

Abstract
Protein tyrosine phosphatase 1B (PTP1B) has anti-inflammatory potential but PTP1B responses are desensitized in the lung by prolonged cigarette smoke exposure. Here we investigate whether PTP1B expression impacts lung disease severity during respiratory syncytial viral (RSV) exacerbations of chronic obstructive pulmonary disease (COPD). Ptp1b-/- mice infected with RSV exhibit exaggerated immune cell infiltration, damaged epithelial cell barriers, cytokine production and increased apoptosis. Elevated expression of S100A9, a damage-associated molecular pattern molecule, was observed in the lungs of Ptp1b-/- mice during RSV infection. Utilizing a neutralizing anti-S100A9 IgG antibody, it was determined that extracellular S100A9 signaling significantly impacts lung damage during RSV infection. Pre-exposure to cigarette smoke desensitized PTP1B activity, which coincided with enhanced S100A9 secretion and inflammation in wild type animals during RSV infection. S100A9 levels in human bronchoalveolar lavage fluid had an inverse relationship with lung function in healthy subjects, smokers and COPD subjects. Fully differentiated human bronchial epithelial cells isolated from COPD donors cultured at the air liquid interface secreted more S100A9 than cells from healthy donors or smokers following RSV infection. Together, these findings show that reduced PTP1B responses contribute to disease symptoms in part by enhancing S100A9 expression during viral-associated COPD exacerbations.
Keywords: respiratory syncytial virus, apoptosis, damage-associated molecular patterns, phosphatase, chronic obstructive pulmonary disease
Introduction
Protein tyrosine phosphatase 1B (PTP1B) is primarily known to negatively regulate insulin receptor signaling, with Ptp1b-/- mice displaying hypersensitivity to insulin1. PTP1B regulates several other receptors, such as the Ephrin receptor, toll-like receptor (TLR) signaling and leptin receptor signaling2. In addition to these receptors, PTP1B has multiple other substrates, including c-Src, protein phosphatase 2A (PP2A) and Bcr-Abl2. Though PTP1B is the most studied tyrosine phosphatase, the role of PTP1B in the lung is not well defined, especially in infection models. PTP1B expression can influence cell recruitment and expansion, with Ptp1b-/- mice having increased B cell numbers in the bone marrow and lymph nodes3, and increased leukocytes in the lungs in an animal asthma model4. Also, the loss of Ptp1b in mice compromises macrophage viability5. Our group has determined that enhancing PTP1B expression in cigarette smoke conditions can resolve inflammation and protease production in the lungs6. Since PTP1B regulates immune cell responses and is altered during lung diseases, investigating the impact of PTP1B deficiency during viral infection is of great importance.
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death in the US7 and the majority of COPD subjects experience at least one exacerbation per year that results in a substantial number of hospital admissions8. Despite improving detection methods utilized to identify pulmonary pathogens in hospitalized patients, the majority of infections are not detectable in adults with community-acquired pneumonia and viruses are detected more frequently than bacteria9. Thus, viral infections are considered a major driving factor of COPD exacerbations and thus contribute disease morbidity and mortality. Rhinovirus, influenza and respiratory syncytial virus (RSV) are frequently detected in the respiratory tract of COPD patients during an exacerbation10. Despite the high frequency of RSV detected during a COPD exacerbation11, few studies have examined the pathogenicity of RSV in COPD animal models12, 13. Patients infected with RSV, usually infants, the elderly and immunocompromised patients but also healthy adults14, develop mild to severe symptoms, including fever, mucus production and wheezing. RSV infection triggers several pathogen-associated molecular pattern (PAMP) receptors that are regulated by PTP1B15, 16. However the ability of RSV to elicit a damage-associated molecular pattern (DAMP) response and the potential regulation of DAMPs by PTP1B are not known. DAMPs are produced from infected, damaged or dead cells and induce a potent inflammatory response17 that could enhance the severity of a COPD exacerbation.
In this study, we determined that a DAMP protein, S100A9, is negatively regulated by PTP1B and loss of PTP1B expression enhances S100A9 expression and worsens lung injury during RSV infection. S100A9 protein induced potent inflammatory responses and enhances lung cell death during RSV infection. Bronchoalveolar lavage fluid (BALF) from healthy human subjects, smokers and COPD patients showed an inverse related relation of S100A9 levels with lung function. Utilizing an in vitro model of viral exacerbations in primary airway epithelial cells from patients, we have demonstrated that cells from COPD patients have reduced PTP1B activity, which coincided with heightened S100A9 secretion. The activation of DAMP responses contributes to viral clearance18. However unregulated and sustained DAMP signaling could play a part in lung disease exacerbations that enhance the loss of lung function. Therefore maintaining an effective lung PTP1B response aids in minimizing lung damage induced by S100A9 expression.
Results
Cigarette smoke exposure desensitizes lung PTP1B responses and deficiency of PTP1B expression enhances susceptibility to cigarette smoke in mice
We have previously observed that PTP1B counters lung inflammation6 and reduced PTP1B lung activity is observed during RSV infection12. To investigate the effect of acute and chronic cigarette smoke exposure on PTP1B activity, FVB/NJ mice were exposed to daily cigarette smoke exposure for 2 weeks (acute) or 6 months (chronic). Acute smoke exposure resulted in a robust PTP1B response in the lungs, which was desensitized following chronic smoke exposure (Figure 1A). Further studies were performed to determine how the loss of PTP1B impacts on the lungs during smoke exposure. Ptp1b-/- mice and their wild type littermates were exposed to 4 months of smoke and immune cell infiltration and lung remodeling were examined. Loss of Ptp1b expression in mice increased BALF immune cell infiltration (Figure 1B) and lung remodeling, as determined by mean linear intercept (MLI) analysis (Figure 1C). These results establish that the loss of PTP1B enhanced the susceptibility to smoke-induced lung damage. Thus, the desensitizing of PTP1B activity by cigarette smoke exposure could be a major contributory factor to disease progression.
Figure 1.
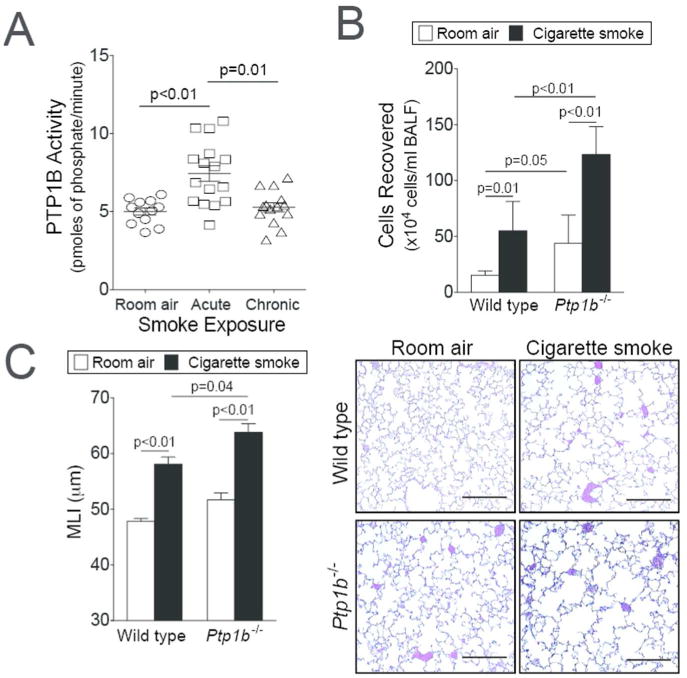
Loss of PTP1B expression enhances lung remodeling. (A) Enhanced PTP1B activity is observed in the lungs of FVB/NJ mice exposed to 2 weeks (acute) of cigarette smoke exposure but not following 6 months (chronic) of exposure. (B) Ptp1b-/- mice and their FVB/NJ wild type littermates were exposed to four months of cigarette smoke or room air. Bronchoalveolar lavage fluid immune cellularity was enhanced by smoke exposure and was further enhanced by loss of Ptp1b expression. (C) Mean linear intercepts (MLI) were measured in the lungs of the mice to assess lung remodeling and comparative histology images of the four mouse groups are presented here (scale bar=40 μm). Slides were randomized, read blindly and scored for MLI. Graphs are represented as mean ± S.E.M, where n= at least 10 animals per group for each time point. p values are shown comparing both groups connected by a line.
Loss of Ptp1b expression results in increased lung damage during RSV infection in mice
Ptp1b-/- mice and their wild type littermates were infected with RSV. For the first 3 days post infection (dpi), Ptp1b-/- and wild type mice had comparable immune cell infiltration. As the infection progressed Ptp1b-/- mice had elevated macrophage, neutrophil, lymphocyte and eosinophil infiltration into their lungs and airways compared to wild type animals (Figure 2A). RSV infected mice exhibited perivascular lymphocytic inflammation, increased inflammatory cell infiltration around the bronchial airways and within the alveolar spaces, which was significantly elevated in Ptp1b-/- mice 7 dpi, compared to wild type mice (Figure 2B). RSV readily infected the airway epithelium in large and small airways, determined by the detection of RSV antigen in both mouse groups (Figure 2C). Ptp1b-/- and wild type mice had comparable RSV titer and weight changes during infection (Figure 2D). Therefore, loss of Ptp1b expression during RSV infection leads to exaggerated inflammation, without impacting viral infectivity.
Figure 2.
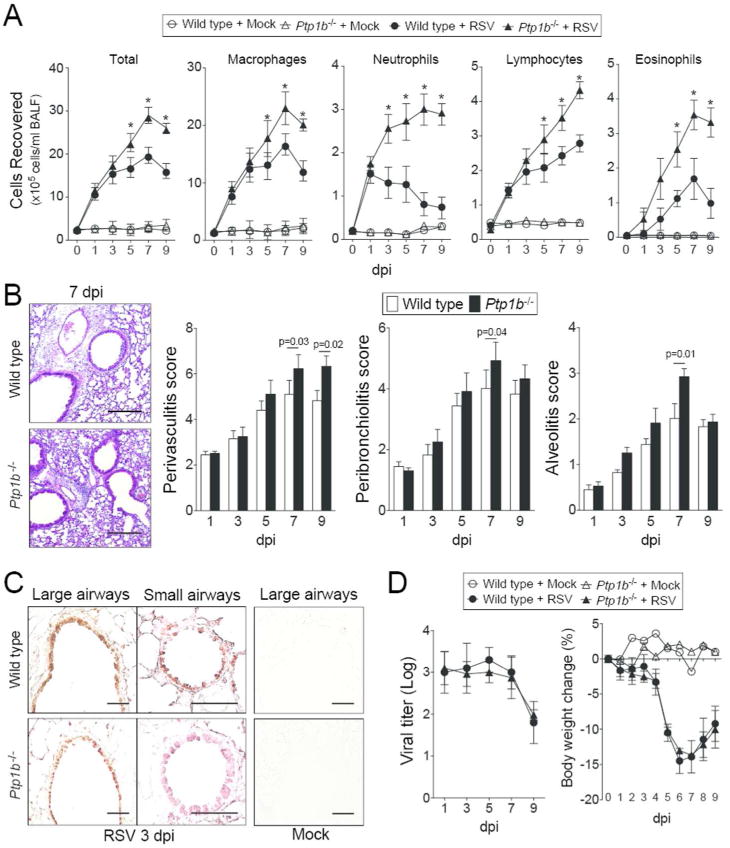
Ptp1b deficient mice display enhanced lung immune cell infiltration during RSV infection. (A) Ptp1b-/- mice (triangle) and their FVB/NJ wild-type littermates (circle) were infected with 1×106 pfu of RSV (closed) or mock control (open) and animals were euthanized on days 0, 1, 3, 5, 7 and 9-dpi. BALF immune cellularity was determined for total immune cell number, macrophages, neutrophils, lymphocytes and eosinophils. *Represents a p-value < 0.05 comparing Ptp1b-/- mice to wild-type mice 9 days post RSV challenge. (B) Comparative histology images of infected animals from each background 7 dpi are presented here (scale bar=50 μm). Histopathology (Peribronchiolitis, perivasculitis and alveolitis scoring) was recorded in mice for each group. Slides were randomized, read blindly and scored for each parameter. p values shown, comparing both treatments connected by a line. (C) Immunohistochemistry was performed on lung tissue from both mouse groups 3 dpi with an antibody that recognizes RSV antigen (brown). Mock treated animals demonstrate negative staining for RSV antigen. (D) RSV infectivity and animal body weight was comparable in wild type and Ptp1b-/- mice. Each graph is represented as mean ± S.E.M. where each measurement was performed on 10 animals/group, with 6 replicates/animal.
Simultaneously, Ptp1b-/- mice had significantly higher protein concentration (Figure 3A) in the BALF compared to infected wild type mice, suggesting potentially increased damage and disruption of the epithelial cell barrier. Enhanced apoptosis and caspase-3 cleavage were observed in the Ptp1b-/- mice 5 dpi (Figure 3A-B). Equally, PTP1B expression negatively regulated the expression of RANTES, PDGF-bb, G-CSF, CXCL1, CXCL9, CXCL10, MCP-1, Eotaxin, IL-4, IL-5 and IL-13, observed 5 dpi (Figure 3C). These inflammation changes may have been influenced by enhanced expression of MyD88 and TRIF and the phosphorylation of IRF3, IRF7 and TBK1 and activation of NFκB during infection, in Ptp1b deficiency compared to wild type mice (Supplemental Figure S1). Therefore PTP1B regulates cytokine signaling associated with lung damage.
Figure 3.
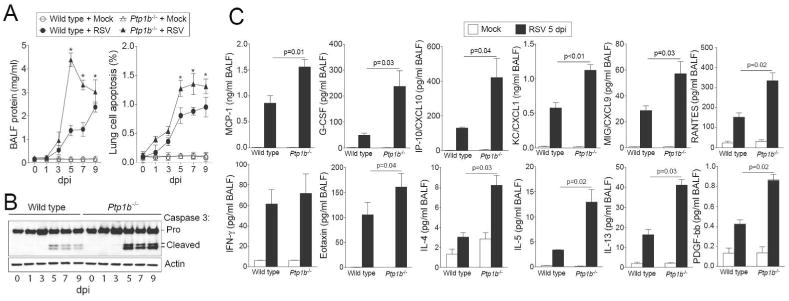
Ptp1b deficient mice display enhanced lung damage and cytokines during RSV infection. (A) Ptp1b-/- mice (triangle) and their FVB/NJ wild-type littermates (circle) were infected with 1×106 pfu of RSV (closed) or mock control (open) and animals were euthanized on days 0, 1, 3, 5, 7 and 9-dpi. Ptp1b-/- mice have enhanced protein content in BALF compared to wild-type mice. Ptp1b-/- mice display greater lung apoptosis. * p < 0.05 compared to wild-type mice on each corresponding time point. (B) Loss of PTP1B expression enhances caspase-3 cleavage and activation as determined by caspase-3 immunoblots conducted on lung tissue lysates. (C) BALF levels of MCP-1, G-CSF, CXCL10, CXCL1, CXCL9, PDGF-bb, RANTES, IFN-γ, Eotaxin, IL-4, IL-5 and IL-13 were determined in both mouse genotypes following 5 dpi. Graphs are represented as mean ± S.E.M, where n= at least 10 per group for each time point. p values shown, comparing both treatments connected by a line.
PTP1B suppresses S100A9 expression during RSV infection
Quantitative PCR (qPCR) arrays were performed to investigate the key Ptp1b-regulated genes during RSV infection that contribute to enhanced apoptosis. Over 350 genes associated with cell death were screened and 24 of these genes were regulated by Ptp1b expression (Figure 4A-B). One of these 24 genes, S100A9, was further investigated as S100A9 regulates inflammation and disease progression during an animal influenza infection model17 and is also regulated by TRIF17 and can activate TLR419. PTP1B can directly impact these signaling processes15, 16. In humans, S100A9 levels are elevated in the serum of COPD patients during an exacerbation20. We detected enhanced S100A9 levels in the BALF (Figure 4C) and increased S100A9 in lung tissue (Figure 4D) during RSV infection in mice. Lung tissue was examined 5 dpi as animals had significantly heightened inflammation (Figure 3C), apoptosis (Figure 3B) and maximum viral load (Figure 2D) in their lungs on this day. The induction of S100A9 during RSV infection was further confirmed in human primary small airway epithelial (SAE) cells and mouse bone marrow derived macrophages (BMDM) (Figure 4E). Airway epithelial cells were utilized, as this was the primary region of the lungs infected by RSV in the mouse model (Figure 2C). Interestingly, stimulation with S100A9 protein induces enhanced gene expression of PTP1B-negatively regulated cytokines in SAE cells (Figure 4F). Indeed, silencing of PTP1B expression (see silencing siRNA efficiency in Supplemental Figure S2A) enhances the expression of MCP-1, PDGF-bb, CXCL10, CXCL1 and CXCL9 following S100A9 stimulation (Figure 4F). Therefore S100A9 signaling could influence lung inflammation during RSV infection.
Figure 4.
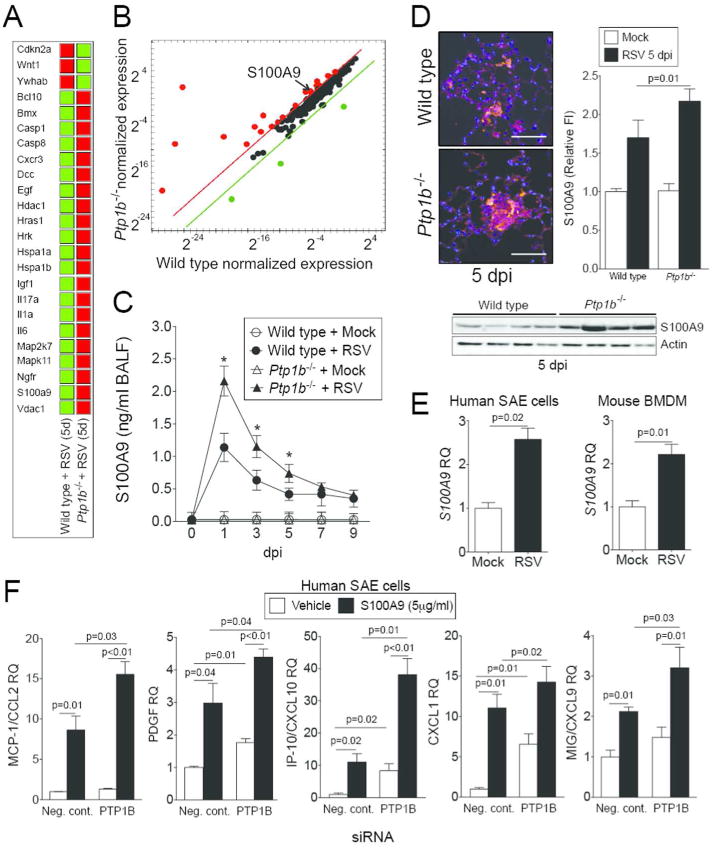
PTP1B hinders S100A9 expression during RSV infection. Gene expression profiling of lung tissue from wild type and Ptp1b-deficient mice was performed 5 dpi. qPCR arrays were performed using the BioRad PrimePCR Apoptosis and Survival panel to examine over 350 genes associated with cell survival and apoptosis. (A) Data were analyzed with the BioRad PrimePCR analysis software. Color code in the clustergram shows standardized gene expression responses from the microarray probes of samples from RSV infected wild type and Ptp1b-/- mice. Red indicates higher expression and green lower expression. (B) Expression values were log2 transformed and normalized to compare wild type and Ptp1b-/- gene expression. Threshold lines demonstrate significantly greater differences in expression between both groups. S100A9, a gene negatively regulated by PTP1B, was examined in the (C) BALF and (D) lung tissue of wild type and Ptp1b-/- mice. (D) Tissue was stained for RSV antigen (red) and fluorescent intensity quantified. S100A9 immunoblots were also conducted on lung tissue. (E) Gene expression of S100A9 is inducible in human SAE cells and murine BMDM following RSV infection. (F) Stimulus of S100A9 protein induces release of MCP-1, PDGF, CXCL10, CXCL1 and CXCL9 from SAE human cells transfected with negative/scrambled (Scr.) or PTP1B siRNA. Graphs are represented as mean ± S.E.M, where n=10 per group. qPCR is represented as relative quantification (RQ). (C) * p < 0.05 compared to wild-type mice on each corresponding time point. (E-F) p values shown, comparing both treatments connected by a line.
Extracellular S100A9 regulates inflammation and apoptosis during RSV infection
Since extracellular S100A9 induces similar inflammatory responses compared to PTP1B deficiency in SAE cells, we hypothesized that extracellular S100A9 significantly contributes to the lung inflammation observed in Ptp1b-/- mice. To investigate this, we used an anti-S100A9 blocking antibody, which neutralizes extracellular S100A9 signaling. Two hours prior to RSV infection animals were intraperitoneal (IP) administered either anti-S100A9 IgG or isotype control IgG, as depicted in Figure 5A. Ptp1b-/- and wild type mice that received anti-S100A9 IgG had little to no weight changes during infection unlike animals that received control IgG (Figure 5B). Blocking extracellular S100A9 signaling also subdued immune cell infiltration into the lungs (Figure 5C) and perivascular lymphocytic inflammation, inflammatory cell infiltration around the bronchial airways and within the alveolar spaces of both animal backgrounds, 5 dpi (Figure 5D).
Figure 5.
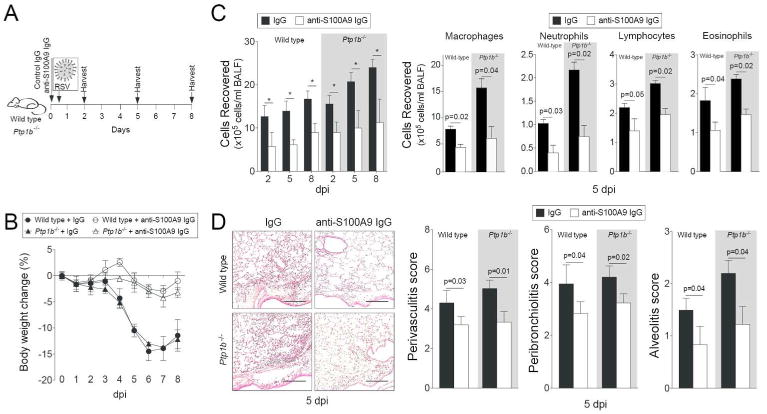
Infiltration of immune cells following RSV-infection is diminished upon neutralization of extracellular S100A9 signaling. (A) Wild type and Ptp1b-/- mice were IP injected with isotype control IgG or anti-S100A9 IgG prior to intranasal infection with 1×106 pfu of RSV and animals were euthanized 2, 5 and 8-days post infections. (B) Animal body weight and (C) BALF cellularity (total BAL cells, macrophages, neutrophils, lymphocytes and eosinophils) were determined in each group (white background = wild type and grey area = Ptp1b-/- mice). (D) Comparative histology images of infected animals 5 dpi are presented here (scale bar=50 μm). Histopathology (Peribronchiolitis, perivasculitis and alveolitis scoring) was recorded in mice for each group. Slides were randomized, read blindly and scored for each parameter. Graphs are represented as mean ± S.E.M., where each measurement was performed 3 times on 10 animals/group. * p < 0.05 or p values shown, comparing both treatments connected by a line.
Subduing S100A9 signaling also diminished protein concentration in the BALF compared to control IgG treated animals (Figure 6A) and reduced RSV infection-induced apoptosis (Figure 6B). Diminished cleavage of caspase-3 was observed in animals treated with the S100A9 neutralizing antibody compared to IgG treated animals (Supplemental Figure S3). Blocking extracellular S100A9 signaling had little to no effect on RSV infectivity (Figure 6C) but significantly decreased MCP-1, Eotaxin, IL-4, IL-5 and IL-13 release in both wild type and Ptp1b-/- animals that received anti-S100A9 IgG (Figure 6D). These results demonstrated that PTP1B-regulated S100A9 contributes to the severity of lung inflammation and apoptosis during RSV infection.
Figure 6.
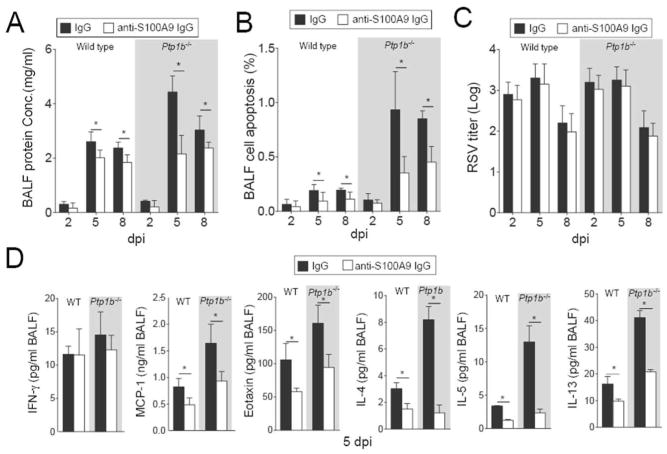
Extracellular S100A9 signaling neutralization reduces lung damage and cytokine production during RSV infection. Wild type and Ptp1b-/- mice were IP injected with isotype control IgG or anti-S100A9 IgG prior to intranasal infected with 1×106 pfu of RSV and animals were euthanized 2, 5 and 8-days post infections. (A) BALF protein content, (B) BALF cells undergoing apoptosis and (C) lung viral titer were determined in each group (white background = wild type and grey area = Ptp1b-/- mice). (D) MCP-1/CCL2, IFN-γ, Eotaxin, IL-4, IL-5 and IL-13 levels were determined in BALF by luminex assays, 5 dpi. Graphs are represented as mean ± S.E.M., where each measurement was performed on 10 animals/group, with 6 replicates/animal. * p < 0.05, comparing both treatments connected by a line.
Pre-exposure to cigarette smoke enhances S100A9 expression during an RSV infection
Here we examined whether chronic smoke exposure prior to RSV infection would impact on S100A9-associated inflammation and apoptosis in the lungs. Animals were exposed to either room air or cigarette smoke for 4 hours daily for 4 months prior to RSV infection as depicted in Figure 7A. Elevated viral titers were observed in smoke exposed animals from both wild type and Ptp1b-/- mice 9 dpi, compared to room air treated mice (Figure 7B). Exposure to 4 months of smoke prior to RSV infection desensitized PTP1B activity further in wild type mice, at 9 dpi (Figure 7C). Unlike animals exposed to room air, BALF S100A9 levels were similarly increased in smoke-exposed wild type and Ptp1b-/- animals during RSV infection (Figure 7D). Likewise, immune cell infiltration and protein concentration in the BALF were comparably increased in wild type and Ptp1b-/- mice during RSV infection following smoke exposure (Figure 7E-F). In contrast, enhanced apoptosis was still observed in Ptp1b-/- animals compared to their wild type littermates independent of their smoke exposure status (Figure 7G). Cigarette smoke and PTP1B both regulate other apoptosis-associated genes, in addition to S100A9. Thus, other factors outside of extracellular S100A9 may play a role in enhancing the apoptosis in Ptp1b-/- mice following smoke and RSV infection. Interestingly, MCP-1 secretion was also enhanced in smoke exposed wild type mice to levels observed in Ptp1b-/- mice (Figure 7H). Therefore cigarette smoke preconditions the lung for heightened S100A9 expression and aggravate viral exacerbations.
Figure 7.
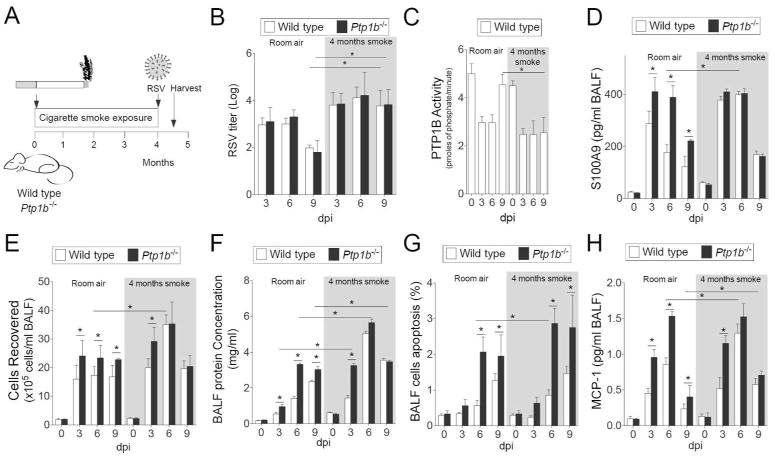
Cigarette smoke exposure enhances RSV infection induced lung damage. (A) Wild type and Ptp1b-/- mice were exposed to cigarette smoke for 4 months prior to intranasal infection with 1×106 pfu of RSV and animals were euthanized 0, 3, 6 and 9-days post infection. (B) Lung RSV titer, (C) lung PTP1B activity, (D) BALF levels of S100A9, (E) BALF cellularity, (F) BALF protein content, (G) BALF cells undergoing apoptosis and (H) BALF MCP-1 levels were determined (white background = room air and grey area = smoke exposed for 4 months). Graphs are represented as mean ± S.E.M., where each measurement was performed on 10 animals/group, with 6 replicates/animal. * p < 0.05, comparing both treatments connected by a line.
Human BALF S100A9 levels have an inverse relationship with lung function
S100A9 was measured in lung BALF from age-matched healthy control subjects, smokers, and subjects with COPD (Figure 8A). There is a significantly higher concentration of S100A9 in the BALF of COPD patients compared to healthy patients (Figure 8A). Importantly, S100A9 BALF levels have an inverse relationship with pulmonary function, determined by FEV1 (percent predicted) (Figure 8B). To determine whether cells from COPD lungs have an exaggerated S100A9 response during RSV infection, human bronchial epithelial cells isolated from healthy, smoking and COPD donors were fully differentiated and cultured at the air liquid interface (ALI) and were infected with RSV (Figure 9A). Cells from COPD subjects secreted more S100A9 (Figure 9B) and MCP-1 (Figure 9C) onto the apical surface of the cells (air exposed) than cells from healthy or smoking donors. A significant reduction in PTP1B activity was observed in the cells from COPD patients compared to cells from healthy and smoker donors (Figure 9D). Cells from COPD donors had a higher viral titer than cells from healthy smokers and non-smokers (Figure 9E). To determine whether cells from COPD patients were more sensitive to S100A9 stimuli, S100A9 protein was added to the apical surface of cells at the ALI (Figure 9F). The cells from COPD subjects had significant increases in MCP-1 and G-CSF secretion compared to the cells from the other subject cohorts (Figure 9F). Elevated levels of lactate dehydrogenase (LDH) were observed on the apical surface of COPD cells compared to healthy cells following S100A9 protein stimulation (Figure 9F). Therefore enhanced extracellular S100A9 in the lungs of COPD patients could exaggerate inflammatory mediators, which contribute to the lung damage observed during RSV infection.
Figure 8.
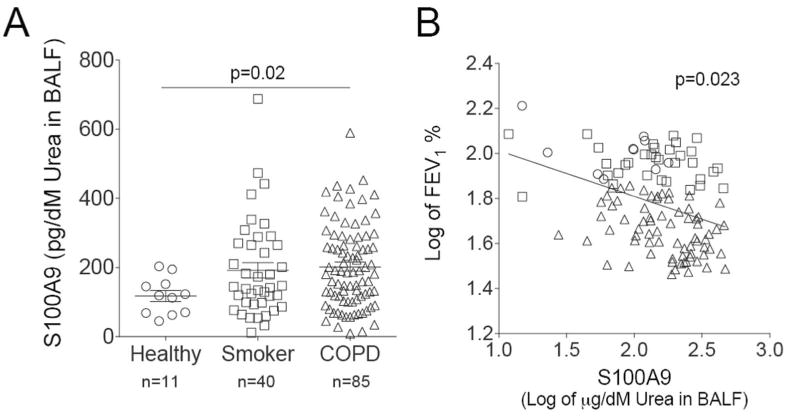
Extracellular S100A9 is increased in human COPD BALF samples. (A) S100A9 was measured in lung BALF from age-matched healthy control subjects (n=11), smokers (n=40), and subjects with COPD (n=85). BALF was standardized to urea levels. (B) BALF S100A9 levels had an inversely relationship with the FEV1% predicted ratio. Data is represented as log of mean ± S.E.M., where each measurement was performed 3 times. p values shown, comparing both treatments connected by a line.
Figure 9.
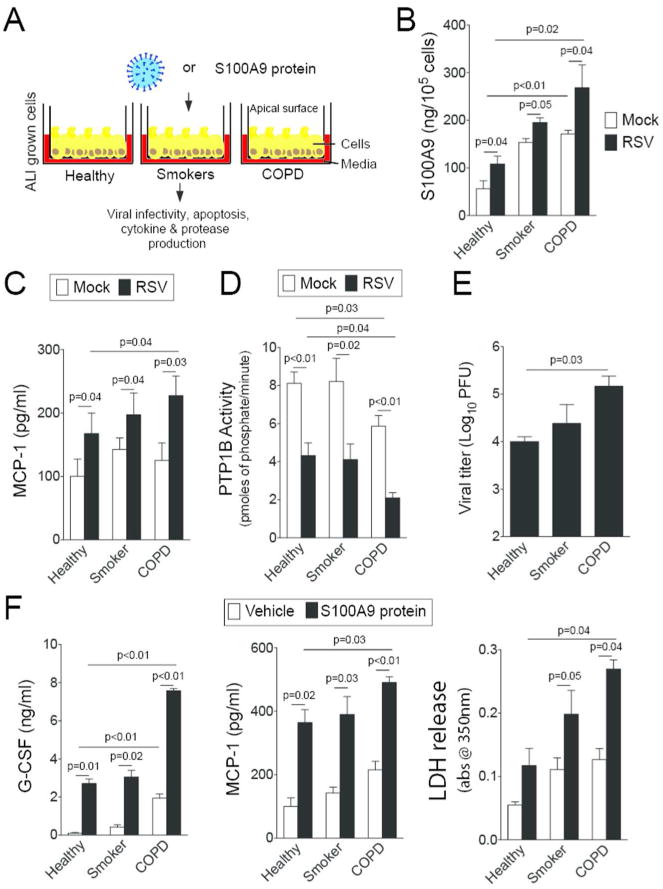
S100A9 signaling is heightened in airway epithelial cells from COPD patients. (A) Fully differentiated human airway epithelial cells grown at the ALI from healthy, smokers and COPD individuals (n=4, 3 and 4, respectively) were infected with (B-E) RSV or (F) treated with S100A9 protein for 2 hours and washed with PBS. Apical surface washes were taken 24 hours later. RSV infection enhanced (B) S100A9 and (C) MCP-1 secretions. COPD cells had reduced (D) PTP1B activity and (E) higher viral titer compared to cells from healthy and smoker donors. (F) Extracellular S100A9 stimuli increased MCP-1 and G-CSF secretion greater in COPD cells compared to cells from healthy and smoker donors. Cells from COPD donors also released greater LDH levels onto the apical surface of cells than the other groups. Graphs are represented as mean ± S.E.M., where each measurement was performed 3 independent days on 3-4 donors/group. p values shown, comparing both treatments connected by a line.
TRIF signaling influences S100A9 expression
Recently TRIF signaling was shown to regulate S100A9 expression during influenza infection17. To determine whether TRIF expression influences S100A9 expression during RSV infection, BMDM from wild type, Trif-/- and Ptp1b-/- animals were infected with RSV for 24 hours. Similar to our in vivo data, RSV infection resulted in S100A9 release, which was enhanced in macrophages deficient for Ptp1b expression (Figure 10A). Loss of Trif expression reduced S100A9 release following RSV infection (Figure 10A). We further confirmed these data in human primary SAE cells using siRNA (Figure 10B and Supplemental Figure S2).
Figure 10.
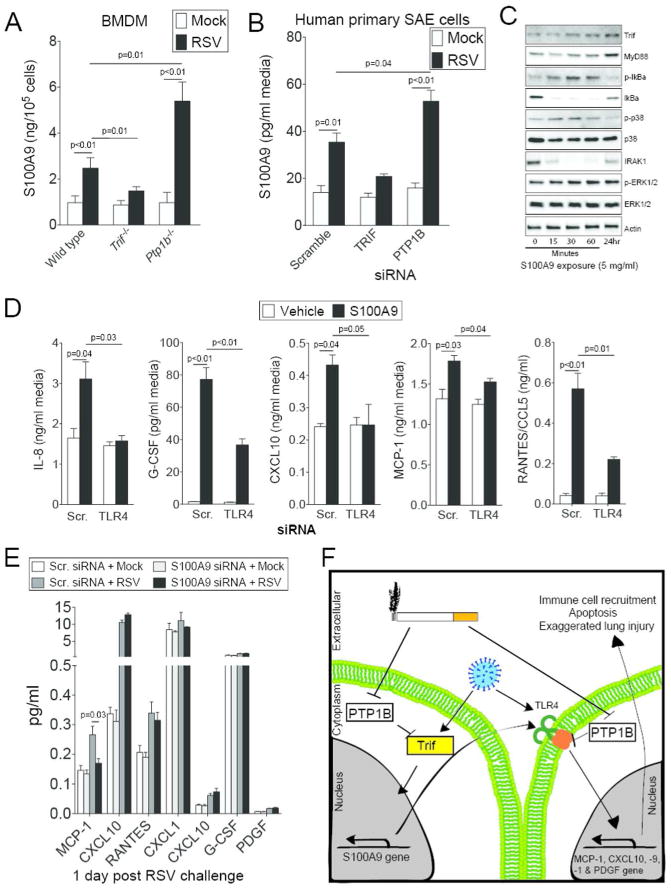
S100A9 induces TLR mediated cytokine responses. (A) Primary bone marrow derived macrophages (BMDM) isolated from wild type, Trif-/- and Ptp1b-/- mice were infected with RSV and ELISA determined extracellular S100A9 protein levels, 24 hours later. (B) Expression of TRIF and PTP1B were silenced in human SAE cells and cells were infected with RSV. ELISA determined extracellular S100A9 levels compared to cells transfected with Scr. siRNA. (C) SAE cells were treated with 5 μg/ml S100A9 protein for 0, 15, 30 and 60 minutes and 24 hours. Trif, MyD88, IκBα, p38, ERK and β-Actin immunoblots were performed. Representative immunoblots demonstrate typical results from 3 separate assays. (D) S100A9 protein induced release of MCP-1, CXCL10, IL-8, RANTES and G-CSF from SAE human cells in a TLR4 dependent manner confirmed in SAE cells transfected with either Scr. siRNA or siRNA specific for TLR4. (E) S100A9 expression was silenced in human SAE cells and cells were treated with RSV to determine the impact of RSV-induced S100A9 on cytokine expression. Graphs are represented as mean ± S.E.M., where each measurement was performed 3 times on samples from 3 independent experiments. p values shown, comparing both treatments connected by a line. (F) Possible pathway for the regulation of S100A9-mediated lung damage during RSV-associated COPD exacerbations.
Treatment of SAE cells with S100A9 protein resulted in the phosphorylation of p38, ERK and IκBα and the induction of TRIF and MyD88 expression (Figure 10C), which suggests activation of TLR4 signaling. Therefore, we determined that extracellular S100A9 could induce cytokine secretion, via TLR4 signaling. SAE cells were transfected with TLR4 siRNA prior to S100A9 protein stimulation, which demonstrated that TLR4 mediates S100A9-induction of IL-8, G-CSF, CXCL10, MCP-1 and CCL5 (Figure 10D). To determine whether induction of S100A9 expression during RSV infection was altering cytokines expression, S100A9 was silenced in SAE cells prior to RSV infection. Silencing S100A9 in human SAE cells reduced RSV infection-induced MCP-1 expression (Figure 10E). Therefore we proposed that PTP1B might facilitate TRIF regulation of S100A9 expression, which can contribute to TLR4 mediated inflammation (Figure 10F).
Discussion
Respiratory viruses are a major cause of COPD exacerbations and these events play a critical role in disease progression and patient hospitalization21. Thus, it is important to decipher how viral infections alter immune responses in the lung that can result in further loss of lung function in COPD patients. In this study, we demonstrated that PTP1B responses are desensitized in smoke exposed lungs, which leaves the lungs prone to enhanced S100A9 expression and consequent heightened lung damage during RSV infection. Ptp1b deficient mice have enhanced immune cell recruitment to the lung, apoptosis and cytokine production during RSV infection. Similar to influenza studies17, RSV infection induces significant lung damage in a S100A9–dependent manner. Importantly, S100A9 responses are heightened in COPD lungs, with epithelial cells from COPD subjects exhibiting an intensified response to S100A9 protein compared to cells isolated from healthy donors. Also, there is a negative relationship with BALF levels of S100A9 and lung function in patients. Extracellular S100A9 signaling is also critical in MCP-1 expression during RSV infection, which could further impact immune cell infiltration22. Overall, our study identifies that smoke-induced desensitization of PTP1B has the potential to enhance lung disease progression, in part, by augmenting the damaging effects of S100A9 expression in the lung during disease exacerbations caused by RSV. It is possible that a similar signaling cascade is used to promote loss of lung function in COPD subjects during infection with other viruses, such as rhinoviruses and influenza.
PTP1B undergoes activation following stimuli with inflammatory factors, such as TNF-α23. Once activated, PTP1B acts as a common negative regulator of NFκB, AKT and MAPK activities5 and can protect mice against injury5. Prior to this study, little was known about how PTP1B could protect against lung inflammation and remodeling. The ability of PTP1B to regulate immune cell infiltration and cell expansion is vitally important. Ptp1b-deficient mice have increased numbers of monocyte/macrophages in the spleen and the bone marrow, which have increased activation due to increased expression of CD805. PTP1B deficiency alters the immune responses to macrophages and apoptosis5 and increases susceptibility to smoke induced COPD-symptoms6. In our study, we observed enhanced macrophage numbers in the BALF. The fate of other cell types are altered by PTP1B expression, with loss of PTP1B in B cells16 or its pharmacological inhibition24 enhancing AKT phosphorylation. In a mouse ovalbumin (OVA) asthma model, loss of Ptp1b elevates the frequency of eosinophils and eosinophil progenitors compared to wild type mice25. In our RSV model, elevated eosinophil infiltration and Th2 cytokines were observed in Ptp1b-/- mice during RSV infection compared to wild type mice. Inhibition of S100A9 subdued these responses. Therefore regulation of S100A9 expression by PTP1B functions as a negative mediator of lung allergic responses. S100 proteins have also been associated with neutrophil activation26. S100A8 and S100A9 directly bind to p67phox and p47phox26, critical components of the NADPH complex, and potentate NADPH oxidase activation in neutrophils. PTP1B regulation of PAMPs is not restricted to TLR4 signaling, as loss of PTP1B heightens TLR2 (lipoteichoic acid, LTA) and TLR3 (polyI:C) signaling in macrophages5. Therefore, PTP1B modulates PAMP and DAMP signaling and immune cell responses in several key cell types observed in COPD exacerbation.
Considering our data, we propose the signaling cascade outlined in Figure 10F, where smoke and RSV infection subdue PTP1B responses and thereby enhance PAMP and DAMP signaling. The exaggerated S100A9 response subsequently activates signaling cascades19, such as TLR4, to enhance lung damage. Of note, PTP1B is oxidized in COPD lungs6 and increased S100A9 secretion is detection in the lungs of COPD subjects27, which could contribute to enhance loss of lung function. Enhancing PTP1B levels in patients may not be the best means of treatment as PTP1B negatively regulates insulin-signaling28 and enhancing PTP1B could have several secondary systemic effects in the body. However targeting specific PTP1B-dependent signaling, such as S100A9, may be feasible considering our S100A9 neutralizing data. Equally PTP1B regulated IL-17 expression during RSV infection, which has been associated with lung pathology in COPD29 and in RSV infections30. S100A9 regulation of key cytokines has previously been identified17, 31-33 but we observed that MCP-1 induction during an RSV infection is dependent on S100A9 expression. The exact role of MCP-1 during viral infections is limited. MCP-1 protects the lung in murine aspiration pneumonitis models34 but it enhances cigarette smoke-induced emphysema in mice35. Induction of MCP-1 by S100A9 may be a recovery response to protect the lung from further S100A9-associated damage, as MCP-1 aids in closure of mechanical wounds36 and inhibiting MCP-1 signaling enhances lung injury during influenza infection37. Equally MCP-1-activated immune cells could alter apoptosis in the lungs38. Identifying the role of MCP-1 in viral-associated COPD exacerbations will be a major future endeavor for our group.
Extracellular S100A9 up regulates apoptosis during RSV infection. However, following smoke exposure Ptp1b-/- mice still experience-enhanced apoptosis that appears to be independent of S100A9 signaling. We identified several other PTP1B-regulated genes that can play a significant role in cell death and survival. Cell death, which is induced by S100A9, can intensify inflammation in the lungs39 and culminate in the exacerbation of the underlying lung disease. S100 proteins can induce cell death by various mechanisms17, 31-33 and these processes could contribute to S100A9-mediated cell-death in COPD lungs following a viral exacerbation. Intracellular S100A9 may be involved in negatively regulating antiviral response or aid in viral replication17. However we have not observed changes in viral replication between wild type and Ptp1b-/- mice while observing higher S100A9 expression in Ptp1b-/- mice. We principally observe RSV infection in airway epithelial cells that are susceptible to S100A9 stimuli for cytokine production and apoptosis and can influence microenvironment changes in the lungs to enhance allergic responses. However we cannot exclude the role of other cells types in disease progression. Recently S100A9 was determined to be required for the maturation of TLR3 and signaling of TLR340. In fact, S100A9 colocalizes with TLR3 to facilitate TLR3 localizing with its agonist40. This S100A9 interaction with TLR3 mediates IFN-β and IL-12 production40. PTP1B can regulate downstream TLR3 signaling, such as TRIF and IRF3, which can regulate TLR3 signaling in addition to S100A9 expression. But PTP1B can modulate other TLRs as maintaining PTP1B activity attenuates the development of LPS-mediated lung injury in mice41. This may be partially dependent on the expression of S100A9 as both S100A9 and LPS induce TLR4 but each in a distinct manner42. However, S100A9 protects against Klebsiella pneumoniae infection, as S100a9-/- mice have enhanced bacterial load and lung damage compared to their wild type littermates43. Therefore additional information is required on the antimicrobial potential of both intracellular and extracellular S100A9 to numerous pathogens and whether dysregulation of S100A9 contributes to lung remodeling and disease progression in other lung infections.
S100A9 expression is also regulated by STAT3 signaling in cancer cells44. Interesting, JAK2 and STAT3 are substrates of PTP1B45; with PTP1B deficiency significantly enhancing the phosphorylation of JAK2 and STAT345. Phosphorylated STAT3 binds directly to the S100A9 promoter46. S100A9 also inhibits the differentiation of dendritic cells and macrophages and induces accumulation of myeloid-derived suppressor cells (MDSCs)47. Ptp1b-/- mice have an expansion of MDSCs, which is linked to STAT3 expression. Thus, it is conceivable that the inactivation of PTP1B during RSV infection could impact on the resolution of inflammation post infection by enhancing MDSC infiltration in the lung. However, this response needs to be tightly controlled as the complete lack of PTP1B expression clearly accentuated lung damage in our RSV infection models. The full impact of unregulated S100A9 in COPD viral exacerbations and the influence of STAT3 signaling require further investigation.
In summary, we have identified that loss of lung PTP1B activity leads to enhanced lung remodeling in animal COPD and RSV infection models by enhancing extracellular S100A9 levels. S100A9 regulates inflammation processes during virus infection and S100A9 expression appears to be TRIF dependent during RSV infection. Future studies targeting PTP1B dependent genes may lead to the development of measures to combat infection-associated exacerbations in COPD and other lung diseases.
Methods
Virus and submerged cell culture
Human RSV strain A2 (ATCC, Manassas, VA; #VR-1540) was used throughout study and was maintained as previously described12. Monolayers of human small airway epithelial (SAE) cells from healthy subjects (Lonza, Walkersville, MD) were cultured under submerged conditions. SAE cells were only used for experiments at passages 3-6 and at a confluency of approximately 70%. Cells were treated with RSV at a multiplicity of infection (MOI) of 0.3 for 24 hours. Cells were also treated with mock control12 or 5 μg/ml S100A9 protein. Cells were transiently transfected with siRNA for TRIF, TLR4, S100A9, PTP1B and scrambled control siRNA (QIAGEN Inc., Valencia, CA) prior to RSV infection. See Figure S2 for silencing efficiency of siRNA.
Animal models
Ptp1b (Ptpn1 gene) knockout (-/-) mice (FVB.129S4(B6)-Ptpn1tm1Bbk/Mmjax), on a FVB/NJ background, were purchased from the Mutant Mouse Resource Center at Jackson Laboratories (Bar Harbor, ME). All mice were maintained in a specific pathogen-free facility at Mount Sinai Roosevelt Hospital. 8-week-old mice were used at the initiation point for all experiments and each experimental parameter had at least 10 animals per group/time point. Mice were exposed to cigarette smoke in a chamber (Teague Enterprises, Davis, CA) for four hours a day, 5 days per week at a total particulate matter concentration of 80 mg/m3. The University of Kentucky reference research cigarettes 3R4F (Lexington, KY) were used to generate cigarette smoke. Smoke exposure was continued for 4 months after RSV infection or mock administered 24 hours after the final day of smoke exposure. Mice were anesthetized by intraperitoneal injection of a mixture of ketamine and xylazine (Sigma Aldrich, St. Louis, MO). Animals were intranasally administered 1×106 PFU RSV or mock, as previously described48. Mice were weighed daily and were euthanized on day's 0, 1, 3, 5, 7 and 9 post RSV or mock administration. BALF and tissues were collected for analysis. Total BALF protein was quantified by bicinchoninic acid assay (Thermo Fisher, Waltham, MA). The lungs underwent pressure-fixation and morphometric analysis in accordance with our previously published protocol12 and in accordance with the ATS/ERS issue statement on quantitative assessment of lung structure49. Fixed sections (6 μm) of paraffin-embedded lungs were H&E stained and were scored independently from 0 to 9 for each section12: peribronchiolitis, perivasculitis and alveolitis for inflammatory cells surrounding a bronchiole, a blood vessel or within alveolar spaces, respectively. 0, was no inflammation; 1–3, was scant cells but not forming a defined layer; 4–6, one to three layers of cells surrounding the vessel; 7–9, four or greater layers of cells surrounding the vessel or bronchial. Every vascular vessel and bronchus was measured on multiple lung lobes from 3 different depths of sectioned tissue. Slides were randomized, read blindly and scored for each parameter. Another group of FVB/NJ and Ptp1b-/- mice were IP injected with 100 μg of normal goat IgG (Santa Cruz Biotechnologies, Santa Cruz, CA) or a neutralizing goat polyclonal IgG targeting murine anti-S100A9 (Santa Cruz Biotechnologies) 2 hours prior to RSV administration. This overall study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and Institutional Animal Care and Use Committee (IACUC) guidelines.
Bone marrow derived macrophages (BMDM) were obtained from mice (wild type, Trif-/- and Ptp1b-/-) by flushing mouse tibiae and femurs with ice-cold PBS through a 70 μm-wide cell strainer and seeding cells at 37°C for 2 hours in a humidified atmosphere with 5% CO2 and then removing non-adherent cells. Cells were incubated in growth medium (MEM, 10% FBS, 2 mM glutamine, 100 IU/ml of penicillin, 100 μg/ml of streptomycin and 20 ng/ml M-CSF) for 7 days prior to RSV infection.
Lung viral titer
Infectious virus titers were determined by plaque assays. Lungs of infected mice were excised and homogenized using a mechanical homogenizer (Kinematica, Bohemia, NY, USA). The viral titers in the homogenates were quantified by plaque assay on epithelial cells using methyl cellulose overlay media (R&D Systems) and staining with 0.5 mg/ml thiazolyl blue tetrazolium bromide (MTT; Sigma Aldrich) solution in PBS for 3 hours at 37°C. Paraffin-embedded lung samples were sectioned at 5 μm, and stained with polyclonal anti-RSV antibody that recognizes RSV antigen from RSV A and B isolates (Abcam) and antibodies using an immunoperoxidase technique (Vectastain ABC Elite kit, Vector Laboratories, Burlingame, CA) to determine areas of lung infected.
Apoptosis measurements
BALF cells were analyzed for apoptosis utilizing the LIVE/DEAD cell viability assay from Life Technologies (Carlsbad, CA) on the Guava easyCyte flow cytometer from EMD Millipore (Temecula, CA). The apoptotic cells were expressed as a percentage of total BALF cells. BALF cells were further characterized in macrophage, neutrophil, eosinophil and lymphocyte cell populations by flow cytometry.
Cytokine measurements
Cytokine gene expression from mouse whole lung tissue or human cells was performed by quantitative PCR (qPCR) using Taqman probes (Life technologies/Applied Biosystems). Human and mouse cytokines were examined in human cell media and mouse BALF, respectively, using beads assays (Bio-Rad Magnetic Cytokine Bead Panels) with the BioRad Bio-Plex 200 system (Bio-Rad, Hercules, CA). Mouse and human S100A9 levels were determined using a bead assay and ELISA (both from R&D Systems), respectively. BALF was standardized for each assay to urea levels, as determined by a commercially available assay as instructed by the manufacturer (Abnova, Walnut, CA, USA). S100A9 fluorescence levels were determined in lung sections from both genotypes 5 dpi and each field of the microscope was quantified using ImageJ software (http://imagej.nih.gov/ij/docs/index.html). Goat polyclonal anti-S100A9 antibody (Santa Cruz Biotechnologies) was utilized to stain sections. All regions of the lung sections were utilized to avoid selection bias. Relative fluorescence intensity was determined (n=10 animals per group).
Intracellular signaling
PTP1B activity was determined as previously described [35]. Cells or tissue were lysed in radio-immunoprecipitation assay (RIPA) buffer, centrifuged at 13,000 x g for 10 minutes and supernatants were collected. Immunoblots were conducted to determine levels of caspase-3, p-IκBα, IκBα (Ser32), p-p38 (Thr180/Tyr182), p38, TRIF, MyD88, IRAK1, p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), ERK1/2, β-actin (all antibodies from Cell Signaling Technologies, Beverly, MA) and S100A9 (Santa Cruz Biotechnologies).
Apoptosis array
qPCR arrays were performed using the Bio-Rad PrimePCR apoptosis and survival panel (Bio-Rad; Apoptosis and Survival Tier 1-4 H384) to examine over 350 genes associated with cell survival and apoptosis. Data was analyzed and normalized with the BioRad PrimePCR analysis software. Several PTP1B regulated genes were validated by qPCR, bead assays and immunoblots.
Human Samples
BALF was obtained from healthy never smokers, smokers, and COPD patients (see Supplemental Tables S1-S2 for demographics). Written consent was obtained from all study participants and was approved by the institutional review board of the University of Miami. Human lungs were obtained from organ donors whose lungs were rejected for transplant. Since some of these patients had a smoking history with macro-pathological evidence of emphysema, tissues from these donors were labeled as tissue from COPD patients. Consent for research was obtained by the Life Alliance Organ Recovery Agency of the University of Miami. All consents were IRB-approved and conformed to the Declaration of Helsinki.
Airway epithelial cells were isolated from healthy, smokers and COPD lungs, de-differentiated through expansion and re-differentiated at an air-liquid interface (ALI) on 24mm T-clear filters (Costar Corning, Corning, NY) as previously described50. RSV, mock or S100A9 protein were added to the apical surface of the cultures and incubated for 2 hours at 37 °C, 5% CO2. Subsequently, the apical surface was rinsed five times with PBS and ALI conditions were restored. Twenty-four hours later the apical surface was rinsed with 600 μl PBS and the rinse was harvested and investigated for cytokine production. The cells were collected for protein and RNA.
Statistical analyses
For statistical analysis, data from 10 animals or three separate cell experiments were pooled. Data are expressed as mean ± S.E.M. Differences between groups of mice over time were compared by two-way analysis of variance (ANOVA). Individual differences between groups were tested by multiple comparison and analysis using the Bonferroni post-test analysis. Pairs of groups were compared by Student's t test (two tailed). p values for significance were set at 0.05. All analysis was performed using GraphPad Prism Software (Version 5 for Mac OS X; La Jolla, CA).
Supplementary Material
Supplemental Figure S1 PTP1B expression regulates NFκB signaling and cytokine response to RSV in Ptp1b deficient mice. (A) Immunblots were performed to examine levels of actin, MyD88 and TRIF protein and IRF3, IRF7 and TBK1 phosphorylation in lung tissue of RSV infected animals. Densitometry analysis was performed against actin or total protein (TBK1, IRF7 and IRF3) levels. (B) Lung NFκB activity were determined following mock and RSV treatment in wild type and Ptp1b-/- mice. Graphs are represented as mean ± S.E.M, where n=10 per group. *Represents a p value less than 0.05 compared to wild type mice on each corresponding time point.
Supplemental Figure S2 Confirmation that expression of Trif and RLRs was silenced in A549 cells. SAE cells were transfected with siRNA targeting PTP1B, TRIF, TLR4 and S100A9 or a negative sequence control (scrambled). Immunoblots were performed to determine PTP1B, TRIF, TLR4 and S100A9 protein levels compared to β-actin.
Supplemental Figure S3 Lung caspase-3 cleavage in RSV-infection mice is diminished upon neutralization of extracellular S100A9 signaling. Wild type and Ptp1b-/- mice were IP injected with isotype control IgG or anti-S100A9 IgG prior to intranasal infection with 1×106 pfu of RSV and animals were euthanized 2, 5 and 8-days post infections. Caspase-3 cleavage was determined by caspase-3 immunoblots conducted on lung tissue lysates.
Supplemental Table S1 Patient demographics for donors
Supplemental Table S2 Patient demographics for epithelial cell donors
Acknowledgments
This work was supported by grants made available to P.G. (Flight Attendant Medical Research Institute (YCSA 113380), R.F. (US National Institutes of Health 5R01HL098528-04) and M.S. (Flight Attendant Medical Research Institute 103027). The authors would like to thank the James P. Mara Center for Lung Disease of the Pulmonary Division of Mount Sinai Roosevelt for their support and Dr. Gerard Turino and Dr. Charles Powell. The authors also thank the patients who participated in this study.
Abbreviations
- PTP1B
Protein tyrosine phosphatase 1B
- RSV
Respiratory syncytial virus
- COPD
chronic obstructive pulmonary disease
- BALF
bronchoalveolar lavage fluid
- SAE
small airway epithelial
- ALI
air liquid interface
- DAMP
damage-associated molecular pattern
- PAMP
pathogen-associated molecular pattern
- dpi
days post infection
Footnotes
Disclosure. The authors declare no financial conflicts of interest.
Supplementary Material is linked to the online version of the paper at http://www.nature.com/mi
References
- 1.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283(5407):1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 2.Feldhammer M, Uetani N, Miranda-Saavedra D, Tremblay ML. PTP1B: a simple enzyme for a complex world. Critical reviews in biochemistry and molecular biology. 2013;48(5):430–445. doi: 10.3109/10409238.2013.819830. [DOI] [PubMed] [Google Scholar]
- 3.Dube N, Bourdeau A, Heinonen KM, Cheng A, Loy AL, Tremblay ML. Genetic ablation of protein tyrosine phosphatase 1B accelerates lymphomagenesis of p53-null mice through the regulation of B-cell development. Cancer Res. 2005;65(21):10088–10095. doi: 10.1158/0008-5472.CAN-05-1353. [DOI] [PubMed] [Google Scholar]
- 4.Berdnikovs S, Abdala-Valencia H, Cook-Mills JM. Endothelial cell PTP1B regulates leukocyte recruitment during allergic inflammation. Am J Physiol Lung Cell Mol Physiol. 2013;304(4):L240–249. doi: 10.1152/ajplung.00375.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traves PG, Pardo V, Pimentel-Santillana M, Gonzalez-Rodriguez A, Mojena M, Rico D, et al. Pivotal role of protein tyrosine phosphatase 1B (PTP1B) in the macrophage response to pro-inflammatory and anti-inflammatory challenge. Cell death & disease. 2014;5:e1125. doi: 10.1038/cddis.2014.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geraghty P, Hardigan AA, Wallace AM, Mirochnitchenko O, Thankachen J, Arellanos L, et al. The glutathione peroxidase 1-protein tyrosine phosphatase 1B-protein phosphatase 2A axis. A key determinant of airway inflammation and alveolar destruction. American journal of respiratory cell and molecular biology. 2013;49(5):721–730. doi: 10.1165/rcmb.2013-0026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miniño AM, Xu J, Kochanek KD. Deaths: Preliminary Data for 2008. National Vital Statistics Reports. 2010;59(2):1–72. [PubMed] [Google Scholar]
- 8.Miravitlles M, Anzueto A, Legnani D, Forstmeier L, Fargel M. Patient's perception of exacerbations of COPD--the PERCEIVE study. Respir Med. 2007;101(3):453–460. doi: 10.1016/j.rmed.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Jain S, Self WH, Wunderink RG, Fakhran S, Balk R, Bramley AM, et al. Community-Acquired Pneumonia Requiring Hospitalization among U.S. Adults. The New England journal of medicine. 2015;373(5):415–427. doi: 10.1056/NEJMoa1500245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walsh EE, Falsey AR, Hennessey PA. Respiratory syncytial and other virus infections in persons with chronic cardiopulmonary disease. Am J Respir Crit Care Med. 1999;160(3):791–795. doi: 10.1164/ajrccm.160.3.9901004. [DOI] [PubMed] [Google Scholar]
- 11.Zwaans WA, Mallia P, van Winden ME, Rohde GG. The relevance of respiratory viral infections in the exacerbations of chronic obstructive pulmonary disease-a systematic review. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology. 2014;61(2):181–188. doi: 10.1016/j.jcv.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foronjy RF, Dabo AJ, Taggart CC, Weldon S, Geraghty P. Respiratory syncytial virus infections enhance cigarette smoke induced COPD in mice. PloS one. 2014;9(2):e90567. doi: 10.1371/journal.pone.0090567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phaybouth V, Wang SZ, Hutt JA, McDonald JD, Harrod KS, Barrett EG. Cigarette smoke suppresses Th1 cytokine production and increases RSV expression in a neonatal model. Am J Physiol Lung Cell Mol Physiol. 2006;290(2):L222–231. doi: 10.1152/ajplung.00148.2005. [DOI] [PubMed] [Google Scholar]
- 14.Borchers AT, Chang C, Gershwin ME, Gershwin LJ. Respiratory syncytial virus--a comprehensive review. Clinical reviews in allergy & immunology. 2013;45(3):331–379. doi: 10.1007/s12016-013-8368-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu H, An H, Hou J, Han C, Wang P, Yu Y, et al. Phosphatase PTP1B negatively regulates MyD88- and TRIF-dependent proinflammatory cytokine and type I interferon production in TLR-triggered macrophages. Mol Immunol. 2008;45(13):3545–3552. doi: 10.1016/j.molimm.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Medgyesi D, Hobeika E, Biesen R, Kollert F, Taddeo A, Voll RE, et al. The protein tyrosine phosphatase PTP1B is a negative regulator of CD40 and BAFF-R signaling and controls B cell autoimmunity. The Journal of experimental medicine. 2014;211(3):427–440. doi: 10.1084/jem.20131196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai SY, Segovia JA, Chang TH, Morris IR, Berton MT, Tessier PA, et al. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS pathogens. 2014;10(1):e1003848. doi: 10.1371/journal.ppat.1003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nature immunology. 2000;1(5):398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 19.Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nature medicine. 2007;13(9):1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 20.Pouwels SD, Nawijn MC, Bathoorn E, Riezebos-Brilman A, van Oosterhout AJ, Kerstjens HA, et al. Increased serum levels of LL37, HMGB1 and S100A9 during exacerbation in COPD patients. The European respiratory journal. 2015 doi: 10.1183/09031936.00158414. [DOI] [PubMed] [Google Scholar]
- 21.Rohde G, Wiethege A, Borg I, Kauth M, Bauer TT, Gillissen A, et al. Respiratory viruses in exacerbations of chronic obstructive pulmonary disease requiring hospitalisation: a case-control study. Thorax. 2003;58(1):37–42. doi: 10.1136/thorax.58.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belperio JA, Keane MP, Burdick MD, Lynch JP, 3rd, Xue YY, Berlin A, et al. Critical role for the chemokine MCP-1/CCR2 in the pathogenesis of bronchiolitis obliterans syndrome. The Journal of clinical investigation. 2001;108(4):547–556. doi: 10.1172/JCI12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. 2008;283(21):14230–14241. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie L, Lee SY, Andersen JN, Waters S, Shen K, Guo XL, et al. Cellular effects of small molecule PTP1B inhibitors on insulin signaling. Biochemistry. 2003;42(44):12792–12804. doi: 10.1021/bi035238p. [DOI] [PubMed] [Google Scholar]
- 25.Berdnikovs S, Pavlov VI, Abdala-Valencia H, McCary CA, Klumpp DJ, Tremblay ML, et al. PTP1B deficiency exacerbates inflammation and accelerates leukocyte trafficking in vivo. Journal of immunology. 2012;188(2):874–884. doi: 10.4049/jimmunol.1004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doussiere J, Bouzidi F, Vignais PV. The S100A8/A9 protein as a partner for the cytosolic factors of NADPH oxidase activation in neutrophils. Eur J Biochem. 2002;269(13):3246–3255. doi: 10.1046/j.1432-1033.2002.03002.x. [DOI] [PubMed] [Google Scholar]
- 27.Merkel D, Rist W, Seither P, Weith A, Lenter MC. Proteomic study of human bronchoalveolar lavage fluids from smokers with chronic obstructive pulmonary disease by combining surface-enhanced laser desorption/ionization-mass spectrometry profiling with mass spectrometric protein identification. Proteomics. 2005;5(11):2972–2980. doi: 10.1002/pmic.200401180. [DOI] [PubMed] [Google Scholar]
- 28.Seely BL, Staubs PA, Reichart DR, Berhanu P, Milarski KL, Saltiel AR, et al. Protein tyrosine phosphatase 1B interacts with the activated insulin receptor. Diabetes. 1996;45(10):1379–1385. doi: 10.2337/diab.45.10.1379. [DOI] [PubMed] [Google Scholar]
- 29.Linden A, Hoshino H, Laan M. Airway neutrophils and interleukin-17. The European respiratory journal. 2000;15(5):973–977. doi: 10.1034/j.1399-3003.2000.15e28.x. [DOI] [PubMed] [Google Scholar]
- 30.Newcomb DC, Boswell MG, Reiss S, Zhou W, Goleniewska K, Toki S, et al. IL-17A inhibits airway reactivity induced by respiratory syncytial virus infection during allergic airway inflammation. Thorax. 2013;68(8):717–723. doi: 10.1136/thoraxjnl-2012-202404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atallah M, Krispin A, Trahtemberg U, Ben-Hamron S, Grau A, Verbovetski I, et al. Constitutive neutrophil apoptosis: regulation by cell concentration via S100 A8/9 and the MEK-ERK pathway. PloS one. 2012;7(2):e29333. doi: 10.1371/journal.pone.0029333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghavami S, Eshragi M, Ande SR, Chazin WJ, Klonisch T, Halayko AJ, et al. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell research. 2010;20(3):314–331. doi: 10.1038/cr.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li C, Chen H, Ding F, Zhang Y, Luo A, Wang M, et al. A novel p53 target gene, S100A9, induces p53-dependent cellular apoptosis and mediates the p53 apoptosis pathway. The Biochemical journal. 2009;422(2):363–372. doi: 10.1042/BJ20090465. [DOI] [PubMed] [Google Scholar]
- 34.Raghavendran K, Davidson BA, Mullan BA, Hutson AD, Russo TA, Manderscheid PA, et al. Acid and particulate-induced aspiration lung injury in mice: importance of MCP-1. American journal of physiology Lung cellular and molecular physiology. 2005;289(1):L134–143. doi: 10.1152/ajplung.00390.2004. [DOI] [PubMed] [Google Scholar]
- 35.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 36.Christensen PJ, Du M, Moore B, Morris S, Toews GB, Paine R., 3rd Expression and functional implications of CCR2 expression on murine alveolar epithelial cells. American journal of physiology Lung cellular and molecular physiology. 2004;286(1):L68–72. doi: 10.1152/ajplung.00079.2003. [DOI] [PubMed] [Google Scholar]
- 37.Narasaraju T, Ng HH, Phoon MC, Chow VT. MCP-1 antibody treatment enhances damage and impedes repair of the alveolar epithelium in influenza pneumonitis. American journal of respiratory cell and molecular biology. 2010;42(6):732–743. doi: 10.1165/rcmb.2008-0423OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang D, Elner SG, Chen X, Field MG, Petty HR, Elner VM. MCP-1-activated monocytes induce apoptosis in human retinal pigment epithelium. Investigative ophthalmology & visual science. 2011;52(8):6026–6034. doi: 10.1167/iovs.10-7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nature reviews Microbiology. 2009;7(2):99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai SY, Segovia JA, Chang TH, Shil NK, Pokharel SM, Kannan TR, et al. Regulation of TLR3 Activation by S100A9. Journal of immunology. 2015 doi: 10.4049/jimmunol.1500378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grinnell KL, Chichger H, Braza J, Duong H, Harrington EO. Protection against LPS-induced pulmonary edema through the attenuation of protein tyrosine phosphatase-1B oxidation. Am J Respir Cell Mol Biol. 2012;46(5):623–632. doi: 10.1165/rcmb.2011-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riva M, Kallberg E, Bjork P, Hancz D, Vogl T, Roth J, et al. Induction of nuclear factor-kappaB responses by the S100A9 protein is Toll-like receptor-4-dependent. Immunology. 2012;137(2):172–182. doi: 10.1111/j.1365-2567.2012.03619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Achouiti A, Vogl T, Urban CF, Rohm M, Hommes TJ, van Zoelen MA, et al. Myeloid-related protein-14 contributes to protective immunity in gram-negative pneumonia derived sepsis. PLoS pathogens. 2012;8(10):e1002987. doi: 10.1371/journal.ppat.1002987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li C, Zhang F, Lin M, Liu J. Induction of S100A9 gene expression by cytokine oncostatin M in breast cancer cells through the STAT3 signaling cascade. Breast cancer research and treatment. 2004;87(2):123–134. doi: 10.1023/B:BREA.0000041594.36418.f6. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Wang B, Zhang W, Wei Y, Bian Z, Zhang CY, et al. Protein tyrosine phosphatase 1B deficiency ameliorates murine experimental colitis via the expansion of myeloid-derived suppressor cells. PloS one. 2013;8(8):e70828. doi: 10.1371/journal.pone.0070828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee MJ, Lee JK, Choi JW, Lee CS, Sim JH, Cho CH, et al. Interleukin-6 induces S100A9 expression in colonic epithelial cells through STAT3 activation in experimental ulcerative colitis. PloS one. 2012;7(9):e38801. doi: 10.1371/journal.pone.0038801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205(10):2235–2249. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foronjy RF, Taggart CC, Dabo AJ, Weldon S, Cummins N, Geraghty P. Type-I interferons induce lung protease responses following respiratory syncytial virus infection via RIG-I-like receptors. Mucosal immunology. 2015;8(1):161–175. doi: 10.1038/mi.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsia CCW, Hyde DM, Ochs M, Weibel ER on behalf of the ATS/ERS Joint Task Force on the Quantitative Assessment of Lung Structure. An Official Research Policy Statement of the American Thoracic Society/European Respiratory Society: Standards for Quantitative Assessment of Lung Structure. Am J Respir Crit Care Med. 2010;181(4):394–418. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nlend MC, Bookman RJ, Conner GE, Salathe M. Regulator of G-protein signaling protein 2 modulates purinergic calcium and ciliary beat frequency responses in airway epithelia. Am J Respir Cell Mol Biol. 2002;27(4):436–445. doi: 10.1165/rcmb.2002-0012OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1 PTP1B expression regulates NFκB signaling and cytokine response to RSV in Ptp1b deficient mice. (A) Immunblots were performed to examine levels of actin, MyD88 and TRIF protein and IRF3, IRF7 and TBK1 phosphorylation in lung tissue of RSV infected animals. Densitometry analysis was performed against actin or total protein (TBK1, IRF7 and IRF3) levels. (B) Lung NFκB activity were determined following mock and RSV treatment in wild type and Ptp1b-/- mice. Graphs are represented as mean ± S.E.M, where n=10 per group. *Represents a p value less than 0.05 compared to wild type mice on each corresponding time point.
Supplemental Figure S2 Confirmation that expression of Trif and RLRs was silenced in A549 cells. SAE cells were transfected with siRNA targeting PTP1B, TRIF, TLR4 and S100A9 or a negative sequence control (scrambled). Immunoblots were performed to determine PTP1B, TRIF, TLR4 and S100A9 protein levels compared to β-actin.
Supplemental Figure S3 Lung caspase-3 cleavage in RSV-infection mice is diminished upon neutralization of extracellular S100A9 signaling. Wild type and Ptp1b-/- mice were IP injected with isotype control IgG or anti-S100A9 IgG prior to intranasal infection with 1×106 pfu of RSV and animals were euthanized 2, 5 and 8-days post infections. Caspase-3 cleavage was determined by caspase-3 immunoblots conducted on lung tissue lysates.
Supplemental Table S1 Patient demographics for donors
Supplemental Table S2 Patient demographics for epithelial cell donors