements such as promoters, RBSs and terminators can be replaced with well-characterized counterparts that function predictably in the heterologous host.248, 249 Additionally, with the decreasing costs of de novo DNA synthesis, genes can be codon-optimized to avoid possible negative effects of codon bias in the heterologous host, especially when the genomic GC- content is drastically different between the native host from which the BGC is obtained and the heterologous production host. Coupled with the increasing availability of standardized genetic parts for various heterologous host systems, including Streptomyces spp. (Section 3.1), automation of these assembly techniques will undoubtedly accelerate the pathway engineering process of large gene clusters to achieve optimal performance of the refactored BGCs in the final production host.250
Table 2.
Strategies for assembling BGCs.
| Strategy (Max size assembled) | Basic principles | Advantages | Limitations |
|---|---|---|---|
| MoClo243,245 (50 kb) | Type IIs restriction enzymes, Golden Gate technology |
|
|
| MASTER ligation244 (29 kb) | MspJI and Golden Gate cloning |
|
|
| DNA assembler246,247 (50 kb) | Yeast HR |
|
|
| Reiterative recombinalion241 | Endonuclease-induced HR paired with recyclable markers |
|
|
| LCR-DNA assembler248 (44 kb) | Ligase cycling reaction followed by in vivo yeast assembly |
|
|
6. Tools and strategies for heterologous host engineering
6.1. Static and dynamic regulation of heterologous metabolic pathways
Once the BGC of interest is transferred into the chosen heterologous host, native host engineering strategies to improve yield and productivity (Section 4) are also applicable in heterologous host systems..251–253 However, taking a BGC out of its native context for expression in a heterologous host can result in unregulated expression of biosynthetic genes and negatively impact metabolite production. The flux of heterologous biosynthetic pathways can be balanced through static control of key biosynthetic genes at the transcription level by tuning promoter strength,254–257 mRNA stability,258, 259 or at the translation level by RBS engineering (Figure 6A).260, 261 E. coli is widely used for the production of malonyl-CoA-derived natural products including coumarins, stilbenes, flavonoids, tetracyclines and polyketides.262 To circumvent the low cellular concentration of malonyl-CoA in E. coli,263–268 various strategies were employed to boost its biosynthesis, including overexpressing acetyl-CoA carboxylase to stimulate conversion of acetyl-CoA into malonyl-CoA,269, 270 upregulating acetyl-CoA synthase to enhance acetyl-CoA availability and disrupting acetyl-CoA consumption pathways.271, 272 Combining these strategies yielded an E. coli strain with 15-fold higher malonyl-CoA levels and a 4-fold improvement in phloroglucinol titer.268 In another study, redirection of carbon flux towards malonyl-CoA increased flavanoid production by 66-fold.262 Disruption of essential fatty acid biosynthesis pathways that compete for malonyl-CoA is more challenging. Chemical inhibition of fatty acid biosynthesis improves polyketide production but is economically inviable for scale-up production.273, 274 Using a synthetic antisense RNA to conditionally downregulate fatty acid biosynthesis in E. coli, the Yan group successfully improved malonyl-CoA concentrations by 4.5-fold and production titers of 4-hydroxycoumarin, resveratrol, and naringenin by 2.53-, 1.70-, and 1.53-fold respectively.275 Similar strategies have been used to increase the production of desired polyketides in E. coli, including those made up of methylmalonyl-CoA, ethylmalonyl-CoA and isobutyryl-CoA building blocks.276, 277
Fig. 6. Static and dynamic regulation of metabolic pathways for secondary metabolite production.

(A) The flux of heterologous metabolite pathways can be balanced through static controls at the transcription level by tuning the strength of promoters and stability of mRNA or at the translation level by modifying the RBSs or interfering the binding of ribosomes to RBSs. (B) Metabolic pathways can be regulated by inducible promoters that respond to extracellular signals or dynamically tuned by promoters that respond to stress-inducing metabolite intermediates of heterologous pathways. Sensor-actuator pairs can also be used to control gene expression levels based on intracellular signals.
As culture conditions change with different growth stages, it will be valuable to incorporate dynamic control circuits to sense these changes and actuate appropriate responses that maximize production yields (Figure 6B). To overcome the metabolic burden of acetyl-CoA overproduction and its negative impact on cell growth in production hosts for fatty acid-derived metabolites, the Keasling group developed a dynamic sensor-regulator system (DSRS) using a transcription factor that senses key acyl-CoA intermediates and regulates expression of the biosynthetic genes, improving biodiesel production titers in E. coli by 300%.278 Malonyl-CoA sensor-actuators that dynamically regulate enzymes involved in malonyl-CoA supply and consumption to maintain appropriate malonyl-CoA concentrations help alleviate cellular toxicity and improve fatty acid production in E. coli.269, 272 It is conceivable that similar strategies can be employed to enhance the production of secondary metabolites from biosynthetic precursors derived from primary metabolism in hosts with robust metabolic models. Due to limited knowledge of their complex metabolism and regulatory networks, it may be challenging to build sensor-actuator circuits in Streptomyces spp. for the dynamic control of metabolic pathways. Nonetheless, general stress-responsive promoters can be employed to dynamically limit the accumulation of toxic pathway intermediates and improve metabolite production without the need for elaborate metabolite-specific sensor-actuator systems.194
6.2. Genome-scale engineering for natural product biosynthesis
For large-scale microbial fermentation of natural products, it is imperative to maintain stable overexpression of the biosynthetic genes and pathways. Plasmid-based expression systems require continuous costly selections and are prone to rapid loss in productivity due to allele segregation. Using RecA-mediated HR to increase the copy-number of genome-integrated pathways up to 40 copies, chemically inducible chromosomal evolution (CIChE) enables the generation of robust production microbes with stable high-level expression of heterologous genes without selection (Figure 7).279 Production yield of the nutraceutical lycopene was increased by 60% by ensuring stable high pathway copy number. Highlighting the genetic stability of CIChE-engineered strains, productivity of the high metabolic burden-biopolymer poly-3-hydroxybutyrate was maintained selection-free for >35 generations longer than plasmid-based systems with selection.279 This method can be used for organisms with genomic integration methods and functional RecA homologs. Given the high metabolic demands and possible generation of toxic compounds during secondary metabolite biosynthesis, CIChE should be useful for the engineering of microbial hosts for the stable overproduction of natural products.
Fig. 7. Chemically inducible chromosomal evolution (CIChE) enables stable high-level expression of heterologous genes without selection.

CIChE enables the creation of multiple chromosomal copies of a recombinant allele based on recA-mediated crossover between leading and trailing homologous regions on different DNA strands. The process is terminated by recA deletion. Reproduced with permissions.279
Additional genetic modifications that cannot be rationally determined may be required to further optimize production. Aided by advances in DNA synthesis, sequencing and high throughput screening technologies, multiplex genome-scale engineering technologies facilitate the elucidation of causal genotype-phenotype relationships and boost efforts to design and reprogram organisms.280 Here we highlight technologies that can be employed to generate genetic diversity in production hosts in a directed and combinatorial manner that can be screened for overproduction phenotypes. While many of these strategies are developed in model organisms like E. coli, they are increasingly adapted to other organisms, including those relevant for heterologous production of bacterial natural products.
Reminiscent of “ribosome engineering” for native actinomycetes (Section 4.1), global transcriptional machinery engineering (gTME) has been successfully employed in E. coli and yeast for phenotype improvement.281–283 Screening of two mutagenesis libraries yielded specific mutations in genes encoding for RNAP subunit (rpoA) and its principal sigma factor (rpoD) that confer a further 2-fold improvement and up to 13.8 g/L L-tyrosine production in engineered E. coli strains.281 Interestingly, the beneficial effects of the rpoA/rpoD mutations on L-tyrosine production, which implicates acid stress resistance and stringent response pathways, are only apparent in mutated genetic backgrounds that likely contain additional synergistic mutations. Although endogenous pathways were used to demonstrate the technology, strategies such as gTME that introduce global perturbation to cellular functions show great potential in improving metabolite production in heterologous host systems that permit efficient combinatorial introduction of directed genomic mutations or plasmid-encoded mutagenesis libraries. Additionally, given the observations in actinomycetes, it is likely that engineering of ribosomes and associated translational machinery will have equally beneficial impacts on the production titers and productivities.
Recombineering with phage-derived proteins enables the combinatorial in vivo introduction of precise alterations to genomes.284 An oligonucleotide-mediated allelic replacement method catalyzed by the λ Red recombinase, MAGE (multiplex automated genome engineering) enables the rapid generation of targeted, multi-site genetic diversity in E. coli with billions of variants created per day (Figure 8A).285, 286 By targeting the RBSs of 20 endogenous genes in the 1-deoxy-D-xylulose-5-phosphate pathway with oligonucleotides containing partial degenerate RBSs biased towards the consensus Shine-Dalgarno sequence, lycopene production was improved by over 5 fold.285 The allelic-replacement efficiency can be further improved by employing co-selection markers around target sites,287, 288 which enabled the simultaneous insertion of T7 promoters in front of 12 genomic operons.289 Oligonucleotide-mediated engineering has also been demonstrated to optimize expression of plasmid-encoded biosynthetic pathways and improve production, as demonstrated with riboflavin.290 Though recombineering is currently limited to a few organisms,284, 291 when applied to hosts expressing heterologous BGCs, these powerful tools could be used to rapidly generate genetic variants with improved overproduction phenotypes.
Fig. 8. Genome-scale engineering tools for optimization of natural product biosynthesis.
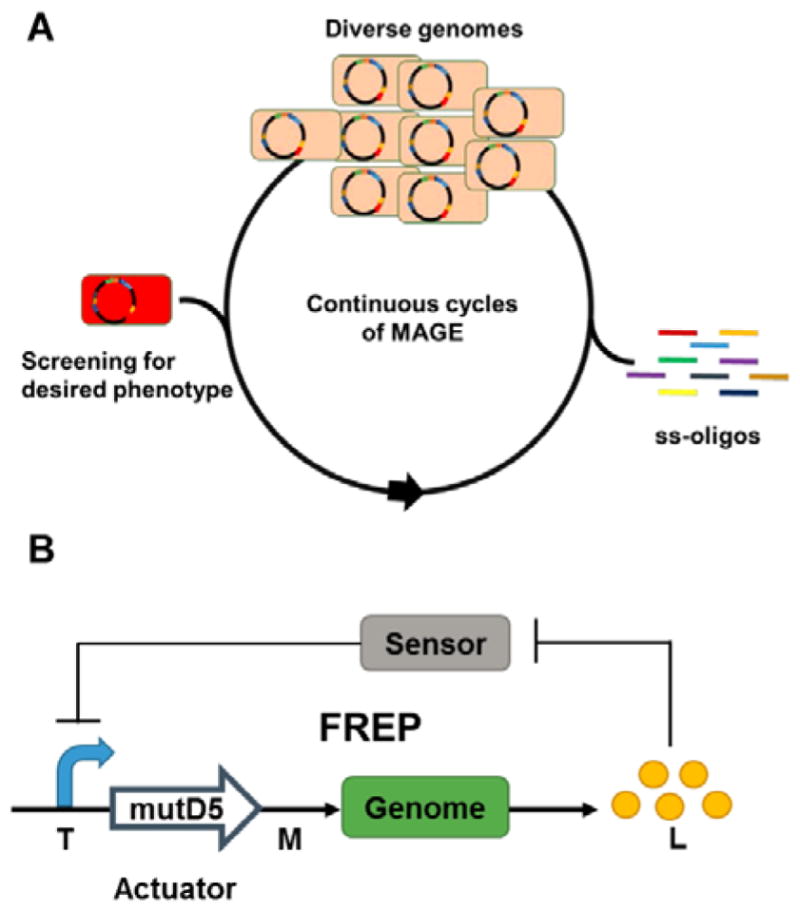
(A) MAGE enables the continuous generation of multi-loci multi-allelic genomic diversity by repeated introduction synthetic single-stranded oligos into E. coli cells.285 (B) FREP consists of a two-module genetic circuit that dynamically controls the genomic mutation rate (M) based on the target metabolite concentration (L).293 The sensor controls the transcriptional level (T) in the system via the actuator, which converts the T into M.
For effective strain improvement, it is important to navigate the complexities of biological systems and evaluate the functional contributions of candidate pathways or genes towards desired phenotypes. Trackable multiplex recombineering (TRMR) oligo-mediated engineering uses barcoded oligonucleotides to introduce expression-modulating cassettes across the E. coli genome and deep-sequencing to map up to 95% of the genes to specific cellular traits.292 In an alternative strategy to mine complex biological systems for engineering targets, feedback-regulated evolution of phenotype (FREP) involves a two-module genetic circuit to dynamically control the genome mutation rate in response to concentrations of a target metabolite (Figure 8B).293 When the concentration of the target metabolite is low, high mutation rates introduce phenotypic diversity whereas lower mutation rates with increasing target metabolite concentrations increase the probability that beneficial mutations are fixed within the evolving population. In the same study, Chou and Keasling also established a general framework for the development of synthetic sensors based on metabolic enzymes for metabolites without known biosensors.293 Overall, these strategies help bridge rational and random engineering methods by accelerating the identification of unpredictable mutations or candidates that are linked to desired traits.
6.3. Genome-minimized hosts
Genome minimization of existing heterologous hosts aims to rationally reduce the chemical and biologically complexities that confound high-level production of target metabolites. Deletion of non-essential genes and endogenous secondary metabolite biosynthetic pathways remove competing metabolic sinks, and facilitate downstream metabolic engineering efforts by simplifying metabolic networks. In addition, cleaner chemical backgrounds in these engineered hosts expedite downstream analysis and purification of target natural product. Moreover, physiological improvements associated with genome-minimization such as faster growth rate, longer stationary phase, higher final cell density and greater genetic stability also contribute to the utility of a heterologous host for secondary metabolite production.294–297
Using Cre-loxP recombination, Komatsu and colleagues deleted 1.4 Mbp of subtelomeric regions harboring endogenous BGCs and non-essential genes such as transposons and insertion sequences from S. avermitlis.226 Streptomycin productivity in the genome-minimized SUKA5 strain is approximately 3.5 and 4 times higher than that in native S. griseus and wild type S. avermitilis strains respectively. Another genome-minimized strain SUKA17, with faster growth and sporulation rate, was able to continuously produce cephamycin C for a longer time without product degradation compared to the native S. clavuligerus host.226, 227 Devoid of terpene synthase activity, the SUKA22 strain (isogenic to SUKA17) was used to heterologously express 29 terpene synthases from actinomycetes, leading to the identification of 13 novel terpenoids.230 The generality of these engineered S. avermitilis strains was demonstrated by the heterologous expression of 21 out of 26 BGCs with production titers ranging from 1–360 mg/L.298
Bibb and coworkers created a S. coelicolor strain devoid of antibiotic activity by deleting 1.73 Mbp of DNA harboring four known active BGCs. Subsequent introduction of rpoB and rpsL mutations yielded the S. coelicolor M1154 strain that produced 20–40 times more antibiotics, congocidine and chloramphenicol, than the parent M145 strain.225 M1154 and related strains have since been used to produce diverse metabolites from Streptomyces and phylogenetically distant actinomycetes.223 Nonetheless, genome minimization for heterologous secondary metabolite production remains an empirical process. A distinct genome-minimized S. coelicolor ZM12 strain that has all 10 PKS/NRPS clusters and 900 kb of its subtelomeric regions deleted failed to express a heterologous nikkomycin BGC that was functionally expressed in the parent M145 strain.299
Other target Streptomyces hosts for genome minimization include S. ambofaciens, S. roseosporus,300 S. toyocaensis301 and S. cinnamonesis302 due to their relatively small genomes encoding multiple PPTase genes and MbtH homologs, which are positive correlates of good heterologous producers. While the production titers of heterologous production systems generally fall in the mg/L range, the ~5 g/L tetracenomycin production achieved in S. cinnamonesis demonstrates the potential of heterologous Streptomyces expression systems for high level production.302 The development of efficient multiplex genetic engineering technologies (Section 3.2) promises the development of more genome-minimized Streptomyces strains for the high-level production of different natural products classes.
7. Concluding remarks
The “golden age” of natural products from the 1930s to 1960s saw the discovery of numerous secondary metabolites that revolutionized medicine and changed the course of human history, dramatically reducing global death rates and enabling treatments of previously incurable diseases and infections.303 Yet by the 1990s, high rediscovery rates, low production titers, challenging product isolation and structural identification as well as impractical chemical synthesis and derivatization prompted pharmaceutical companies to scale down their natural product discovery programs in favor of synthetic combinatorial libraries, resulting in the waning of drug and drug lead pipelines today. Propelled by high throughput and low costs of next-generation sequencing technologies, metagenomic and genomic approaches have revealed immense microbial biodiversity and biosynthetic capability. In this rapidly growing body of genomic information lies a vast unexplored chemical diversity that may be mined for societal needs. Many of the genetic tools and host engineering strategies for improving natural product titers presented in this review can also be employed for the activation of cryptic BGCs and the discovery of novel natural products.4
While our chemical synthetic abilities continue to advance, microbial fermentation is increasingly explored as an alternative approach to produce structurally complex natural products. With the host engineering tools and strategies presented in this review, it is possible to significantly improve production although the integration of different strategies will be needed to achieve industrially relevant titers (Figure 3B). Coupled with better understanding of the determinants of a good overproduction host,183 emerging technologies in diverse fields such as computational modelling and synthetic biology promise to accelerate the process of natural product discovery and development. Advances in DNA assembly and synthetic biology technologies drive the use of modular genetic parts for BGC refactoring and constructing of synthetic regulatory circuits for the precise and dynamic control of gene function. Technical breakthroughs in recombineering and genome editing pave the way for genome-scale perturbations of biological systems and the creation of genetically recoded organisms with programmable functions.304, 305 Although mainly demonstrated in model organisms like E. coli and yeast, these technologies promise unprecedented access to genome-encoded natural products when adapted to natural product-relevant organisms such as actinomycetes. The recent development of single-cell screening of Streptomyces allows high-throughput characterization of genetic parts and genetic circuits to enable more complex engineering of natural product producers.21 When used in conjunction with biosensors, the same technique can be used to rapidly screen for overproduction phenotypes. Additionally, with genome editing tools such as the CRISPR-Cas9 technology, it is conceivable that selected non-model native hosts can be strategically engineered to be heterologous production hosts for specialized classes of secondary metabolites or cellular platforms for combinatorial biosynthesis to create natural product derivatives and “unnatural” natural products.5, 6, 306 Continued technological and conceptual advances in these areas will open up opportunities to fully explore and harness Nature’s immensely diverse chemical repertoire.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (GM077596) (H.Z.), the Visiting Investigator Programme of A*STAR (H.Z.), and the National Research Foundation, Singapore (M.M.Z.). We thank members of the Metabolic Engineering Research Laboratory in A*STAR and the Zhao laboratory in UIUC for constructive comments.
References
- 1.Ji HF, Li XJ, Zhang HY. EMBO Rep. 2009;10:194–200. doi: 10.1038/embor.2009.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newman DJ, Cragg GM. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlson EE. ACS Chem Biol. 2010;5:639–653. doi: 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rutledge PJ, Challis GL. Nat Rev Microbiol. 2015 doi: 10.1038/nrmicro3496. [DOI] [PubMed] [Google Scholar]
- 5.Kim E, Moore BS, Yoon YJ. Nat Chem Biol. 2015;11:649–659. doi: 10.1038/nchembio.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun H, Liu Z, Zhao H, Ang EL. Drug Design Dev Ther. 2015;9:823. doi: 10.2147/DDDT.S63023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corre C, Challis GL. Nat Prod Rep. 2009;26:977–986. doi: 10.1039/b713024b. [DOI] [PubMed] [Google Scholar]
- 8.Challis GL. Microbiology. 2008;154:1555–1569. doi: 10.1099/mic.0.2008/018523-0. [DOI] [PubMed] [Google Scholar]
- 9.Nett M, Ikeda H, Moore BS. Nat Prod Rep. 2009;26:1362–1384. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziemert N, Jensen PR. Methods Enzymol. 2012;517:161–182. doi: 10.1016/B978-0-12-404634-4.00008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich: 2000. [Google Scholar]
- 12.Suzuki H. Host-Mimicking Strategies in DNA Methylation for Improved Bacterial Transformation. INTECH Open Access Publisher; 2012. [Google Scholar]
- 13.Kim JY, Wang Y, Park MS, Ji GE. J Microbiol Biotechnol. 2010;20:1022–1026. doi: 10.4014/jmb.1003.03014. [DOI] [PubMed] [Google Scholar]
- 14.Chater KF, Wilde LC. J Gen Microbiol. 1980;116:323–334. doi: 10.1099/00221287-116-2-323. [DOI] [PubMed] [Google Scholar]
- 15.Medema MH, Breitling R, Takano E. Methods Enzymol. 2011;497:485–502. doi: 10.1016/B978-0-12-385075-1.00021-4. [DOI] [PubMed] [Google Scholar]
- 16.Seghezzi N, Amar P, Koebmann B, Jensen PR, Virolle MJ. Appl Microbiol Biotechnol. 2011;90:615–623. doi: 10.1007/s00253-010-3018-0. [DOI] [PubMed] [Google Scholar]
- 17.Bibb MJ, Janssen GR, Ward JM. Gene. 1986;41:E357–E368. [Google Scholar]
- 18.Luo Y, Zhang L, Barton KW, Zhao H. ACS Synth Biol. 2015;4:1001–1010. doi: 10.1021/acssynbio.5b00016. [DOI] [PubMed] [Google Scholar]
- 19.Siegl T, Tokovenko B, Myronovskyi M, Luzhetskyy A. Metab Eng. 2013;19:98–106. doi: 10.1016/j.ymben.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Wang W, Li X, Wang J, Xiang S, Feng X, Yang K. Appl Environ Microbiol. 2013;79:4484–4492. doi: 10.1128/AEM.00985-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai C, Zhang Y, Zhao X, Hu Y, Xiang S, Miao J, Lou C, Zhang L. Proc Natl Acad Sci U S A. 2015;112:12181–12186. doi: 10.1073/pnas.1511027112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S, Wang J, Li X, Yin S, Wang W, Yang K. Microb Cell Fact. 2015;14:1–11. doi: 10.1186/s12934-015-0351-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lussier FX, Denis F, Shareck F. Applied and environmental microbiology. 2010;76:967–970. doi: 10.1128/AEM.02186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herai S, Hashimoto Y, Higashibata H, Maseda H, Ikeda H, Omura S, Kobayashi M. Proc Natl Acad Sci U S A. 2004;101:14031–14035. doi: 10.1073/pnas.0406058101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez-Garcia A, Combes P, Perez-Redondo R, Smith MC, Smith MC. Nucleic Acids Res. 2005;33:e87. doi: 10.1093/nar/gni086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudolph MM, Vockenhuber MP, Suess B. Microbiology. 2013;159:1416–1422. doi: 10.1099/mic.0.067322-0. [DOI] [PubMed] [Google Scholar]
- 27.Jia X, Zhang J, Sun W, He W, Jiang H, Chen D, Murchie AI. Cell. 2013;152:68–81. doi: 10.1016/j.cell.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 28.Weigand JE, Sanchez M, Gunnesch EB, Zeiher S, Schroeder R, Suess B. RNA. 2008;14:89–97. doi: 10.1261/rna.772408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Alia D, Nieselt K, Steigele S, Muller J, Verburg I, Takano E. J Bacteriol. 2010;192:1160–1164. doi: 10.1128/JB.01374-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uguru GC, Mondhe M, Goh S, Hesketh A, Bibb MJ, Good L, Stach JE. PLoS One. 2013;8:e67509. doi: 10.1371/journal.pone.0067509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Longstaff DG, Larue RC, Faust JE, Mahapatra A, Zhang L, Green-Church KB, Krzycki JA. Proc Natl Acad Sci U S A. 2007;104:1021–1026. doi: 10.1073/pnas.0610294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myronovskyi M, Welle E, Fedorenko V, Luzhetskyy A. Appl Environ Microbiol. 2011;77:5370–5383. doi: 10.1128/AEM.00434-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ingram C, Brawner M, Youngman P, Westpheling J. J Bacteriol. 1989;171:6617–6624. doi: 10.1128/jb.171.12.6617-6624.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Craney A, Hohenauer T, Xu Y, Navani NK, Li Y, Nodwell J. Nucleic Acids Res. 2007;35:e46. doi: 10.1093/nar/gkm086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li P, Li J, Guo Z, Tang W, Han J, Meng X, Hao T, Zhu Y, Zhang L, Chen Y. Appl Microbiol Biotechnol. 2015;99:1923–1933. doi: 10.1007/s00253-014-6369-0. [DOI] [PubMed] [Google Scholar]
- 36.Dubeau MP, Ghinet MG, Jacques PE, Clermont N, Beaulieu C, Brzezinski R. Appl Environ Microbiol. 2009;75:1211–1214. doi: 10.1128/AEM.02139-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeng H, Wen S, Xu W, He Z, Zhai G, Liu Y, Deng Z, Sun Y. Appl Microbiol Biotechnol. 2015 doi: 10.1007/s00253-015-6931-4. [DOI] [PubMed] [Google Scholar]
- 38.Hosted TJ, Baltz RH. J Bacteriol. 1997;179:180–186. doi: 10.1128/jb.179.1.180-186.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fisher SH, Bruton CJ, Chater KF. Mol Gen Genet. 1987;206:35–44. doi: 10.1007/BF00326533. [DOI] [PubMed] [Google Scholar]
- 40.Fernández-Martínez LT, Bibb MJ. Sci Rep. 2014:4. doi: 10.1038/srep07100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu Z, Xie P, Qin Z. Acta Biochim Biophys Sin. 2010:gmq080. doi: 10.1093/abbs/gmq080. [DOI] [PubMed] [Google Scholar]
- 42.Thorpe HM, Smith MC. Proc Natl Acad Sci U S A. 1998;95:5505–5510. doi: 10.1073/pnas.95.10.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L, Ou X, Zhao G, Ding X. J Bacteriol. 2008;190:6392–6397. doi: 10.1128/JB.00777-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gregory MA, Till R, Smith MC. J Bacteriol. 2003;185:5320–5323. doi: 10.1128/JB.185.17.5320-5323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baltz RH. J Ind Microbiol Biotechnol. 2012;39:661–672. doi: 10.1007/s10295-011-1069-6. [DOI] [PubMed] [Google Scholar]
- 46.Morita K, Yamamoto T, Fusada N, Komatsu M, Ikeda H, Hirano N, Takahashi H. FEMS Microbiol Lett. 2009;297:234–240. doi: 10.1111/j.1574-6968.2009.01683.x. [DOI] [PubMed] [Google Scholar]
- 47.Olivares EC, Hollis RP, Calos MP. Gene. 2001;278:167–176. doi: 10.1016/s0378-1119(01)00711-9. [DOI] [PubMed] [Google Scholar]
- 48.Fayed B, Younger E, Taylor G, Smith MC. BMC Biotechnol. 2014;14:51. doi: 10.1186/1472-6750-14-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fedoryshyn M, Petzke L, Welle E, Bechthold A, Luzhetskyy A. Gene. 2008;419:43–47. doi: 10.1016/j.gene.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 50.Khodakaramian G, Lissenden S, Gust B, Moir L, Hoskisson PA, Chater KF, Smith MC. Nucleic Acids Res. 2006;34:e20–e20. doi: 10.1093/nar/gnj019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zelyas N, Tahlan K, Jensen SE. Gene. 2009;443:48–54. doi: 10.1016/j.gene.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 52.Herrmann S, Siegl T, Luzhetska M, Petzke L, Jilg C, Welle E, Erb A, Leadlay PF, Bechthold A, Luzhetskyy A. Appl Environ Microbiol. 2012;78:1804–1812. doi: 10.1128/AEM.06054-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fogg PC, Colloms S, Rosser S, Stark M, Smith MC. J Mol Biol. 2014;426:2703–2716. doi: 10.1016/j.jmb.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang B, Zhang L, Dai R, Yu M, Zhao G, Ding X. PLoS One. 2013;8 doi: 10.1371/journal.pone.0055906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Du D, Wang L, Tian Y, Liu H, Tan H, Niu G. Sci Rep. 2015;5 doi: 10.1038/srep08740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colloms SD, Merrick CA, Olorunniji FJ, Stark WM, Smith MC, Osbourn A, Keasling JD, Rosser SJ. Nucleic Acids Res. 2014;42:e23–e23. doi: 10.1093/nar/gkt1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang L, Wang L, Wang J, Ou X, Zhao G, Ding X. J Mol Cell Biol. 2010;2:264–275. doi: 10.1093/jmcb/mjq025. [DOI] [PubMed] [Google Scholar]
- 58.Murakami T, Burian J, Yanai K, Bibb MJ, Thompson CJ. Proc Natl Acad Sci U S A. 2011;108:16020–16025. doi: 10.1073/pnas.1108124108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou TC, Kim BG, Zhong JJ. Appl Microbiol Biotechnol. 2014;98:7911–7922. doi: 10.1007/s00253-014-5943-9. [DOI] [PubMed] [Google Scholar]
- 60.Cobb RE, Wang Y, Zhao H. ACS Synth Biol. 2015;4:723–728. doi: 10.1021/sb500351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tong Y, Charusanti P, Zhang L, Weber T, Lee SY. ACS Synth Biol. 2015;4:1020–1029. doi: 10.1021/acssynbio.5b00038. [DOI] [PubMed] [Google Scholar]
- 62.Huang H, Zheng G, Jiang W, Hu H, Lu Y. Acta Biochim Biophys Sin (Shanghai) 2015;47:231–243. doi: 10.1093/abbs/gmv007. [DOI] [PubMed] [Google Scholar]
- 63.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H. Nature. 2014 doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doudna JA, Charpentier E. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 65.Dymond J, Boeke J. Bioengineered. 2012;3:170–173. doi: 10.4161/bbug.19543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Demain AL. J Ind Microbiol Biotechnol. 2006;33:486–495. doi: 10.1007/s10295-005-0076-x. [DOI] [PubMed] [Google Scholar]
- 67.Paradkar A. The Journal of antibiotics. 2013;66:411–420. doi: 10.1038/ja.2013.26. [DOI] [PubMed] [Google Scholar]
- 68.Katz L, Khosla C. Nature biotechnology. 2007;25:428. doi: 10.1038/nbt0407-428. [DOI] [PubMed] [Google Scholar]
- 69.Xu B, Jin Z, Wang H, Jin Q, Jin X, Cen P. Appl Microbiol Biotechnol. 2008;80:261–267. doi: 10.1007/s00253-008-1540-0. [DOI] [PubMed] [Google Scholar]
- 70.Jin Z, Xu B, Lin S, Jin Q, Cen P. Appl Microbiol Biotechnol. 2009;159:655–663. doi: 10.1007/s12010-008-8500-0. [DOI] [PubMed] [Google Scholar]
- 71.Lv XA, Jin YY, Li YD, Zhang H, Liang XL. Appl Microbiol Biotechnol. 2013;97:641–648. doi: 10.1007/s00253-012-4322-7. [DOI] [PubMed] [Google Scholar]
- 72.Luo JM, Li JS, Liu D, Liu F, Wang YT, Song XR, Wang M. J Agri Food Chem. 2012;60:6026–6036. doi: 10.1021/jf300663w. [DOI] [PubMed] [Google Scholar]
- 73.Stutzman-Engwall K, Conlon S, Fedechko R, McArthur H, Pekrun K, Chen Y, Jenne S, La C, Trinh N, Kim S. Metab Eng. 2005;7:27–37. doi: 10.1016/j.ymben.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 74.Stephanopoulos G. Nat Biotechnol. 2002;20:666–668. doi: 10.1038/nbt0702-666. [DOI] [PubMed] [Google Scholar]
- 75.Zhang YX, Perry K, Vinci VA, Powell K, Stemmer WP, del Cardayré SB. Nature. 2002;415:644–646. doi: 10.1038/415644a. [DOI] [PubMed] [Google Scholar]
- 76.Gong J, Zheng H, Wu Z, Chen T, Zhao X. Biotechnol Adv. 2009;27:996–1005. doi: 10.1016/j.biotechadv.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 77.Ochi K, Hosaka T. Appl Microbiol Biotechnol. 2013;97:87–98. doi: 10.1007/s00253-012-4551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ochi K. Bioscience, biotechnology, and biochemistry. 2007;71:1373–1386. doi: 10.1271/bbb.70007. [DOI] [PubMed] [Google Scholar]
- 79.Ochi K, Tanaka Y, Tojo S. J Ind Microbiol Biotechnol. 2014;41:403–414. doi: 10.1007/s10295-013-1349-4. [DOI] [PubMed] [Google Scholar]
- 80.Wang L, Zhao Y, Liu Q, Huang Y, Hu C, Liao G. Journal of applied microbiology. 2012;112:1095–1101. doi: 10.1111/j.1365-2672.2012.05302.x. [DOI] [PubMed] [Google Scholar]
- 81.Baltz RH. Journal of industrial microbiology & biotechnology. 2011;38:657–666. doi: 10.1007/s10295-010-0934-z. [DOI] [PubMed] [Google Scholar]
- 82.Shima J, Hesketh A, Okamoto S, Kawamoto S, Ochi K. J Bacteriol. 1996;178:7276–7284. doi: 10.1128/jb.178.24.7276-7284.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hu H, Ochi K. Appl Environ Microbiol. 2001;67:1885–1892. doi: 10.1128/AEM.67.4.1885-1892.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tanaka Y, Kasahara K, Hirose Y, Murakami K, Kugimiya R, Ochi K. J Bacteriol. 2013;195:2959–2970. doi: 10.1128/JB.00147-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Imai Y, Fujiwara T, Ochi K, Hosaka T. J Antibiot (Tokyo) 2012;65:323–326. doi: 10.1038/ja.2012.16. [DOI] [PubMed] [Google Scholar]
- 86.Wang G, Hosaka T, Ochi K. Appl Environ Microbiol. 2008;74:2834–2840. doi: 10.1128/AEM.02800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tamehiro N, Hosaka T, Xu J, Hu H, Otake N, Ochi K. Appl Environ Microbiol. 2003;69:6412–6417. doi: 10.1128/AEM.69.11.6412-6417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Beltrametti F, Rossi R, Selva E, Marinelli F. J Ind Microbiol Biotechnol. 2006;33:283–288. doi: 10.1007/s10295-005-0061-4. [DOI] [PubMed] [Google Scholar]
- 89.Derewacz DK, Goodwin CR, McNees CR, McLean JA, Bachmann BO. Proc Natl Acad Sci U S A. 2013;110:2336–2341. doi: 10.1073/pnas.1218524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hosaka T, Ohnishi-Kameyama M, Muramatsu H, Murakami K, Tsurumi Y, Kodani S, Yoshida M, Fujie A, Ochi K. Nat Biotechnol. 2009;27:462–464. doi: 10.1038/nbt.1538. [DOI] [PubMed] [Google Scholar]
- 91.Nishimura K, Hosaka T, Tokuyama S, Okamoto S, Ochi K. J Bacteriol. 2007;189:3876–3883. doi: 10.1128/JB.01776-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hosaka T, Xu J, Ochi K. Mol Microbiol. 2006;61:883–897. doi: 10.1111/j.1365-2958.2006.05285.x. [DOI] [PubMed] [Google Scholar]
- 93.Li L, Guo J, Wen Y, Chen Z, Song Y, Li J. J Ind Microbiol Biotechnol. 2010;37:673–679. doi: 10.1007/s10295-010-0710-0. [DOI] [PubMed] [Google Scholar]
- 94.Pan Y, Lu C, Dong H, Yu L, Liu G, Tan H. Microb Cell Fact. 2013;12:65. doi: 10.1186/1475-2859-12-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carata E, Peano C, Tredici SM, Ferrari F, Talà A, Corti G, Bicciato S, De Bellis G, Alifano P. Microb Cell Fact. 2009;8:18. doi: 10.1186/1475-2859-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tanaka Y, Komatsu M, Okamoto S, Tokuyama S, Kaji A, Ikeda H, Ochi K. Appl Environ Microbiol. 2009;75:4919–4922. doi: 10.1128/AEM.00681-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hu H, Zhang Q, Ochi K. J Bacteriol. 2002;184:3984–3991. doi: 10.1128/JB.184.14.3984-3991.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu J, Tozawa Y, Lai C, Hayashi H, Ochi K. Mol Genet Genom. 2002;268:179–189. doi: 10.1007/s00438-002-0730-1. [DOI] [PubMed] [Google Scholar]
- 99.Ishikawa J, Chiba K, Kurita H, Satoh H. Antimicrob Agents Chemother. 2006;50:1342–1346. doi: 10.1128/AAC.50.4.1342-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tala A, Wang G, Zemanova M, Okamoto S, Ochi K, Alifano P. J Bacteriol. 2009;191:805–814. doi: 10.1128/JB.01311-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vigliotta G, Tredici SM, Damiano F, Montinaro MR, Pulimeno R, Di Summa R, Massardo DR, Gnoni GV, Alifano P. Mol Microbiol. 2005;55:396–412. doi: 10.1111/j.1365-2958.2004.04406.x. [DOI] [PubMed] [Google Scholar]
- 102.Okamoto-Hosoya Y, Okamoto S, Ochi K. Appl Environ Microbiol. 2003;69:4256–4259. doi: 10.1128/AEM.69.7.4256-4259.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Orelle C, Carlson ED, Szal T, Florin T, Jewett MC, Mankin AS. Nature. 2015;524:119–124. doi: 10.1038/nature14862. [DOI] [PubMed] [Google Scholar]
- 104.Fried SD, Schmied WH, Uttamapinant C, Chin JW. Angewandte Chemie International Edition. 2015;54:12791–12794. doi: 10.1002/anie.201506311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fierro F, Barredo JL, Diez B, Gutierrez S, Fernandez FJ, Martin JF. Proc Natl Acad Sci U S A. 1995;92:6200–6204. doi: 10.1073/pnas.92.13.6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Peschke U, Schmidt H, Zhang HZ, Piepersberg W. Mol Microbiol. 1995;16:1137–1156. doi: 10.1111/j.1365-2958.1995.tb02338.x. [DOI] [PubMed] [Google Scholar]
- 107.Yanai K, Murakami T, Bibb M. Proc Natl Acad Sci U S A. 2006;103:9661–9666. doi: 10.1073/pnas.0603251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liao G, Li J, Li L, Yang H, Tian Y, Tan H. Microb Cell Fact. 2010;9:6. doi: 10.1186/1475-2859-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jiang L, Wei J, Li L, Niu G, Tan H. Appl Microbiol Biotechnol. 2013;97:10469–10477. doi: 10.1007/s00253-013-5270-6. [DOI] [PubMed] [Google Scholar]
- 110.Du D, Zhu Y, Wei J, Tian Y, Niu G, Tan H. Appl Microbiol Biotechnol. 2013;97:6383–6396. doi: 10.1007/s00253-013-4836-7. [DOI] [PubMed] [Google Scholar]
- 111.Yu L, Cao N, Wang L, Xiao C, Guo M, Chu J, Zhuang Y, Zhang S. Enzyme and microbial technology. 2012;50:318–324. doi: 10.1016/j.enzmictec.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 112.Zhu T, Cheng X, Liu Y, Deng Z, You D. Metab Eng. 2013;19:69–78. doi: 10.1016/j.ymben.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 113.Qiu J, Zhuo Y, Zhu D, Zhou X, Zhang L, Bai L, Deng Z. Appl Microbiol Biotechnol. 2011;92:337–345. doi: 10.1007/s00253-011-3439-4. [DOI] [PubMed] [Google Scholar]
- 114.Malla S, Niraula NP, Liou K, Sohng JK. Microbiol Res. 2010;165:259–267. doi: 10.1016/j.micres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 115.Xu Y, Willems A, Au-yeung C, Tahlan K, Nodwell JR. MBio. 2012;3:e00191–00112. doi: 10.1128/mBio.00191-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bibb MJ. Curr Opin Microbiol. 2005;8:208–215. doi: 10.1016/j.mib.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 117.van Wezel GP, McDowall KJ. Nat Prod Rep. 2011;28:1311–1333. doi: 10.1039/c1np00003a. [DOI] [PubMed] [Google Scholar]
- 118.Romero-Rodríguez A, Robledo-Casados I, Sánchez S. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2015;1849:1017–1039. doi: 10.1016/j.bbagrm.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 119.Olano C, Lombó F, Méndez C, Salas JA. Metab Eng. 2008;10:281–292. doi: 10.1016/j.ymben.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 120.Chen Y, Smanski MJ, Shen B. Appl Microbiol Biotechnol. 2010;86:19–25. doi: 10.1007/s00253-009-2428-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wietzorrek A, Bibb M. Mol Microbiol. 1997;25:1181–1184. doi: 10.1046/j.1365-2958.1997.5421903.x. [DOI] [PubMed] [Google Scholar]
- 122.Sheldon PJ, Busarow SB, Hutchinson CR. Mol Microbiol. 2002;44:449–460. doi: 10.1046/j.1365-2958.2002.02886.x. [DOI] [PubMed] [Google Scholar]
- 123.Liu G, Tian Y, Yang H, Tan H. Mol Microbiol. 2005;55:1855–1866. doi: 10.1111/j.1365-2958.2005.04512.x. [DOI] [PubMed] [Google Scholar]
- 124.Chen Y, Wendt-Pienkowski E, Shen B. J Bacteriol. 2008;190:5587–5596. doi: 10.1128/JB.00592-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bate N, Bignell DR, Cundliffe E. Mol Microbiol. 2006;62:148–156. doi: 10.1111/j.1365-2958.2006.05338.x. [DOI] [PubMed] [Google Scholar]
- 126.Stratigopoulos G, Bate N, Cundliffe E. Mol Microbiol. 2004;54:1326–1334. doi: 10.1111/j.1365-2958.2004.04347.x. [DOI] [PubMed] [Google Scholar]
- 127.Laureti L, Song L, Huang S, Corre C, Leblond P, Challis GL, Aigle B. Proc Natl Acad Sci U S A. 2011;108:6258–6263. doi: 10.1073/pnas.1019077108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Olano C, García I, González A, Rodriguez M, Rozas D, Rubio J, Sánchez-Hidalgo M, Braña AF, Méndez C, Salas JA. Microb Biotechnol. 2014;7:242–256. doi: 10.1111/1751-7915.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Franke J, Ishida K, Hertweck C. Angewandte Chemie International Edition. 2012;51:11611–11615. doi: 10.1002/anie.201205566. [DOI] [PubMed] [Google Scholar]
- 130.Goranovič D, Blažič M, Magdevska V, Horvat J, Kuščer E, Polak T, Santos-Aberturas J, Martínez-Castro M, Barreiro C, Mrak P. BMC Microbiol. 2012;12:238. doi: 10.1186/1471-2180-12-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kuščer E, Coates N, Challis I, Gregory M, Wilkinson B, Sheridan R, Petković H. J Bacteriol. 2007;189:4756–4763. doi: 10.1128/JB.00129-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Santos-Aberturas J, Payero TD, Vicente CM, Guerra SM, Cañibano C, Martín JF, Aparicio JF. Metab Eng. 2011;13:756–767. doi: 10.1016/j.ymben.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 133.Antón N, Santos-Aberturas J, Mendes MV, Guerra SM, Martín JF, Aparicio JF. Microbiology. 2007;153:3174–3183. doi: 10.1099/mic.0.2007/009126-0. [DOI] [PubMed] [Google Scholar]
- 134.Du YL, Chen SF, Cheng LY, Shen XL, Tian Y, Li YQ. J Microbiol. 2009;47:506–513. doi: 10.1007/s12275-009-0014-0. [DOI] [PubMed] [Google Scholar]
- 135.Wu H, Liu W, Dong D, Li J, Zhang D, Lu C. J Ind Microbiol Biotechnol. 2014;41:163–172. doi: 10.1007/s10295-013-1370-7. [DOI] [PubMed] [Google Scholar]
- 136.Vicente CM, Payero TD, Santos-Aberturas J, Barreales EG, de Pedro A, Aparicio JF. Appl Microbiol Biotechnol. 2015;99:5123–5135. doi: 10.1007/s00253-015-6472-x. [DOI] [PubMed] [Google Scholar]
- 137.Smanski MJ, Peterson RM, Rajski SR, Shen B. Antimicrob Agents Chemother. 2009;53:1299–1304. doi: 10.1128/AAC.01358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maddocks SE, Oyston PC. Microbiology. 2008;154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 139.Challis GL, Hopwood DA. Proc Natl Acad Sci U S A. 2003;100:14555–14561. doi: 10.1073/pnas.1934677100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kitani S, Ikeda H, Sakamoto T, Noguchi S, Nihira T. Appl Microbiol Biotechnol. 2009;82:1089–1096. doi: 10.1007/s00253-008-1850-2. [DOI] [PubMed] [Google Scholar]
- 141.Chen Y, Yin M, Horsman GP, Huang S, Shen B. J Antibiot. 2010;63:482–485. doi: 10.1038/ja.2010.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chen Y, Yin M, Horsman GP, Shen B. J Nat Prod. 2011;74:420–424. doi: 10.1021/np100825y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li L, Zhao Y, Ruan L, Yang S, Ge M, Jiang W, Lu Y. Metab Eng. 2015;29:12–25. doi: 10.1016/j.ymben.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 144.Kitani S, Miyamoto KT, Takamatsu S, Herawati E, Iguchi H, Nishitomi K, Uchida M, Nagamitsu T, Omura S, Ikeda H. Proc Natl Acad Sci U S A. 2011;108:16410–16415. doi: 10.1073/pnas.1113908108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hsiao NH, Nakayama S, Merlo ME, de Vries M, Bunet R, Kitani S, Nihira T, Takano E. Chem Biol. 2009;16:951–960. doi: 10.1016/j.chembiol.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 146.Tan GY, Bai L, Zhong JJ. Biotechnol Bioeng. 2013;110:2984–2993. doi: 10.1002/bit.24965. [DOI] [PubMed] [Google Scholar]
- 147.Nodwell JR. Mol Microbiol. 2014;94:483–485. doi: 10.1111/mmi.12787. [DOI] [PubMed] [Google Scholar]
- 148.Takano E. Curr Opin Microbiol. 2006;9:287–294. doi: 10.1016/j.mib.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 149.Sidda JD, Song L, Poon V, Al-Bassam M, Lazos O, Buttner MJ, Challis GL, Corre C. Chem Sci. 2013;5:86–89. [Google Scholar]
- 150.Gottelt M, Kol S, Gomez-Escribano JP, Bibb M, Takano E. Microbiology. 2010;156:2343–2353. doi: 10.1099/mic.0.038281-0. [DOI] [PubMed] [Google Scholar]
- 151.Tan GY, Peng Y, Lu C, Bai L, Zhong JJ. Metab Eng. 2015;28:74–81. doi: 10.1016/j.ymben.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 152.Biarnes-Carrera M, Breitling R, Takano E. Curr Opin Chem Biol. 2015;28:91–98. doi: 10.1016/j.cbpa.2015.06.024. [DOI] [PubMed] [Google Scholar]
- 153.Zhuo Y, Zhang W, Chen D, Gao H, Tao J, Liu M, Gou Z, Zhou X, Ye BC, Zhang Q. Proc Natl Acad Sci U S A. 2010;107:11250–11254. doi: 10.1073/pnas.1006085107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang H, Yang L, Wu K, Li G. Microb Cell Fact. 2014;13 doi: 10.1186/1475-2859-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu SP, Yu P, Yuan PH, Zhou ZX, Bu QT, Mao XM, Li YQ. Appl Microbiol Biotechnol. 2015;99:2715–2726. doi: 10.1007/s00253-014-6307-1. [DOI] [PubMed] [Google Scholar]
- 156.Foulston LC, Bibb MJ. Proc Natl Acad Sci U S A. 2010;107:13461–13466. doi: 10.1073/pnas.1008285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sherwood EJ, Bibb MJ. Proc Natl Acad Sci U S A. 2013;110:E2500–E2509. doi: 10.1073/pnas.1305392110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Seipke RF, Patrick E, Hutchings MI. PeerJ. 2014;2:e253. doi: 10.7717/peerj.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Luo S, Sun D, Zhu J, Chen Z, Wen Y, Li J. Appl Microbiol Biotechnol. 2014;98:7097–7112. doi: 10.1007/s00253-014-5759-7. [DOI] [PubMed] [Google Scholar]
- 160.Segall-Shapiro TH, Meyer AJ, Ellington AD, Sontag ED, Voigt CA. Molecular systems biology. 2014;10:742. doi: 10.15252/msb.20145299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ryu YG, Butler MJ, Chater KF, Lee KJ. Appl Environ Microbiol. 2006;72:7132–7139. doi: 10.1128/AEM.01308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Butler MJ, Bruheim P, Jovetic S, Marinelli F, Postma PW, Bibb MJ. Appl Environ Microbiol. 2002;68:4731–4739. doi: 10.1128/AEM.68.10.4731-4739.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Borodina I, Siebring J, Zhang J, Smith CP, van Keulen G, Dijkhuizen L, Nielsen J. J Biol Chem. 2008;283:25186–25199. doi: 10.1074/jbc.M803105200. [DOI] [PubMed] [Google Scholar]
- 164.Tang Z, Xiao C, Zhuang Y, Chu J, Zhang S, Herron PR, Hunter IS, Guo M. Enzyme and Microbial Technology. 2011;49:17–24. doi: 10.1016/j.enzmictec.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 165.Li R, Townsend CA. Metab Eng. 2006;8:240–252. doi: 10.1016/j.ymben.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 166.Reeves AR, Brikun IA, Cernota WH, Leach BI, Gonzalez MC, Weber JM. Metab Eng. 2007;9:293. doi: 10.1016/j.ymben.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Reeves A, Brikun I, Cernota W, Leach B, Gonzalez M, Weber JM. J Ind Microbiol Biotechnol. 2006;33:600–609. doi: 10.1007/s10295-006-0094-3. [DOI] [PubMed] [Google Scholar]
- 168.Zabala D, Braña AF, Flórez AB, Salas JA, Méndez C. Metab Eng. 2013;20:187–197. doi: 10.1016/j.ymben.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 169.Zabala D, Brana AF, Salas JA, Mendez C. J Antibiot. 2015 doi: 10.1038/ja.2015.104. [DOI] [PubMed] [Google Scholar]
- 170.Zhang L, Li Y, Wang Z, Xia Y, Chen W, Tang K. Biotechnol Adv. 2007;25:123–136. doi: 10.1016/j.biotechadv.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 171.Stark BC, Dikshit KL, Pagilla KR. Comput Struct Biotechnol J. 2012;3:1–8. doi: 10.5936/csbj.201210002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mehmood N, Olmos E, Goergen JL, Blanchard F, Ullisch D, Klöckner W, Büchs J, Delaunay S. Biotechnol Bioeng. 2011;108:2151–2161. doi: 10.1002/bit.23177. [DOI] [PubMed] [Google Scholar]
- 173.Wang S, Liu F, Hou Z, Zong G, Zhu X, Ling P. World J Microbiol Biotechnol. 2014;30:1369–1376. doi: 10.1007/s11274-013-1561-4. [DOI] [PubMed] [Google Scholar]
- 174.Wu H, Li J, Dong D, Liu T, Zhang T, Zhang D, Liu W. Enzyme and microbial technology. 2015;81:80–87. doi: 10.1016/j.enzmictec.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 175.Magnolo SK, Leenutaphong DL, DeModena JA, Curtis JE, Bailey JE, Galazzo JL, Hughes DE. Nat Biotechnol. 1991;9:473–476. doi: 10.1038/nbt0591-473. [DOI] [PubMed] [Google Scholar]
- 176.Wang T, Bai L, Zhu D, Lei X, Liu G, Deng Z, You D. Enzyme and microbial technology. 2012;50:5–9. doi: 10.1016/j.enzmictec.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 177.Kim YJ, Sa SO, Chang YK, Hong S, Hong Y. J Microbiol Biotechnol. 2007;17:2066. [PubMed] [Google Scholar]
- 178.Lu Y-W, San Roman AK, Gehring AM. J Bacteriol. 2008;190:6903–6908. doi: 10.1128/JB.00865-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sánchez C, Du L, Edwards DJ, Toney MD, Shen B. Chem Biol. 2001;8:725–738. doi: 10.1016/s1074-5521(01)00047-3. [DOI] [PubMed] [Google Scholar]
- 180.Jiang H, Wang YY, Ran XX, Fan WM, Jiang XH, Guan WJ, Li YQ. Appl Environ Microbiol. 2013;79:3346–3354. doi: 10.1128/AEM.00099-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bunet R, Riclea R, Laureti L, Hôtel L, Paris C, Girardet JM, Spiteller D, Dickschat JS, Leblond P, Aigle B. PLoS One. 2014;9:e87607. doi: 10.1371/journal.pone.0087607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Medema MH, Alam MT, Heijne WH, van den Berg MA, Müller U, Trefzer A, Bovenberg RA, Breitling R, Takano E. Microbial biotechnology. 2011;4:300–305. doi: 10.1111/j.1751-7915.2010.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Medema MH, Alam MT, Heijne WH, van den Berg MA, Müller U, Trefzer A, Bovenberg RA, Breitling R, Takano E. Microb Biotechnol. 2011;4:300–305. doi: 10.1111/j.1751-7915.2010.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yang Q, Ding X, Liu X, Liu S, Sun Y, Yu Z, Hu S, Rang J, He H, He L. Microbial cell factories. 2014;13:1. doi: 10.1186/1475-2859-13-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Alam MT, Merlo ME, Hodgson DA, Wellington EM, Takano E, Breitling R. BMC genomics. 2010;11:1. [Google Scholar]
- 186.Kim M, Sang Yi J, Kim J, Kim JN, Kim MW, Kim BG. Biotechnology journal. 2014;9:1185–1194. doi: 10.1002/biot.201300539. [DOI] [PubMed] [Google Scholar]
- 187.Borodina I, Krabben P, Nielsen J. Genome research. 2005;15:820–829. doi: 10.1101/gr.3364705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Baltz RH. Curr Opin Pharmacol. 2008;8:557–563. doi: 10.1016/j.coph.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 189.Fenical W, Jensen PR. Nat Chem Biol. 2006;2:666–673. doi: 10.1038/nchembio841. [DOI] [PubMed] [Google Scholar]
- 190.Wenzel SC, Muller R. Nat Prod Rep. 2009;26:1385–1407. doi: 10.1039/b817073h. [DOI] [PubMed] [Google Scholar]
- 191.Milshteyn A, Schneider JS, Brady SF. Chem Biol. 2014;21:1211–1223. doi: 10.1016/j.chembiol.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Copp JN, Neilan BA. Appl Environ Microbiol. 2006;72:2298–2305. doi: 10.1128/AEM.72.4.2298-2305.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Roberts AA, Copp JN, Marahiel MA, Neilan BA. Chembiochem. 2009;10:1869–1877. doi: 10.1002/cbic.200900095. [DOI] [PubMed] [Google Scholar]
- 194.Dahl RH, Zhang F, Alonso-Gutierrez J, Baidoo E, Batth TS, Redding-Johanson AM, Petzold CJ, Mukhopadhyay A, Lee TS, Adams PD, Keasling JD. Nat Biotechnol. 2013;31:1039–1046. doi: 10.1038/nbt.2689. [DOI] [PubMed] [Google Scholar]
- 195.Pickens LB, Tang Y, Chooi YH. Annu Rev Chem Biomol Eng. 2011;2:211–236. doi: 10.1146/annurev-chembioeng-061010-114209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mutka SC, Carney JR, Liu Y, Kennedy J. Biochemistry. 2006;45:1321–1330. doi: 10.1021/bi052075r. [DOI] [PubMed] [Google Scholar]
- 197.Pfeifer BA, Wang CC, Walsh CT, Khosla C. Appl Environ Microbiol. 2003;69:6698–6702. doi: 10.1128/AEM.69.11.6698-6702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhang H, Wang Y, Wu J, Skalina K, Pfeifer BA. Chem Biol. 2010;17:1232–1240. doi: 10.1016/j.chembiol.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 199.Zhang H, Skalina K, Jiang M, Pfeifer BA. Biotechnol Prog. 2012;28:292–296. doi: 10.1002/btpr.702. [DOI] [PubMed] [Google Scholar]
- 200.Gross F, Gottschalk D, Muller R. Appl Microbiol Biotechnol. 2005;68:66–74. doi: 10.1007/s00253-004-1836-7. [DOI] [PubMed] [Google Scholar]
- 201.Owen JG, Copp JN, Ackerley DF. Biochem J. 2011;436:709–717. doi: 10.1042/BJ20110321. [DOI] [PubMed] [Google Scholar]
- 202.Blank LM, Ebert BE, Buehler K, Buhler B. Antioxid Redox Signal. 2010;13:349–394. doi: 10.1089/ars.2009.2931. [DOI] [PubMed] [Google Scholar]
- 203.Fernandez M, Duque P, Pizarro-Tobias P, Van Dillewijn R-M, Ramos JL. Microb Biotechnol. 2009;2:1751–7915. doi: 10.1111/j.1751-7915.2009.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Simon O, Klaiber I, Huber A, Pfannstiel J. J Proteomics. 2014;109:1876–7737. doi: 10.1016/j.jprot.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 205.Blank LM, Ionidis G, Ebert BE, Buhler B, Schmid A. The FEBS journal. 2008;275:5173–5190. doi: 10.1111/j.1742-4658.2008.06648.x. [DOI] [PubMed] [Google Scholar]
- 206.Loeschcke A, Thies S. Appl Microbiol Biotechnol. 2015;99:6197–6214. doi: 10.1007/s00253-015-6745-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tareq FS, Lee MA, Lee HS, Lee YJ, Lee JS, Hasan CM, Islam MT, Shin HJ. Org Lett. 2014;16:928–931. doi: 10.1021/ol403657r. [DOI] [PubMed] [Google Scholar]
- 208.Tareq FS, Kim JH, Lee MA, Lee HS, Lee YJ, Lee JS, Shin HJ. Org Lett. 2012;14:1464–1467. doi: 10.1021/ol300202z. [DOI] [PubMed] [Google Scholar]
- 209.Zhang H, Boghigian BA, Armando J, Pfeifer BA. Nat Prod Rep. 2011;28:125–151. doi: 10.1039/c0np00037j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Stein T. Mol Microbiol. 2005;56:845–857. doi: 10.1111/j.1365-2958.2005.04587.x. [DOI] [PubMed] [Google Scholar]
- 211.Hu Y, Potts MB, Colosimo D, Herrera-Herrera ML, Legako AG, Yousufuddin M, White MA, MacMillan JB. J Am Chem Soc. 2013;135:13387–13392. doi: 10.1021/ja403412y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hamdache A, Lamarti A, Aleu J, Collado IG. J Nat Prod. 2011;74:893–899. doi: 10.1021/np100853e. [DOI] [PubMed] [Google Scholar]
- 213.Bron S, Holsappel S, Venema G, Peeters BP. Mol Gen Genet. 1991;226:88–96. doi: 10.1007/BF00273591. [DOI] [PubMed] [Google Scholar]
- 214.Borisova SA, Circello BT, Zhang JK, van der Donk WA, Metcalf WW. Chem Biol. 2010;17:28–37. doi: 10.1016/j.chembiol.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Choi SK, Park SY, Kim R, Kim SB, Lee CH, Kim JF, Park SH. J Bacteriol. 2009;191:3350–3358. doi: 10.1128/JB.01728-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Herzner AM, Dischinger J, Szekat C, Josten M, Schmitz S, Yakeleba A, Reinartz R, Jansen A, Sahl HG, Piel J, Bierbaum G. PLoS One. 2011;6:e22389. doi: 10.1371/journal.pone.0022389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tsuge K, Inoue S, Ano T, Itaya M, Shoda M. Antimicrob Agents Chemother. 2005;49:4641–4648. doi: 10.1128/AAC.49.11.4641-4648.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Yuksel S, Hansen JN. Appl Microbiol Biotechnol. 2007;74:640–649. doi: 10.1007/s00253-006-0713-y. [DOI] [PubMed] [Google Scholar]
- 219.Tang Y, Frewert S, Harmrolfs K, Herrmann J, Karmann L, Kazmaier U, Xia L, Zhang Y, Muller R. J Biotechnol. 2015;194:112–114. doi: 10.1016/j.jbiotec.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 220.Li Y, Li Z, Yamanaka K, Xu Y, Zhang W, Vlamakis H, Kolter R, Moore BS, Qian PY. Sci Rep. 2015;5:9383. doi: 10.1038/srep09383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Baltz RH. J Ind Microbiol Biotechnol. 2010;37:759–772. doi: 10.1007/s10295-010-0730-9. [DOI] [PubMed] [Google Scholar]
- 222.Luo Y, Li BZ, Liu D, Zhang L, Chen Y, Jia B, Zeng BX, Zhao H, Yuan YJ. Chem Soc Rev. 2015;44:5265–5290. doi: 10.1039/c5cs00025d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Gomez-Escribano JP, Bibb MJ. J Ind Microbiol Biotechnol. 2014;41:425–431. doi: 10.1007/s10295-013-1348-5. [DOI] [PubMed] [Google Scholar]
- 224.Procopio RE, Silva IR, Martins MK, Azevedo JL, Araujo JM. The Brazilian journal of infectious diseases : an official publication of the Brazilian Society of Infectious Diseases. 2012;16:466–471. doi: 10.1016/j.bjid.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 225.Gomez-Escribano JP, Bibb MJ. Microb Biotechnol. 2011;4:207–215. doi: 10.1111/j.1751-7915.2010.00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H. Proc Natl Acad Sci U S A. 2010;107:2646–2651. doi: 10.1073/pnas.0914833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Komatsu M, Komatsu K, Koiwai H, Yamada Y, Kozone I, Izumikawa M, Hashimoto J, Takagi M, Omura S, Shin-ya K, Cane DE, Ikeda H. ACS Synth Biol. 2013;2:384–396. doi: 10.1021/sb3001003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Jung WS, Lee SK, Hong JSJ, Park SR, Jeong SJ, Han AR, Sohng JK, Kim BG, Choi CY, Sherman DH. Appl Microbiol Biotechnol. 2006;72:763–769. doi: 10.1007/s00253-006-0318-5. [DOI] [PubMed] [Google Scholar]
- 229.Park SR, Ahn MS, Han AR, Park JW, Yoon YJ. J Microbiol Biotechnol. 2011;21:1143–1146. doi: 10.4014/jmb.1108.08012. [DOI] [PubMed] [Google Scholar]
- 230.Yamada Y, Kuzuyama T, Komatsu M, Shin-ya K, Omura S, Cane DE, Ikeda H. Proc Natl Acad Sci U S A. 2015;112:857–862. doi: 10.1073/pnas.1422108112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Siddiqui MS, Thodey K, Trenchard I, Smolke CD. FEMS yeast research. 2012;12:144–170. doi: 10.1111/j.1567-1364.2011.00774.x. [DOI] [PubMed] [Google Scholar]
- 232.Ross AC, Gulland LE, Dorrestein PC, Moore BS. ACS Synth Biol. 2015;4:414–420. doi: 10.1021/sb500280q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Fu J, Bian X, Hu S, Wang H, Huang F, Seibert PM, Plaza A, Xia L, Müller R, Stewart AF. Nat Biotechnol. 2012;30:440–446. doi: 10.1038/nbt.2183. [DOI] [PubMed] [Google Scholar]
- 234.Kouprina N, Larionov V. Nat Protoc. 2008;3:371–377. doi: 10.1038/nprot.2008.5. [DOI] [PubMed] [Google Scholar]
- 235.Yamanaka K, Reynolds KA, Kersten RD, Ryan KS, Gonzalez DJ, Nizet V, Dorrestein PC, Moore BS. Proc Natl Acad Sci U S A. 2014;111:1957–1962. doi: 10.1073/pnas.1319584111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lee NC, Larionov V, Kouprina N. Nucleic Acids Res. 2015;43:e55–e55. doi: 10.1093/nar/gkv112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Jiang W, Zhao X, Gabrieli T, Lou C, Ebenstein Y, Zhu TF. Nat Commun. 2015;6 doi: 10.1038/ncomms9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Shao Z, Rao G, Li C, Abil Z, Luo Y, Zhao H. ACS Synth Biol. 2013;2:662–669. doi: 10.1021/sb400058n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Montiel D, Kang HS, Chang FY, Charlop-Powers Z, Brady SF. Proc Natl Acad Sci U S A. 2015;112:8953–8958. doi: 10.1073/pnas.1507606112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 241.Wingler LM, Cornish VW. Proceedings of the National Academy of Sciences. 2011;108:15135–15140. doi: 10.1073/pnas.1100507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Li MZ, Elledge SJ. Nat Methods. 2007;4:251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
- 243.Engler C, Kandzia R, Marillonnet S. PLoS One. 2008;3:e3647. doi: 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chen WH, Qin ZJ, Wang J, Zhao GP. Nucleic Acids Res. 2013;41:e93–e93. doi: 10.1093/nar/gkt122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Werner S, Engler C, Weber E, Gruetzner R, Marillonnet S. Bioengineered. 2012;3:38–43. doi: 10.4161/bbug.3.1.18223. [DOI] [PubMed] [Google Scholar]
- 246.Shao Z, Luo Y, Zhao H. Mol BioSystems. 2011;7:1056–1059. doi: 10.1039/c0mb00338g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Luo Y, Huang H, Liang J, Wang M, Lu L, Shao Z, Cobb RE, Zhao H. Nat Commun. 2013;4 doi: 10.1038/ncomms3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Ongley SE, Bian X, Neilan BA, Muller R. Nat Prod Rep. 2013;30:1121–1138. doi: 10.1039/c3np70034h. [DOI] [PubMed] [Google Scholar]
- 249.Cobb RE, Ning JC, Zhao H. J Ind Microbiol Biotechnol. 2014;41:469–477. doi: 10.1007/s10295-013-1358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Smanski MJ, Bhatia S, Zhao D, Park Y, Woodruff LBA, Giannoukos G, Ciulla D, Busby M, Calderon J, Nicol R, Gordon DB, Densmore D, Voigt CA. Nat Biotechnol. 2014;32:1241–1249. doi: 10.1038/nbt.3063. [DOI] [PubMed] [Google Scholar]
- 251.Keasling JD. Science. 2010;330:1355–1358. doi: 10.1126/science.1193990. [DOI] [PubMed] [Google Scholar]
- 252.Zhang F, Ouellet M, Batth TS, Adams PD, Petzold CJ, Mukhopadhyay A, Keasling JD. Metab Eng. 2012;14:653–660. doi: 10.1016/j.ymben.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 253.Woodruff LB, Boyle NR, Gill RT. Metab Eng. 2013;17:1–11. doi: 10.1016/j.ymben.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 254.Ajikumar PK, Xiao WH, Tyo KE, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G. Science. 2010;330:70–74. doi: 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Yuan LZ, Rouviere PE, Larossa RA, Suh W. Metab Eng. 2006;8:79–90. doi: 10.1016/j.ymben.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 256.De Mey M, Maertens J, Boogmans S, Soetaert WK, Vandamme EJ, Cunin R, Foulquie-Moreno MR. BMC Biotechnol. 2010;10:26. doi: 10.1186/1472-6750-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Proc Natl Acad Sci U S A. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Smolke CD, Martin VJ, Keasling JD. Metab Eng. 2001;3:313–321. doi: 10.1006/mben.2001.0194. [DOI] [PubMed] [Google Scholar]
- 259.Pfleger BF, Pitera DJ, Smolke CD, Keasling JD. Nat Biotechnol. 2006;24:1027–1032. doi: 10.1038/nbt1226. [DOI] [PubMed] [Google Scholar]
- 260.Salis HM, Mirsky EA, Voigt CA. Nat Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Seo SW, Kim SC, Jung GY. Biotechnol Bioprocess Eng. 2012;17:1–7. doi: 10.1007/s12257-012-0242-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Fowler ZL, Gikandi WW, Koffas MA. Appl Environ Microbiol. 2009;75:5831–5839. doi: 10.1128/AEM.00270-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Griffin MO, Fricovsky E, Ceballos G, Villarreal F. Am J Physiol Cell Physiol. 2010;299:C539–548. doi: 10.1152/ajpcell.00047.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Han AR, Park JW, Lee MK, Ban YH, Yoo YJ, Kim EJ, Kim E, Kim BG, Sohng JK, Yoon YJ. Appl Environ Microbiol. 2011;77:4912–4923. doi: 10.1128/AEM.02527-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Koirala N, Pandey RP, Thang DV, Jung HJ, Sohng JK. J Ind Microbiol Biotechnol. 2014;41:1647–1658. doi: 10.1007/s10295-014-1504-6. [DOI] [PubMed] [Google Scholar]
- 266.Malla S, Koffas MA, Kazlauskas RJ, Kim BG. Appl Environ Microbiol. 2012;78:684–694. doi: 10.1128/AEM.06274-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Olano C, Mendez C, Salas JA. Microb Biotechnol. 2011;4:144–164. doi: 10.1111/j.1751-7915.2010.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Zha W, Rubin-Pitel SB, Shao Z, Zhao H. Metab Eng. 2009;11:192–198. doi: 10.1016/j.ymben.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 269.Xu P, Li L, Zhang F, Stephanopoulos G, Koffas M. Proc Natl Acad Sci U S A. 2014;111:11299–11304. doi: 10.1073/pnas.1406401111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Lussier FX, Colatriano D, Wiltshire Z, Page JE, Martin VJ. Comput Struct Biotechnol J. 2012;3:e201210020. doi: 10.5936/csbj.201210020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Causey TB, Shanmugam KT, Yomano LP, Ingram LO. Proc Natl Acad Sci U S A. 2004;101:2235–2240. doi: 10.1073/pnas.0308171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Liu D, Xiao Y, Evans BS, Zhang F. ACS Synth Biol. 2015;4:132–140. doi: 10.1021/sb400158w. [DOI] [PubMed] [Google Scholar]
- 273.Xu P, Gu Q, Wang W, Wong L, Bower AG, Collins CH, Koffas MA. Nat Commun. 2013;4:1409. doi: 10.1038/ncomms2425. [DOI] [PubMed] [Google Scholar]
- 274.Santos CN, Koffas M, Stephanopoulos G. Metab Eng. 2011;13:392–400. doi: 10.1016/j.ymben.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 275.Yang Y, Lin Y, Li L, Linhardt RJ, Yan Y. Metab Eng. 2015;29:217–226. doi: 10.1016/j.ymben.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 276.Gao X, Wang P, Tang Y. Appl Microbiol Biotechnol. 2010;88:1233–1242. doi: 10.1007/s00253-010-2860-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Chan YA, Podevels AM, Kevany BM, Thomas MG. Nat Prod Rep. 2009;26:90–114. doi: 10.1039/b801658p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Zhang F, Carothers JM, Keasling JD. Nat Biotechnol. 2012;30:354–359. doi: 10.1038/nbt.2149. [DOI] [PubMed] [Google Scholar]
- 279.Tyo KE, Ajikumar PK, Stephanopoulos G. Nat Biotechnol. 2009;27:760–765. doi: 10.1038/nbt.1555. [DOI] [PubMed] [Google Scholar]
- 280.Si T, Xiao H, Zhao H. Biotechnol Adv. 2015;33:1420–1432. doi: 10.1016/j.biotechadv.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Santos CN, Xiao W, Stephanopoulos G. Proc Natl Acad Sci U S A. 2012;109:13538–13543. doi: 10.1073/pnas.1206346109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Alper H, Stephanopoulos G. Metab Eng. 2007;9:258–267. doi: 10.1016/j.ymben.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 283.Santos CN, Stephanopoulos G. Curr Opin Chem Biol. 2008;12:168–176. doi: 10.1016/j.cbpa.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 284.Pines G, Freed EF, Winkler JD, Gill RT. ACS Synth Biol. 2015 doi: 10.1021/acssynbio.5b00009. [DOI] [PubMed] [Google Scholar]
- 285.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Gallagher RR, Li Z, Lewis AO, Isaacs FJ. Nat Protocols. 2014;9:2301–2316. doi: 10.1038/nprot.2014.082. [DOI] [PubMed] [Google Scholar]
- 287.Carr PA, Wang HH, Sterling B, Isaacs FJ, Lajoie MJ, Xu G, Church GM, Jacobson JM. Nucleic Acids Res. 2012;40:e132. doi: 10.1093/nar/gks455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Gregg CJ, Lajoie MJ, Napolitano MG, Mosberg JA, Goodman DB, Aach J, Isaacs FJ, Church GM. Nucleic Acids Res. 2014;42:4779–4790. doi: 10.1093/nar/gkt1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Wang HH, Kim H, Cong L, Jeong J, Bang D, Church GM. Nat Methods. 2012;9:591–593. doi: 10.1038/nmeth.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Li Y, Gu Q, Lin Z, Wang Z, Chen T, Zhao X. ACS Synth Biol. 2013;2:651–661. doi: 10.1021/sb400051t. [DOI] [PubMed] [Google Scholar]
- 291.DiCarlo JE, Conley AJ, Penttila M, Jantti J, Wang HH, Church GM. ACS Synth Biol. 2013;2:741–749. doi: 10.1021/sb400117c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Warner JR, Reeder PJ, Karimpour-Fard A, Woodruff LB, Gill RT. Nat Biotechnol. 2010;28:856–862. doi: 10.1038/nbt.1653. [DOI] [PubMed] [Google Scholar]
- 293.Chou HH, Keasling JD. Nat Commun. 2013;4:2595. doi: 10.1038/ncomms3595. [DOI] [PubMed] [Google Scholar]
- 294.Martinez-Garcia E, de Lorenzo V. Environmental microbiology. 2011;13:2702–2716. doi: 10.1111/j.1462-2920.2011.02538.x. [DOI] [PubMed] [Google Scholar]
- 295.Martinez-Garcia E, Nikel PI, Chavarria M, de Lorenzo V. Environmental microbiology. 2014;16:291–303. doi: 10.1111/1462-2920.12309. [DOI] [PubMed] [Google Scholar]
- 296.Mizoguchi H, Mori H, Fujio T. Biotechnol Appl Biochem. 2007;46:157–167. doi: 10.1042/BA20060107. [DOI] [PubMed] [Google Scholar]
- 297.Pósfai G, Plunkett G, Fehér T, Frisch D, Keil GM, Umenhoffer K, Kolisnychenko V, Stahl B, Sharma SS, De Arruda M. Science. 2006;312:1044–1046. doi: 10.1126/science.1126439. [DOI] [PubMed] [Google Scholar]
- 298.Ikeda H, Shin-ya K, Omura S. J Ind Microbiol Biotechnol. 2014;41:233–250. doi: 10.1007/s10295-013-1327-x. [DOI] [PubMed] [Google Scholar]
- 299.Zhou M, Jing X, Xie P, Chen W, Wang T, Xia H, Qin Z. FEMS Microbiol Lett. 2012;333:169–179. doi: 10.1111/j.1574-6968.2012.02609.x. [DOI] [PubMed] [Google Scholar]
- 300.Alexander DC, Rock J, He X, Brian P, Miao V, Baltz RH. Appl Environ Microbiol. 2010;76:6877–6887. doi: 10.1128/AEM.01248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Banik JJ, Craig JW, Calle PY, Brady SF. J Am Chem Soc. 2010;132:15661–15670. doi: 10.1021/ja105825a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Li C, Hazzard C, Florova G, Reynolds KA. Metab Eng. 2009;11:319–327. doi: 10.1016/j.ymben.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 303.Shen B. Cell. 2015;163:1297–1300. doi: 10.1016/j.cell.2015.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA. Science. 2013;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Amiram M, Haimovich AD, Fan C, Wang YS, Aerni HR, Ntai I, Moonan DW, Ma NJ, Rovner AJ, Hong SH. Nature biotechnology. 2015;33:1272–1279. doi: 10.1038/nbt.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Thaker MN, Wright GD. ACS Synth Biol. 2012;4:195–206. doi: 10.1021/sb300092n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.