

Abstract
Terpenoid cyclases catalyze the most complex reactions in biology, in that more than half of the substrate carbon atoms often undergo changes in bonding during the course of a multistep cyclization cascade that proceeds through multiple carbocation intermediates. Many cyclization mechanisms require stereospecific deprotonation and reprotonation steps, and most cyclization cascades are terminated by deprotonation to yield an olefin product. The first bacterial terpenoid cyclase to yield a crystal structure was pentalenene synthase from Streptomyces exfoliatus UC5319. This cyclase generates the hydrocarbon precursor of the pentalenolactone family of antibiotics. The structures of pentalenene synthase and other terpenoid cyclases reveal predominantly nonpolar active sites typically lacking amino acid side chains capable of serving general base-general acid functions. What chemical species, then, enables the Brønsted acid-base chemistry required in the catalytic mechanisms of these enzymes? The most likely candidate for such general base-general acid chemistry is the co-product inorganic pyrophosphate. Here, we briefly review biological and nonbiological systems in which phosphate and its derivatives serve general base and general acid functions in catalysis. These examples highlight the fact that the Brønsted acid-base activities of phosphate derivatives are comparable to the Brønsted acid-base activities of amino acid side chains.
Introduction
The greater family of terpenoids, including steroids and carotenoids, currently includes more than 75,000 members, and new terpenoids continue to be discovered to this day. The vast chemodiversity of this family of natural products arises in large part from the catalytic versatility of terpenoid cyclases. These enzymes catalyze complex cyclization cascades, typically proceeding through multiple carbocation intermediates, to generate myriad hydrocarbon skeletons containing multiple rings and stereocenters.1–5 Terpenoid cyclases generally belong to one of two classes, depending on the chemistry that initiates the cyclization cascade: a class I terpenoid cyclase triggers the metal-dependent ionization of an isoprenoid diphosphate, such as the sesquiterpene (C15) farnesyl diphosphate (FPP), to generate an allylic carbocation plus inorganic pyrophosphate (PPi); a class II terpenoid cyclase generates an initial tertiary carbocation by protonation of a carbon-carbon double bond.6–10 This Review focuses exclusively on class I terpenoid cyclases, specifically with regard to general base-general acid catalysis that facilitates proton transfer steps subsequent to initial carbocation formation.
Pentalenene synthase from Streptomyces exfoliatus UC5319 is a prototypical class I terpenoid cyclase that catalyzes the cyclization of FPP to form pentalenene, which comprises the first committed step in the biosynthesis of the pentalenolactone family of antibiotics (Figure 1).11–13 The biological activity of pentalenolactone against Gram-positive and Gram-negative bacteria was discovered nearly 60 years ago,14 and the molecular basis for this activity is the inhibition of the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase.15,16 The stereospecific formation of the tricyclic pentalenene precursor is critical for the antibacterial activity of pentalenolactone, and the three fused rings and four stereocenters of pentalenene are precisely generated in a single reaction catalyzed by pentalenene synthase. Notably, this is the enzyme that first brought the Cane and Christianson laboratories together for a decades-long collaboration on terpenoid cyclase structure and function.
Figure 1.

Farnesyl diphosphate cyclization reaction catalyzed by pentalenene synthase from Streptomyces exfoliatus UC5319, as initially proposed by Cane and colleagues.18 This reaction is the first step in the biosynthesis of the antibiotic pentalenolactone. The co-product inorganic pyrophosphate (PPO−, i.e., the PPi anion) is proposed to be the general base-general acid that mediates stereospecific proton transfers in catalysis.
Cane and colleagues reported the preparation of cell-free Streptomyces extracts of pentalenene synthase, enabling enzymological studies with isotopically labeled substrates to elucidate the cyclization mechanism.12,13,17 Notable features of the mechanism first proposed for pentalenene synthase include ionization of FPP, cyclization, and stereospecific deprotonation to form the intermediate humulene; this proton is subsequently utilized to reprotonate the adjacent carbon atom to generate the bicyclic protoilludyl cation, which is proposed to undergo hydride transfer, carbon-carbon bond formation, and a final stereospecific proton abstraction to yield pentalenene. As noted by Cane and colleagues,18 a single general base-general acid in the active site would be geometrically competent to mediate the stereospecific deprotonation-reprotonation-deprotonation sequence in the multistep cyclization cascade (Figure 1).
The cloning and expression of pentalenene synthase from S. exfoliatus UC5319 yielded an enzyme identical to the native enzyme in all respects18 and ultimately led to the first crystal structure determination of a bacterial terpenoid cyclase.19 The enzyme active site was found to be predominantly nonpolar, consistent with the accommodation of the lipophilic substrate. Even so, a single polar residue was observed near the mouth of the active site, H309, and this residue was hypothesized to be the stereospecific general base-general acid required for pentalenene formation.19 However, Cane and colleagues demonstrated that H309 mutants of pentalenene synthase exhibit nearly full activity.20 Mutagenesis of other, less conventional general base candidates, e.g., W308 (a tryptophan had been suggested as a catalytic general base in another sesquiterpene cyclase, 5-epi-aristolochene synthase21), also proved fruitless.22 Where, then, is the general base-general acid that mediates stereospecific proton transfer and the final proton abstraction in the cyclization mechanism?
This question has persisted in the analysis of structure-function relationships ever since the first crystal structures of class I terpenoid cyclases were reported.19,21 The first crystal structure determination of a fungal terpenoid cyclase, aristolochene synthase from Penicillium roqueforti, revealed a similar α-helical fold to that of pentalenene synthase, and a similarly barren, nonpolar active site devoid of an obvious general base-general acid.23 While an active site tyrosine was considered as a possible general base-general acid, mutagenesis studies with the related aristolochene synthase from Aspergillus terreus demonstrated that the corresponding tyrosine was not required for catalytic activity.24 Another possible candidate suggested for the general base in P. roqueforti aristolochene synthase was the diphosphate leaving group,23 and structural studies of A. terreus aristolochene synthase provided compelling structural data that implicated the product PPi anion as a general base-general acid.25 Subsequent structural and enzymological studies of aristolochene synthase and other terpenoid synthases likewise implicated the PPi anion in general base-general acid catalysis.26–29
Given that the PPi anion is a co-product for all class I terpenoid cyclase reactions, the possibility of its regular participation in catalysis is intriguing to consider. Following FPP ionization, the PPi anion is locked in place by 3 Mg2+ ions and 3–4 hydrogen bond interactions,30 as first revealed in the crystal structure of the trichodiene synthase-Mg2+3-PPi complex.31 Ordinarily, inorganic phosphate (Pi) or PPi might not be considered as routine participants in general base catalysis, even though the range of pKa values characterizing their various ionization states are comparable to those of amino acid side chains (Figure 2). Moreover, these pKa values can be further modulated by the protein environment, e.g., by hydrogen bond interactions and/or metal ion coordination interactions. However, since a tertiary carbocation is quite acidic, with a pKa of approximately −10 or even lower, a general base in a terpenoid cyclase active site is perhaps not needed so much for activating a sluggish deprotonation step, but instead simply for accepting a highly reactive proton that has nowhere else to go.
Figure 2.

Ionization states and pKa values of phosphoric acid (inorganic phosphate) and pyrophosphoric acid (inorganic pyrophosphate).
Also influencing the stereospecificity of deprotonation, of course, is the conformation of the carbocation intermediate, since the empty 2p orbital of the carbocation must be aligned with the breaking C-H bond to facilitate proton loss and π bond formation. The three-dimensional contour of the terpenoid cyclase active site thus additionally influences carbocation acidity and the stereospecificity of proton transfer steps by controlling the conformations of macrocyclic intermediates.
The first direct structural clue regarding the feasibility of the PPi co-product serving as a general base in terpenoid biosynthesis was provided by the X-ray crystal structure of FPP synthase from Escherichia coli complexed with 3 Mg2+ ions, isopentenyl diphosphate (IPP), and the nonreactive substrate analogue dimethylallyl-S-thiolodiphosphate (DMASPP).32 This enzyme is not a terpenoid cyclase, but instead an isoprenoid chain elongation enzyme: it catalyzes successive coupling reactions of dimethylallyl diphosphate (DMAPP) with 2 molecules of IPP to generate FPP. Even so, the structure of this enzyme is homologous to that of class I terpenoid cyclases.33 The structure of the FPP synthase–Mg2+3–DMSPP–IPP complex revealed that the free (i.e., not complexed to Mg2+) oxygen atom of the DMSPP thiolodiphosphate group is oriented toward the pro-R hydrogen on C2 of IPP, as if poised for the stereospecific proton abstraction that terminates the chain elongation reaction (Figure 3). More recently, the binding of farnesyl-S-thiolodiphosphate as well as aza analogues of carbocation intermediates to aristolochene synthase from Aspergillus terreus revealed that a free oxygen of the PPi anion is ideally positioned for regiospecific and stereospecific proton abstraction from carbocation intermediates,34 consistent with the results of earlier structural studies.25 These results strongly suggest that the substrate itself provides useful chemical functionality – the diphosphate/PPi anion – for general base-general acid chemistry in the active site of a class I terpenoid cyclase.
Figure 3.

The active site of E. coli farnesyl diphosphate synthase complexed with three Mg2+ ions (blue spheres 1, 2, and 3), dimethylallyl-S-thiolodiphosphate (DMSPP, yellow), and isopentenyl diphosphate (IPP, green). Metal coordination and hydrogen bond interactions are indicated by solid blue and dotted magenta lines, respectively. The diphosphate group of DMSPP is oriented for abstraction of the pro-R hydrogen from IPP following the chain elongation reaction. Reprinted with permission from: Hosfield, D. J., Zhang, Y., Dougan, D. R., Broun, A., Tari, L. W., Swanson, R. V. & Finn, J. Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J. Biol. Chem. 279, 8526–8529 (2004). Copyright 2004 The American Society for Biochemistry & Molecular Biology.
PPi and Pi in General Acid-General Base Catalysis
While phosphate or diphosphate anions such as Pi or PPi are not often considered for general base or general acid functions in enzyme active sites, the relative acidities of their conjugate acid forms are comparable to those of amino acid side chains that serve as more traditional general acids or general bases. For example, the carboxylic acid side chains of glutamic acid and aspartic acid have pKa values of ~4, and the imidazolium side chain of histidine has a pKa value of ~6 in aqueous solution. In comparison, the second and third ionizations of pyrophosphoric acid have pKa values of 2.0 and 6.6, and the first and second ionizations of phosphoric acid have pKa values of 2.1 and 7.2 (Figure 2).35
Occasionally participating in enzyme acid-base chemistry are the side chains of cysteine (pKa ~ 8.3), lysine (pKa ~ 10.5), tyrosine (pKa ~ 10.9), and arginine (pKa ~ 12.5), and the pKa values for these basic side chains are comparable to those of the third ionization of phosphoric acid (12.7) or the fourth ionization of pyrophosphoric acid (9.4). As with the pKa values of amino acid side chains, the pKa values of Pi and PPi can be modulated by their environment and optimized for catalytic function. From the perspective of this chemistry, then, it is quite reasonable to consider the possibility of PPi in general base-general acid catalysis in a terpenoid cyclase active site. Indeed, there is ample precedent for comparable functions of phosphate derivatives in biological and nonbiological catalysis. Selected examples are summarized below.
Kynureninase
Also known as L-kynurenine hydrolase, this enzyme utilizes pyridoxal-5′-phosphate (PLP) as a cofactor to catalyze a retro-Claisen reaction yielding products alanine and anthranilic acid (Figure 4).36 The first step of catalysis is a trans-aldimination with the internal aldimine to generate an external aldimine. The side chain of K227 serves as a general base to deprotonate Cα and then reprotonate C-4′ to yield a kynurenine ketamine.37 Subsequent addition of a water molecule to the γ-carbonyl group of ketamine yields a gem-diol; cleavage of the Cβ-Cγ bond results in a carboxylic acid product and an enamine intermediate.38 Protonation of this enamine at Cβ yields pyruvate ketamine, which leads to an alanine quinonoid after deprotonation at C-4′. A final protonation at the quinonoid Cα gives the final products L-alanine and aldimine.39–41
Figure 4.

Proposed catalytic mechanism of kynureninase. The PLP-phosphate serves as a general base-general acid in the activation of Y226 for catalysis. Hydrolysis of the enamine product shown ultimately yields product alanine. Reprinted with permission from: Phillips, R. S. Chemistry and diversity of pyridoxal-5′-phosphate dependent enzymes. Biochim. Biophys. Acta – Prot. Proteomics. 1854, 1167–1174 (2015). Copyright 2015 Elsevier B.V.
Crystal structures of human kynureninase41,42 reveal that N333, Y275, and S332 are located in the active site and serve as potential hydrogen bond partners for the ligand. The hydrophobic side chains of I110, W305, F306, and F314 form a pocket that accommodates the substrate aromatic group. The δ-oxygen of N333 and the backbone amide of S332 both hydrogen bond with the phosphate group of PLP. The imidazole ring of H102 provides a π-stacking partner for the ligand. The structure of the human enzyme complexed with 3-hydroxykynurenine shows that N333 also hydrogen bonds with the C3 hydroxyl group of the inhibitor. Accordingly, the side chains of N333, S332, and H102 dictate specificity for ligand binding.
The position and orientation of a strictly conserved tyrosine, Y275 (or Y226 in the homologous enzyme from Pseudomonas fluorescens43), led to the suggestion that the phosphate group of PLP serves as a general base in catalysis. Given that the side chain of Y226 hydrogen bonds to the phosphate group of PLP, it is hypothesized that the phosphate group of PLP abstracts a proton from Y226 in the first step of the reaction, and donates this proton back to Y226 in a subsequent step (Figure 4).
The Y226F mutant was prepared to test this hypothesis.44 The mutation resulted in a reduction of the kcat by about 2800-fold for the substrate L-kynurenine, from 16 s−1 for the wild-type enzyme to 5.8 × 10−3 s−1 for Y226F kynureninase. The kcat/Km saw a 375-fold reduction from 2.0×105 M−1s−1 for the wild-type enzyme to 530 M−1s−1 for Y226F kynureninase. To study complexation of the wild-type enzyme with 5-bromodihydrokynurenine, 31P-NMR was used to demonstrate upfield spectroscopic shifts from 4.5 ppm to 5.0, 3.3, and 2.0 ppm upon complex formation. These shifts indicated that the phosphate group of PLP undergoes a change in ionization state from dianionic to monoanionic. However, the Y226F mutant does not exhibit these 31P-NMR spectroscopic shifts; coupled with the dramatic reduction in turnover for the mutant enzyme, these results suggest that without the Y226 hydrogen bond the PLP phosphate group cannot acquire a proton.45,46 The residual activity was hypothesized to be due to a water-mediated hydride shift in the absence of Y226.44 These data support the proposed role of the PLP phosphate group as the general base that deprotonates Y226 in catalysis.
L-Serine dehydratase
Eukaryotic L-serine dehydratase (SDH) is a PLP-dependent enzyme that catalyzes the dehydration of L-serine to form pyruvate and ammonia as the final products.47 SDH uses the phosphate group of PLP as a general base in the first step of catalysis.
The catalytic mechanism of SDH is illustrated in Figure 5. First, L-serine enters the active site, with its α-amino group forming a hydrogen bond with the phosphate group of PLP.48 The phosphate group then abstracts a proton from the α-amino group, forming the free base, which then attacks the C4′ atom to form a gem-diamine intermediate. Following release of the PLP cofactor from K41 to form a PLP-serine aldimine intermediate, the PLP phosphate group serves as a general acid and donates a proton to the serine hydroxyl group, thereby enabling its departure as a water molecule in concert with the K41-mediated abstraction of the Cα-H proton. The resulting PLP-aminoacrylate intermediate undergoes nucleophilic attack by K41 at C4′, and the resulting tetrahedral gem-diamine intermediate collapses to yield amino acrylate and enzyme-bound PLP; aminoacrylate then undergoes nonenzymatic hydrolysis to yield pyruvate and ammonia.49
Figure 5.

Proposed catalytic mechanism of serine dehydratase. The serine substrate is red, the PLP cofactor is green, and the catalytic lysine is blue. The PLP phosphate group serves as a general base-general acid during the course of the reaction. Product aminoacrylate (frame 6, red) is ultimately hydrolyzed nonenzymatically to form pyruvate and ammonia. Reprinted with permission from: Yamada, T., Komoto, J., Takata, Y., Ogawa, H., Pitot, H. C., & Takusagawa, F. Crystal structure of serine dehydratase from rat liver. Biochemistry 42, 12854–12865 (2003). Copyright 2015 American Chemical Society.
Comparison of the crystal structure of SDH from rat liver with structures of other PLP-dependent enzymes suggests five distinct fold families that utilize the general acid-general base function of the PLP phosphate group.50 In SDH, the N1 atom of PLP is adjacent to a neutral residue and is not protonated, and the phosphate group sits in a neutral pocket largely defined by a tetraglycine loop. Since the phosphate group does not interact with any charged amino acids, it likely exists predominantly as the monoanion, ROPO3H−. All related PLP-dependent enzymes in this group cleave the Ser/Thr Cβ-Oγ bond of serine or threonine. These enzymes belong to the Type II family, which generally catalyze elimination or substitution reactions at the β-carbon. The Type I family is the largest family and contains enzymes that catalyze transaminations, decarboxylations, and β-eliminations. The remaining three families are much smaller and more specific with Type III containing alanine racemase, Type IV containing D-amino acid aminotransferases, and Type V containing glycogen phosphorylase. It is likely that in these families, too, the PLP phosphate group serves as a general base-general acid in catalysis, e.g., as established for glycogen phosphorylase.51
Aspartate transcarbamoylase
The condensation of L-aspartate and carbamoyl phosphate to form N-carbamoyl-L-aspartate and Pi is the first committed step in the biosynthesis of pyrimidine nucleotides in E. coli, and this reaction is catalyzed by the allosteric enzyme aspartate transcarbamoylase (ATCase).52–54 Numerous crystal structures of ATCase have been determined in both T and R states with various ligands bound. One such ligand, N-phosphonacetyl-L-aspartate (PALA), is a bisubstrate analogue in that it mimics structural features of both substrates (Figure 6). Various details of the catalytic mechanism have been studied and reviewed,52–54 and critical structure-mechanism relationships derive from the crystal structure of the ATCase–PALA complex.55 Gouaux and colleagues56 used this crystal structure as a template to model the binding of the tetrahedral intermediate. Intriguingly, this study suggested that ideal, 6-membered ring chair-like geometry would support intramolecular proton abstraction by the departing phosphate group to facilitate collapse of the tetrahedral intermediate (Figure 7).
Figure 6.

Aspartate transcarbamoylase catalyzes the condensation of carbamoyl phosphate with L-aspartate to yield N-carbamoyl-L-aspartate plus inorganic phosphate (Pi). The bi-substrate analogue N-phosphonacetyl-L-aspartate (PALA) mimics structural features of both substrates and is a potent ATCase inhibitor. The binding of PALA reveals key intermolecular and intramolecular interactions that suggest a catalytic function for the phosphate group (see Figure 7).
Figure 7.
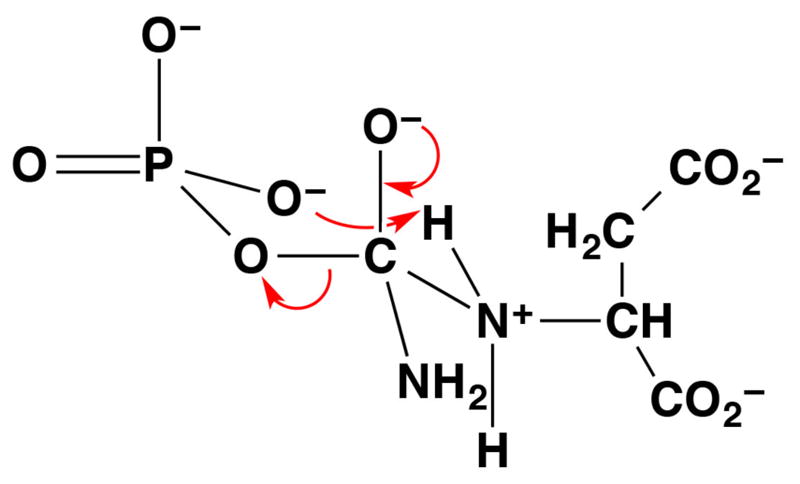
The tetrahedral intermediate in the reaction catalyzed by aspartate transcarbamoylase adopts a 6-membered ring chair-like conformation.56 This conformation enables the phosphate leaving group to serve as a general base, and intermolecular proton transfer facilitates collapse of the tetrahedral intermediate in this example of substrate-assisted catalysis.
Catalysis by phosphate in organic synthesis
The participation of phosphate or phosphoric acid in catalysis is not limited to enzymes, but also includes small molecule systems that function in non-aqueous solvents. Specifically, derivatives of phosphoric acid readily serve as general acid-general base catalysts (i.e., Brønsted acid-Brønsted base catalysts) in organic synthesis. For example, a 1,1′-bi-2-naphthyl (BINOL)-derived phosphoric acid such as that illustrated in Figure 8a can function as a general acid to protonate a reactant, and as a general base to deprotonate a reaction intermediate in a subsequent reaction step.
Figure 8.
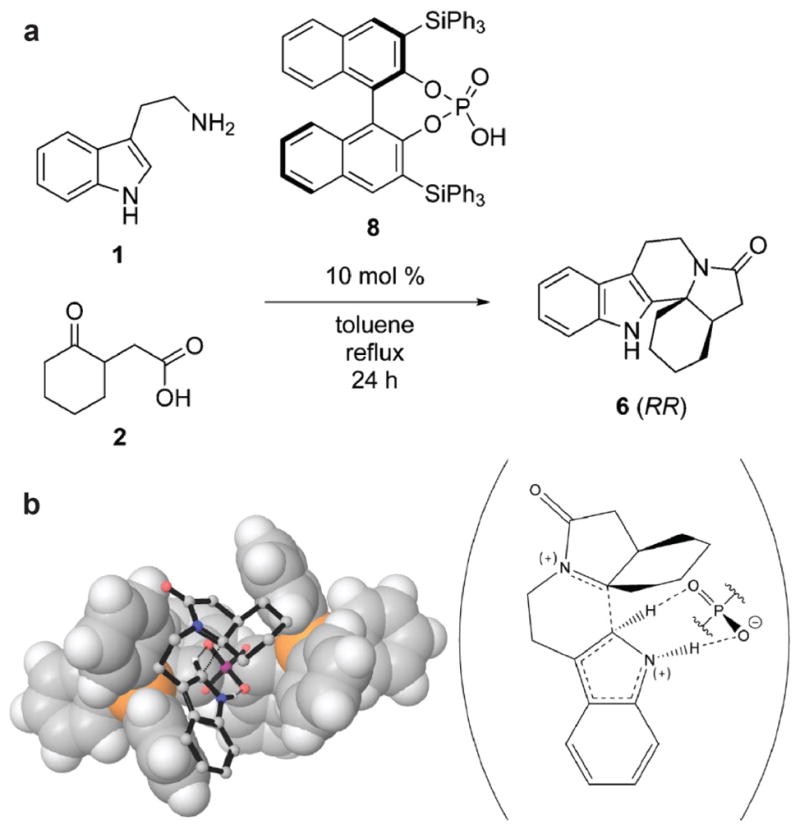
(a) Pictet-Spengler reaction of tryptamine 1 and (2-oxocyclohexyl)acetic acid 2 catalyzed by BINOL derivative 3,3′-bis-(triphenylsilyl)-1,1′-bi-2-naphthol phosphoric acid 8. (b) Calculated transition state structure for the Pictet-Spengler reaction of 1 and 2 (stick figures) catalyzed by 8 (van der Waals surface). The molecular scheme on the right illustrates the proton abstraction by phosphate that quenches the carbocation intermediate. Reprinted with permission from: Overvoorde, L. M., Grayson, M. N., Luo, Y., & Goodman, J. M. (2015) Mechanistic insights into a BINOL-derived phosphoric acid-catalyzed asymmetric Pictet-Spengler reaction. J. Org. Chem. 80, 2634–2640 (2015). Copyright 2015 American Chemical Society.
Consider the Pictet-Spengler reaction (Figure 8a). This is an important reaction used in organic synthesis to generate tetrahydro-β-carboline skeletons found in alkaloid natural products. The mechanism of this reaction requires protonation of an iminium intermediate to facilitate an electrophilic aromatic substitution reaction on the indole ring, and then deprotonation of the tertiary carbocation intermediate to yield the β-carboline product (Figure 8b). Numerous general acids-general bases will catalyze this reaction, such as acetic acid in methylene chloride.57 Interestingly, the Pictet-Spengler reaction is also catalyzed by an enzyme in plant alkaloid biosynthesis. The three-dimensional crystal structure of a “Pictet-Spenglerase”, strictosidine synthase, as well as kinetic isotope effects measured using a specifically deuterated substrate, indicate that E309 serves as a general acid-general base in this reaction (Figure 9).58
Figure 9.

Mechanism of strictosidine synthase, a “Pictet-Spenglerase”, illustrating the general acid-general base function of E309 as determined through kinetic isotope effects using a specifically deuterated substrate. The mechanism of this reaction is identical to that proposed for the general acid-catalyzed reaction in organic synthesis. Reprinted with permission from: Maresh, J. J., Giddings, L.-A., Friedrich, A., Loris, E. A., Panjikar, S., Trout, B. L., Stöckigt, J., Peters, B., & O’Connor, S. E. Strictosidine synthase: mechanism of a Pictet-Spengler catalyzing enzyme. J. Am. Chem. Soc. 130, 710–723 (2008). Copyright 2008 American Chemical Society.
Just as acetic acid or E309 can catalyze the Pictet-Spengler reaction in organic solvent or in an enzyme active site, respectively, so too can phosphoric acid derivatives. For example, BINOL derivatives such as 3,3′-bis-(triphenylsilyl)-1,1′-bi-2-naphthol phosphoric acid catalyze this reaction in organic solvent (Figure 8a).59–61 The final step of this reaction involves deprotonation of a tertiary carbocation (Figure 8b), which is analogous to the final step of a reaction catalyzed by a terpenoid synthase. If a phosphate derivative can direct the final C-H deprotonation in a Pictet-Spengler reaction, then it is reasonable to suggest that the PPi anion could direct the final C-H deprotonation of a terpenoid cyclization cascade, as proposed for pentalenene synthase in Figure 1.
Concluding remarks
Given the precedent for the function of phosphate and its derivatives in general base-general acid catalysis in enzyme and non-enzyme systems, and given the general dearth of amino acid side chains that could serve general base-general acid functions in terpenoid cyclase active sites, it appears more likely than not that the PPi co-product serves this role in terpenoid cyclization cascades.23,25–29 An alternative possibility would be a water molecule trapped in the active site; however, since such a water molecule could prematurely quench carbocation intermediates in catalysis, it would have to be located and restrained so as to be chemically inert. Moreover, often there is insufficient residual volume for water binding once the substrate has bound in a terpenoid cyclase active site: enclosed active site volumes are typically just slightly larger than substrate volumes, thereby ensuring a snug fit between the template and the flexible isoprenoid diphosphate substrate.62,63
Elegant quantum chemical calculations have been used to study the pentalenene synthase mechanism, including the role of PPi as the general base that terminates the cyclization cascade.64 These calculations correctly predict a kinetic isotope effect measured for the partitioning of products pentalenene and Δ6-protoilludene (Figure 10), so a PPi general base can direct the formation of major and minor cyclization products. Notably, these and earlier65 quantum chemical calculations point toward alternative cyclization pathways involving the 7-protoilludyl cation as a critical intermediate (structure C in Figure 10), so the details of the pentalenene synthase mechanism remain a topic of current interest. Net 1,4-, 1,5-, and 1,6-proton transfers may occur in some terpenoid cyclase mechanisms without requiring the participation of PPi or the enzyme,66 so the role of PPi in such systems would be less extensive. It is important to note, too, that intramolecular proton or hydride transfer mechanisms can sometimes be ruled out through experiments demonstrating the direct incorporation of a solvent-derived proton in the cyclization product.67 X-ray crystal structures reveal that proton transfers of this sort could be mediated by the PPi-Mg2+3-complex and Mg2+-bound water molecules.34
Figure 10.

Alternative pentalenene synthase cyclization pathways proceeding through the 7-protoilludyl cation intermediate C predicted by quantum chemical calculations65 and supported by experimentally-measured kinetic isotope effects.64 Reprinted with permission from: Zu, L., Xu, M., Lodewyk, M. W., Cane, D. E., Peters, R. J., & Tantillo, D. J. Effects of isotopically sensitive branching on product distribution for pentalenene synthase: support for a mechanism predicted by quantum chemistry. J. Am. Chem. Soc. 134, 11369–11371 (2012). Copyright 2012 American Chemical Society.
In closing, given the chemical and functional parallels between the phosphate anions of Pi or PPi and the carboxylate anions of aspartate or glutamate side chains, it is somewhat surprising that there are not more examples of general base-general acid catalysis by phosphate-phosphoric acid derivatives in enzyme mechanisms. Such anions, whether generated through the chemistry of catalysis or present simply as buffer components, are not necessarily innocent bystanders in enzyme structure and chemistry. In terpenoid synthase structure and mechanism, the metal-coordinated PPi co-product is the most likely source of Brønsted base-acid functionality in the hydrophobic enzyme active site. Future experimental and computational studies will undoubtedly illuminate further details of this function and its contribution to catalysis.
Acknowledgments
We thank the National Institutes of Health for grant GM56838 in support of this research. D.W.C. thanks the Radcliffe Institute for Advanced Study for the Elizabeth S. and Richard M. Cashin Fellowship.
Footnotes
Dedicated to Professor David E. Cane, with profound gratitude for sharing his wisdom and friendship in more than two decades of collaboration on the structural biology and chemistry of terpenoid biosynthesis.
References
- 1.Cane DE. Isoprenoid biosynthesis. Stereochemistry of the cyclization of allylic pyrophosphates. Acc Chem Res. 1985;18:220–226. [Google Scholar]
- 2.Croteau R, Cane DE. Monoterpene and sesquiterpene cyclases. Methods Enzymol. 1985;110:383–405. [Google Scholar]
- 3.Cane DE. Enzymatic formation of sesquiterpenes. Chem Rev. 1990;90:1089–1103. [Google Scholar]
- 4.Allemann RK. Chemical wizardry? The generation of diversity in terpenoid biosynthesis. Pure Appl Chem. 2008;80:1791–1798. [Google Scholar]
- 5.Austin MB, O’Maille PE, Noel JP. Evolving biosynthetic tangos negotiate mechanistic landscapes. Nat Chem Biol. 2008;4:217–222. doi: 10.1038/nchembio0408-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wendt KU, Schulz GE. Isoprenoid biosynthesis: Manifold chemistry catalyzed by similar enzymes. Structure. 1998;6:127–133. doi: 10.1016/s0969-2126(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 7.Lesburg CA, Caruthers JM, Paschall CM, Christianson DW. Managing and manipulating carbocations in biology: Terpenoid cyclase structure and mechanism. Curr Opin Struct Biol. 1998;8:695–703. doi: 10.1016/s0959-440x(98)80088-2. [DOI] [PubMed] [Google Scholar]
- 8.Tholl D. Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr Opin Plant Biol. 2006;9:297–304. doi: 10.1016/j.pbi.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 10.Christianson DW. Unearthing the roots of the terpenome. Curr Opin Chem Biol. 2008;12:141–150. doi: 10.1016/j.cbpa.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cane DE, Rossi T, Tillman AM, Pachlatko JP. Stereochemical studies of isoprenoid biosynthesis. Biosynthesis of pentalenolactone from [U-13C6]glucose and [6-2H2]glucose. J Am Chem Soc. 1981;103:1838–1843. [Google Scholar]
- 12.Cane DE, Tillman AM. Pentalenene biosynthesis and the enzymatic cyclization of farnesyl pyrophosphate. J Am Chem Soc. 1983;105:122–124. [Google Scholar]
- 13.Cane DE, et al. Biosynthesis of pentalenene and pentalenolactone. J Am Chem Soc. 1990;112:4513–4524. [Google Scholar]
- 14.Koe BK, Sobin BA, Celmer WD. PA 132, a new antibiotic. I. Isolation and chemical properties. Antibiot Annu. 1957:672–675. [PubMed] [Google Scholar]
- 15.Hartmann S, Neeff J, Heer U, Mecke D. Arenaemycin (pentalenolactone): A specific inhibitor of glycolysis. FEBS Lett. 1978;93:339–342. doi: 10.1016/0014-5793(78)81135-1. [DOI] [PubMed] [Google Scholar]
- 16.Mann K, Mecke D. Inhibition of spinach glyceraldehyde-3-phosphate dehydrogenases by pentalenolactone. Nature. 1979;282:535–536. [Google Scholar]
- 17.Cane DE, Abell C, Tillman AM. Pentalenene biosynthesis and the enzymatic cyclization of farnesyl pyrophosphate: Proof that the cyclization is catalyzed by a single enzyme. Bioorg Chem. 1984;12:312–328. [Google Scholar]
- 18.Cane DE, et al. Pentalenene synthase. Purification, molecular cloning, sequencing, and high-level expression in Escherichia coli of a terpenoid cyclase from Streptomyces UC5319. Biochemistry. 1994;33:5846–5857. doi: 10.1021/bi00185a024. [DOI] [PubMed] [Google Scholar]
- 19.Lesburg CA, Zhai G, Cane DE, Christianson DW. Crystal structure of pentalenene synthase: Mechanistic insights on terpenoid cyclization reactions in biology. Science. 1997;277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 20.Seemann M, Zhai G, Umezawa K, Cane D. Pentalenene Synthase. Histidine-309 is not required for catalytic activity. J Am Chem Soc. 1999;121:591–592. [Google Scholar]
- 21.Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 22.Seemann M, et al. Pentalenene synthase. Analysis of active site residues by site-directed mutagenesis. J Am Chem Soc. 2002;124:7681–7689. doi: 10.1021/ja026058q. [DOI] [PubMed] [Google Scholar]
- 23.Caruthers JM, Kang I, Rynkiewicz MJ, Cane DE, Christianson DW. Crystal structure determination of aristolochene synthase from the blue cheese mold, Penicillium roqueforti. J Biol Chem. 2000;275:25533–25539. doi: 10.1074/jbc.M000433200. [DOI] [PubMed] [Google Scholar]
- 24.Felicetti B, Cane DE. Aristolochene synthase: Mechanistic analysis of active site residues by site-directed mutagenesis. J Am Chem Soc. 2004;126:7212–7221. doi: 10.1021/ja0499593. [DOI] [PubMed] [Google Scholar]
- 25.Shishova EY, Di Costanzo L, Cane DE, Christianson DW. X-ray crystal structure of aristolochene synthase from Aspergillus terreus and evolution of templates for the cyclization of farnesyl diphosphate. Biochemistry. 2007;46:1941–1951. doi: 10.1021/bi0622524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller DJ, Allemann RK. Sesquiterpene synthases: passive catalysts or active players? Nat Prod Rep. 2012;29:60–71. doi: 10.1039/c1np00060h. [DOI] [PubMed] [Google Scholar]
- 27.Faraldos JA, Gonzalez V, Allemann RK. The role of aristolochene synthase in diphosphate activation. Chem Commun. 2012;48:3230–3232. doi: 10.1039/c2cc17588f. [DOI] [PubMed] [Google Scholar]
- 28.Köksal M, Zimmer I, Schnitzler JP, Christianson DW. Structure of isoprene synthase illuminates the chemical mechanism of teragram atmospheric carbon emission. J Mol Biol. 2010;402:363–373. doi: 10.1016/j.jmb.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faraldos JA, et al. Probing the mechanism of 1,4-conjugate elimination reactions catalyzed by terpene synthases. J Am Chem Soc. 2012;134:20844–20848. doi: 10.1021/ja311022s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aaron JA, Christianson DW. Trinuclear metal clusters in catalysis by terpenoid synthases. Pure Appl Chem. 2010;82:1585–1597. doi: 10.1351/PAC-CON-09-09-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rynkiewicz MJ, Cane DE, Christianson DW. Structure of trichodiene synthase from Fusarium sporotrichiodes provides mechanistic inferences on the terpene cyclization cascade. Proc Natl Acad Sci USA. 2001;98:13543–13548. doi: 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hosfield DJ, et al. Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J Biol Chem. 2004;279:8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 33.Tarshis LC, Yan M, Poulter CD, Sacchettini JC. Crystal structure of recombinant farnesyl diphosphate synthase at 2.6-Å resolution. Biochemistry. 1994;33:10871–10877. doi: 10.1021/bi00202a004. [DOI] [PubMed] [Google Scholar]
- 34.Chen M, et al. Mechanistic insights from the binding of substrate and carbocation intermediate analogues to aristolochene synthase. Biochemistry. 2013;52:5441–5453. doi: 10.1021/bi400691v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tymoczko JL, Berg JM, Stryer L. Biochemistry: A Short Course. 2. New York: W. H. Freeman and Company; 2013. p. A3. [Google Scholar]
- 36.Phillips RS, Sundararaju B, Koushik SV. The catalytic mechanism of kynureninase from Pseudomonas fluorescens: Evidence for transient quinonoid and ketimine intermediates from rapid-scanning stopped-flow spectrophotometry. Biochemistry. 1998;37:8783–8789. doi: 10.1021/bi980066v. [DOI] [PubMed] [Google Scholar]
- 37.Phillips RS, Dua R. Stereochemistry and mechanism of aldol reactions catalyzed by kynureninase. J Am Chem Soc. 1991;113:7385–7388. [Google Scholar]
- 38.Koushik SV, Moore JA, III, Sundararaju B, Phillips RS. The catalytic mechanism of kynureninase from Pseudomonas fluorescens: insights from the effects of pH and isotopic substitution on steady-state and pre-steady-state kinetics. Biochemistry. 1998;37:1376–1382. doi: 10.1021/bi971130w. [DOI] [PubMed] [Google Scholar]
- 39.Gawandi VB, Liskey D, Lima S, Phillips RS. Reaction of Pseudomonas fluorescens kynureninase with β-benzoyl-L-alanine: Detection of a new reaction intermediate and a change in rate-determining step. Biochemistry. 2004;43:3230–3237. doi: 10.1021/bi036043k. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S, Gawandi VB, Capito N, Phillips RS. Substituent effects on the reaction of β-benzoylalanines with Pseudomonas fluorescens kynureninase. Biochemistry. 2010;49:7913–7919. doi: 10.1021/bi100955b. [DOI] [PubMed] [Google Scholar]
- 41.Lima S, Kumar S, Gawanadi B, Momany C, Phillips RS. Crystal structure of the Homo sapiens kynurenine-3-hydroxyhippuric acid inhibitor complex: Insights into the molecular basis of kynurenine substrate specificity. J Med Chem. 2009;52:389–396. doi: 10.1021/jm8010806. [DOI] [PubMed] [Google Scholar]
- 42.Lima S, Khristoforov R, Momany C, Phillips RS. Crystal structure of Homo sapiens kynureninase. Biochemistry. 2007;46:2735–2744. doi: 10.1021/bi0616697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Momany C, Levdikov V, Blagova L, Lima S, Phillips RS. Three-dimensional structure of kynureninase from Pseudomonas fluorescens. Biochemistry. 2004;43:1193–1203. doi: 10.1021/bi035744e. [DOI] [PubMed] [Google Scholar]
- 44.Phillips RS, Scott I, Paulose R, Patel A, Barron TC. The phosphate of pyridoxal-5′-phosphate is an acid/base catalyst in the mechanism of Pseudomonas fluorescens kynureninase. FEBS J. 2014;281:1100–1109. doi: 10.1111/febs.12671. [DOI] [PubMed] [Google Scholar]
- 45.Phillips RS. Structure and mechanism of kynureninase. Arch Biochem & Biophys. 2014;544:69–74. doi: 10.1016/j.abb.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 46.Phillips RS. Chemistry and diversity of pyridoxal-5′-phosphate dependent enzymes. Biochim Biophys Acta – Prot Proteomics. 2015;1854:1167–1174. doi: 10.1016/j.bbapap.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 47.Flint DH, Allen RM. Iron-sulfur proteins with nonredox functions. Chem Rev. 1996;96:2315–2334. doi: 10.1021/cr950041r. [DOI] [PubMed] [Google Scholar]
- 48.Hwang BY, Cho BK, Yun K, Koteshwar K, Kim BG. Revisit of aminotransferases in the genomic era and its application to biocatalysis. J Mol Catal B: Enzymatic. 2005;37:47–55. [Google Scholar]
- 49.Urusova DV, et al. Crystal Structure of D-serine dehydratase from Escherichia coli. Biochim Biophys Acta – Prot Proteomics. 2012;1824:422–432. doi: 10.1016/j.bbapap.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamada T, et al. Crystal structure of serine dehydratase from rat liver. Biochemistry. 2003;42:12854–12865. doi: 10.1021/bi035324p. [DOI] [PubMed] [Google Scholar]
- 51.Johnson LN, Hu SH, Barford D. Catalytic mechanism of glycogen phosphorylase. Faraday Discuss. 1992;93:131–142. doi: 10.1039/fd9929300131. [DOI] [PubMed] [Google Scholar]
- 52.Kantrowitz ER. Allostery and cooperativity in Escherichia coli aspartate transcarbamoylase. Arch Biochem Biophys. 2012;519:81–90. doi: 10.1016/j.abb.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lipscomb WN, Kantrowitz ER. Structure and mechanisms of Escherichia coli aspartate transcarbamoylase. Acc Chem Res. 2012;45:444–453. doi: 10.1021/ar200166p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lipscomb WN. Aspartate transcarbamylase from Escherichia coli: Activity and regulation. Adv Enzymol Relat Areas Mol Biol. 1994;68:67–151. doi: 10.1002/9780470123140.ch3. [DOI] [PubMed] [Google Scholar]
- 55.Krause KL, Volz KW, Lipscomb WN. 2.5 Å structure of aspartate carbamoyltransferase complexed with the bisubstrate analog N-(phosphonacetyl)-L-aspartate. J Mol Biol. 1987;193:527–553. doi: 10.1016/0022-2836(87)90265-8. [DOI] [PubMed] [Google Scholar]
- 56.Gouaux JE, Krause KL, Lipscomb WN. The catalytic mechanism of Escherichia coli aspartate carbamoyltransferase: A molecular modelling study. Biochem Biophys Res Commun. 1987;142:893–897. doi: 10.1016/0006-291x(87)91497-5. [DOI] [PubMed] [Google Scholar]
- 57.Zhou H, Liao X, Cook JM. Regiospecific, enantiospecific total synthesis of the 12-alkoxy-substituted indole alkaloids, (+)-12-methoxy-Na-methylvellosimine, (+)-12-methoxyaffinisine, and (−)-fuchsiaefoline. Org Lett. 2004;6:249–252. doi: 10.1021/ol0362212. [DOI] [PubMed] [Google Scholar]
- 58.Maresh JJ, et al. Strictosidine synthase: mechanism of a Pictet-Spengler catalyzing enzyme. J Am Chem Soc. 2008;130:710–723. doi: 10.1021/ja077190z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seayad J, Seayad AM, List B. Catalytic asymmetric Pictet-Spengler reaction. J Am Chem Soc. 2006;128:1086–1087. doi: 10.1021/ja057444l. [DOI] [PubMed] [Google Scholar]
- 60.Holloway CA, Muratore ME, Storer RI, Dixon DJ. Direct enantioselective Brønsted acid catalyzed N-acyliminium cyclization cascades of tryptamines and ketoacids. Org Lett. 2010;12:4720–4723. doi: 10.1021/ol101651t. [DOI] [PubMed] [Google Scholar]
- 61.Overvoorde LM, Grayson MN, Luo Y, Goodman JM. Mechanistic insights into a BINOL-derived phosphoric acid-catalyzed asymmetric Pictet-Spengler reaction. J Org Chem. 2015;80:2634–2640. doi: 10.1021/jo5028134. [DOI] [PubMed] [Google Scholar]
- 62.Rynkiewicz MJ, Cane DE, Christianson DW. X-ray crystal structures of D100E trichodiene synthase and its pyrophosphate complex reveal the basis for terpene product diversity. Biochemistry. 2002;41:1732–1741. doi: 10.1021/bi011960g. [DOI] [PubMed] [Google Scholar]
- 63.Köksal M, Jin Y, Coates RM, Croteau R, Christianson DW. Taxadiene synthase structure and evolution of modular architecture in terpene biosynthesis. Nature. 2011;469:116–120. doi: 10.1038/nature09628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zu L, et al. Effect of isotopically sensitive branching on product distribution for pentalenene synthase: support for a mechanism predicted by quantum chemistry. J Am Chem Soc. 2012;134:11369–11371. doi: 10.1021/ja3043245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gutta P, Tantillo DJ. Theoretical studies on farnesyl cation cyclization: pathways to pentalenene. J Am Chem Soc. 2006;128:6172–6179. doi: 10.1021/ja058031n. [DOI] [PubMed] [Google Scholar]
- 66.Hong YJ, Tantillo DJ. Feasibility of intramolecular proton transfers in terpene biosynthesis – guiding principles. J Am Chem Soc. 2015;137:4134–4140. doi: 10.1021/ja512685x. [DOI] [PubMed] [Google Scholar]
- 67.Miller DJ, et al. Stereochemistry of eudesmane cation formation during catalysis by aristolochene synthase from Penicillium roqueforti. Org Biomol Chem. 2008;6:2346–2354. doi: 10.1039/b804198a. [DOI] [PubMed] [Google Scholar]