

Abstract
The hypothalamus is comprised of neuronal clusters that are essential for body weight regulation and other physiological functions. Insights into the complex cellular physiology of this region are critical to understanding the pathogenesis of obesity; however, human hypothalamic cells are largely inaccessible for direct study. Here we describe a technique for generation of hypothalamic arcuate-like neurons from human pluripotent stem (hPS) cells. Early activation of SHH signaling and inhibition of BMP and TGFβ signaling, followed by timed NOTCH inhibition, can efficiently differentiate hPSC into NKX2.1+ hypothalamic progenitors. Subsequent incubation with BDNF induces the differentiation and maturation of POMC and NPY neurons that are major cell types in the arcuate hypothalamus. These neurons have functional characteristics consistent with arcuate neurons.
Keywords: Hypothalamic neurons, human pluripotent stem cells, ARC neurons, feeder-free
Introduction
A major function of the hypothalamus is as central integrator of systemic energy homeostasis; its dysfunction can result in extreme alterations of body weight. This neuroendocrine brain region, located just above the median eminence, is comprised of cell types that subserve specific metabolic and behavioral aspects of the control of body weight, as well as hepatic glucose production, body temperature, autonomic physiology, neuroendocrine axes, serum osmolarity and circadian rhythms. The hypothalamus is comprised of several cell clusters with distinct physiological functions including the arcuate (Marcelle et al.), ventromedial (VMH), dorsal medial (DMH) and paraventricular (PVH) nuclei (Barsh and Schwartz, 2002). ARC neurons, such as those expressing Pro-opiomelanocortin (POMC) and Neuropeptide Y (NPY)/Agouti-related peptide (AGRP), can sense peripheral hormones—insulin, leptin, ghrelin, and secrete neuropeptides αMSH and NPY/AGRP to engage receptors on DMH, PVN and other brain areas that regulate food intake and energy expenditure (Barsh and Schwartz, 2002). Hypomorphic mutations of genes involved in hypothalamic leptin-melanocortin signaling lead to severe monogenic obesity in humans and rodents (van der Klaauw and Farooqi, 2015). Therefore, insights into the complex cellular physiology of hypothalamus are important in a wide range of clinical circumstances, including obesity. However, human hypothalamic cells are not readily available for direct studies.
Based on fundamental aspects of hypothalamic development and neuronal differentiation studies, we have developed a differentiation protocol for efficient and reproducible generation of hypothalamic arcuate-like neurons from human embryonic stem (hES) cells and induced pluripotent stem (iPS) cells (Wang et al., 2015). This protocol is based on the three key steps, composed of neuronal differentiation, ventralization and hypothalamic differentiation. The protocol is based on treatments that have each been shown effective alone, or in a different developmental context and are combined here to specifically induce hypothalamic differentiation.
The first step is the induction of neuronal differentiation from pluripotent stem cells through inhibition of transforming growth factor β (TGFβ) and bone morphogenetic protein (BMP) signaling pathways, as shown by Studer’s lab (Chambers et al., 2009). The second step is the ventralization and induction of Nkx2.1+ neuronal progenitors by activation of sonic hedgehog (SHH) signaling, as shown in the study of medial ganglionic eminence neuron and mouse hypothalamic neuron differentiation (Liu et al., 2013; Wataya et al., 2008). The third is the inhibition of telencephalic development by inhibition of Notch signaling. Notch inhibition has previously been shown to accelerate neuronal development and inhibit Pax6 expression (Chambers et al., 2012). Pax6 and FoxG1 mark the telencephalon, and are not expressed in the diencephalon (Maroof et al., 2013). After 12 days of differentiation, more than 80% cells are Nkx2.1+ hypothalamic neuron progenitors expressing MASH1, NHLH2, SF1, OTP, SIM1, but not Pax6 or FoxG1. After further differentiation for 12 days in the presence of DAPT and brain-derived neurotrophic factor (BDNF), hypothalamic neurons express key markers of different neuron types in the ARC, including POMC, NPY/AGRP and somatostatin.
Here we present detailed protocols for the differentiation of hypothalamic neurons. Basic Protocol 1 describes the preparation of hES or hiPS cells for neuron differentiation. Basic Protocol 2 explains how to generate hypothalamic neuron progenitors from ES or iPS cells, and neuro differentiation and maturation from Nkx2.1+ progenitors is given in Basic Protocol 3.
Basic Protocol 1: Preparation of hES or iPS cells for neuron differentiation
hES or iPS cells (hPS cells) can be cultured on a layer of feeder cells or in a feeder-free system(Kime et al., 2015; Mallon et al., 2006; Thomson et al., 1998). hPS cells cultured on feeder cells or extracellular matrix (e.g. matrigel) show no obvious differences in hypothalamic differentiation with this protocol. In the protocol described here, we maintain and expand hPS cells cultures in 6-well plates with mouse embryonic fibroblasts (MEF). We then seed hPS cells on matrigel-coated plate (feeder-free) to initiate neuron differentiation once each well reaches 95–100% confluence. The hPS cells should be in uniform shape and active proliferation before starting differentiation.
Materials (Commercial information for all reagents are provided in Appendix Tables 1 and 2.)
hES or iPS cell line
Mouse embryonic fibroblasts (MEF)
Matrigel
DMEM:F12 medium
hES medium (see Reagents and Solutions)
mTeSR™1 complete kit
TrypLE Express Enzyme
Rock inhibitor (Y27632)
0.4% Trypan blue
Centrifuge (for 15ml tubes)
6-well culture plate (Nunc™ Cell-Culture Treated Multidishes)
15ml Falcon tubes (ThermoFisher)
50ml Falcon tubes (ThermoFisher)
Falcon® 5ml round bottom polystyrene tube with cell strainer cap (ThermoFisher)
1.5ml micro centrifuge tubes
5% CO2, 37 °C humidified incubator
Cell counting chambers (ThermoFisher)
Inverted microscope (Olympus)
- Countless automated cell counter (ThermoFisher)
- The day before thawing or splitting hES or iPS cell lines, thaw matrigel (store at −80 °C) overnight at 4 °C.
-
Dilute matrigel in cold DMEM/F12 medium (v/v, 1:50) and keep the mixture on ice. Then add 1ml diluted matrigel into each well on 6-well plate and place the plate in 37 °C incubator.Keep matrigel and DMEM/F12 medium on ice when preparing the diluted matrigel. We recommend cooling the pipet (10 ml) and tips (1ml) at −20 °C before use.
-
2–4 hours later, the matrigel plate is ready for use.Using a 20× objective lens, gel-like structure should be visible on the plate. Matrigel should be evenly distributed on the plate. Prolonged incubation may cause uneven distribution of the gel (thicker periphery, thinner center).
-
hES or iPS cells are maintained and expanded in hES medium on MEF plates. When the hES or iPS colonies have reached 95–100% confluency on MEF plates (Figure 1A), detach cells by treating with TrypLE express enzyme (0.5 ml/well) for 4 min in 37 °C incubator, or until morphological changes as shown in Figure 1B occur. For hPS cells cultured on matrigel plates in mTeSR medium, the procedures at this step and following steps are the same.Plates should be inspected microscopically to assess integrity of individual hPSC contacts with neighboring cells.
- Harvest cells by adding 3 ml hES medium to each well and detach all cells from the plate by pipeting up and down. Transfer cell suspension to 15 ml falcon tube.
- Spin down cells at 800 rpm for 4 min at RT.
- Aspirate supernatant and resuspend cell pellet with 2ml hES medium with 10 μM Rock inhibitor.
- Use P1000 Gilson pipet to disaggregate cell clusters completely by pipeting up and down at least 10 times.
- Pipet cell suspension through falcon tube with cell-strainer cap (35 μm mesh size) to make single cell suspension.
- Mix 10 μl cell suspension with 10 μl 0.4% Trypan blue. Add 10 μl cell mixture to cell counting chamber.
- Count cells in the chamber with countless automated cell counter.
- Aspirate matrigel from each well before adding cells.
-
Add 1 million cells to each well on a matrigel-coated 6-well plate and culture in mTeSR1 medium/hES medium plus 10 μM Rock inhibitor. Next day, start neuron induction when cells have reached 95–100% confluence (Figure 1C).May need to culture hPS cells in mTeSR1 medium (for feeder-free culture) until each well reaches 95–100% confluence before inducing neuron differentiation.
Figure 1. Feeder- and feeder-free culture of Human pluripotent stem cells.
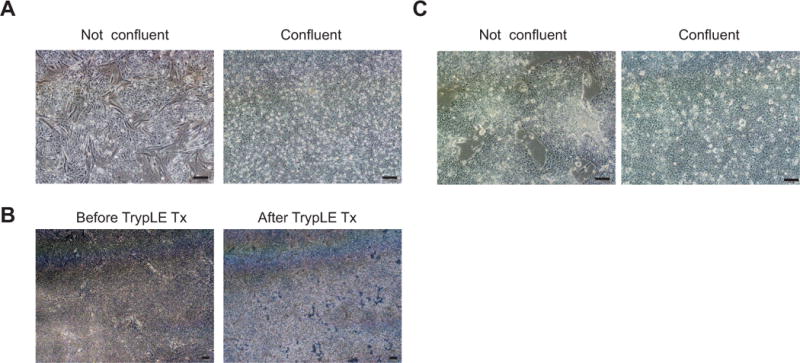
(A) hPSCs grown on mouse embryonic fibroblasts (MEF). Left image indicates non- confluent culture while right image indicates confluent culture; (B) hPS cells grown on MEFs before and after trypsin LE treatment; (C) hPS cells grown on matrigel plate in mTeSR1 medium. Left image indicates non- confluent culture while right image indicates confluent culture. Scale bar represents 100 μm.
Basic Protocol 2. Neuron induction: generation of hypothalamic neuron progenitors from hES or iPS cells
SB 431542 and LDN 193189 are used from day 1 to day 8 to inhibit TGFβ and BMP signaling in order to promote neuron differentiation from human ES/iPS cells (Figure 2A)(Chambers et al., 2009). SHH and Purmophamine are combined from days 1 to 8 to induce ventral brain development and NKX2.1 expression. Further inhibition of Notch signaling by DAPT (days 9 to 12) increases NKX2.1 expression and enriches for neuron precursors of ARC cell types. The Nkx2.1 GFP/W-hES line (Goulburn et al., 2011) can be used to monitor the changes of Nkx2.1 expression during the first 12 days of the differentiation period.
Figure 2. Generation of NKX2.1+ neuron progenitors from hES/iPS cells.
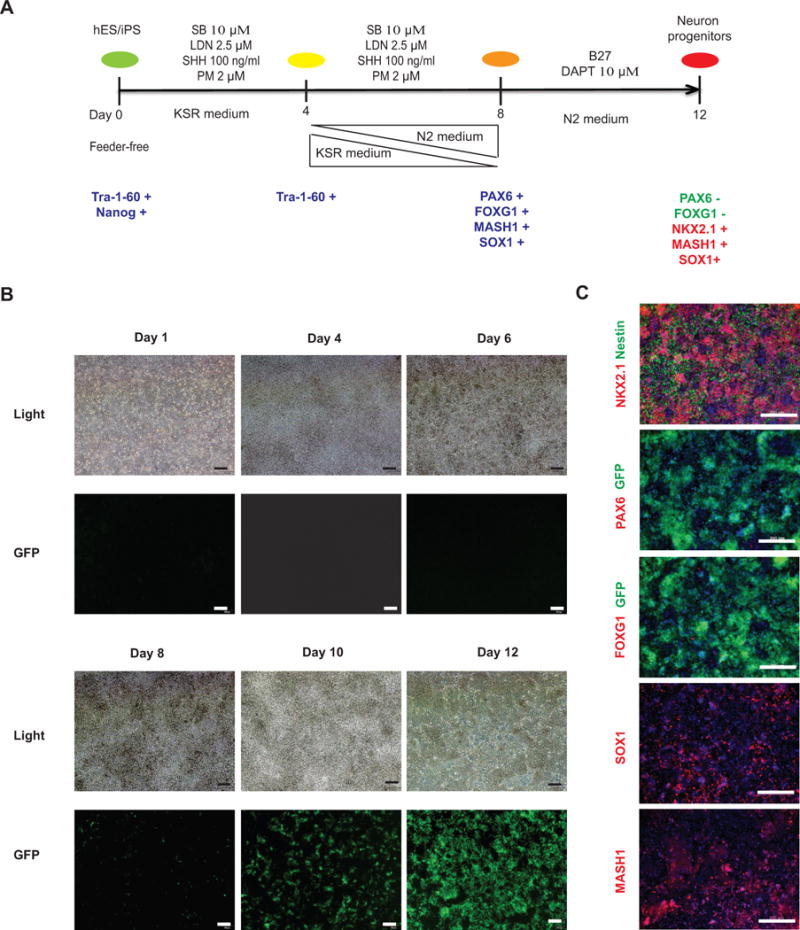
(A) Schematic of the differentiation protocol for the generation of hypothalamic progenitors from hES/iPS cells. (B) Live cell imaging on 1, 4, 6, 8, 10, 12 day differentiated cells during the first 12 days of hypothalamic neuron differentiation. Scale bar, 100 μm. (C) Immunocytochemistry analysis of neuron progenitor markers in 12 days differentiated NKX2.1 W/GFP-hES cells. NKX2.1, Nestin, PAX6, FOXG1, SOX1, MASH1 were analyzed. GFP is an endogenous signal indicating the expression of NKX2.1. Scale bar, 200 μm.
Materials (The commercial information for all reagents are shown in Appendix Tables 1 and 2.)
KSR medium (see Reagents and Solutions)
N2 medium (see Reagents and Solutions)
SHH
Purmorphamine
SB 431542
LDN 193189
DAPT
B27 (50×)
-
Days 1 to Day 4 (Figure 2B):
Day 1: When hES/iPS cells have reached 95–100% confluency on the matrigel plate, switch cells into KSR medium supplemented with SHH (100 ng/ml), purmorphamine (2 μM), 10 μM SB 431542, 2.5 μM LDN 193189. After adding these four differentiation factors (SB, LDN, SHH, Purmorphamine) to the KSR medium, use the differentiation medium immediately. Add 2–3 ml of the differentiation medium (same volume for the following steps) to each well of a 6-well plate. Change medium daily.
We recommend preparing small volumes of differentiation medium (10 ml or 50ml, depending on how many wells are being used) for everyday use. If not used immediately, differentiation medium can be stored at 2–8 °C for up to 2 weeks.
Day 5: Prepare KSR medium and N2 medium in 3:1 ratio. Then add the four differentiation factors mentioned above at the same concentrations. Switch cells into the KSR/N2 medium mixture for 24 hrs.
Day 6 (Figure 2B): Prepare KSR medium and N2 medium in 1:1 ratio. Then add the four differentiation factors mentioned above at the same concentrations. Switch cells into the KSR/N2 medium mixture for 24hrs.
Day 7: Prepare KSR medium and N2 medium in 1:3 ratio. Then add the four differentiation factors mentioned above at the same concentrations. Switch cells into the KSR/N2 medium mixture for 24hrs.
Day 8 (Figure 2B): Add the four differentiation factors mentioned above at the same concentrations into N2 medium. Switch cells into the N2 medium mixture for 24hrs.
Days 9–12 (Figure 2B): Add B27 and 10 μM DAPT to N2 medium. Switch neurons into the N2/B27/DAPT medium. Prepare and change medium daily. By day 12, >80% of cells will become NKX2.1+ neuron progenitors (Figure 2C). These cells also express other progenitor markers including SOX1, MASH1, but not FOXG1 and PAX6, which are markers for dorsal forebrain neuron progenitors (Figure 2C).
Basic Protocol 3. Neuron differentiation and maturation from Nkx2.1+ progenitors
In this protocol, the aim is to create functional neurons from previously generated Nkx2.1+ progenitors (Figure 3A). The extracellular matrix—poly-L-ornithine and laminin are used to enhance the attachment and differentiation of neuron progenitors. At early stages, the Notch inhibitor—DAPT—is used to inhibit the proliferation of progenitor cells and promote further neuron differentiation(Crawford and Roelink, 2007; Nelson et al., 2007). The neurotrophic factor, BDNF, is introduced following DAPT treatment to improve the survival, differentiation, and maturation of these neurons (Figure 3A).
Figure 3. Differentiation of hypothalamic ARC-like neurons from NKX2.1+ progenitors.
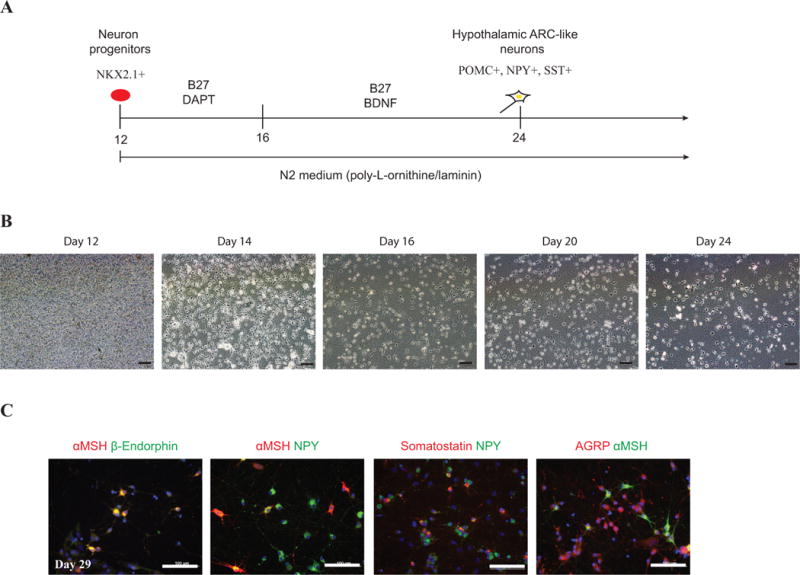
(A) Schematic of the differentiation protocol for generating hypothalamic ARC-like neurons from day 12 neuronal progenitors; (B) Live cell imaging during neuron differentiation on days 12, 14, 16, 20 and 24 on Poly-L-Ornithine and laminin coated plate; Scale bar, 100 μm; (C) Immunocytochemistry of 29 days of differentiated neurons. Neurons were stained with αMSH, β-Endorphin, NPY, Somatostatin and AGRP. Scale bar represents, 100 μm.
Materials
0.01% Poly-L-Ornithine
Mouse natural laminin
N2 medium (see Reagents and Solutions)
TrypLE express enzyme
Rock inhibitor (Y-27632)
B27
DAPT
BDNF
6-well plate, 12-well plate, 24-well plate
5ml Falcon tube with cell-strainer cap (35μm nylon mesh)
0.4% Trypan blue
Cell counting chamber slides
Countless automated cell counter
Centrifuge
Inverted microscope
37°C water bath
- Harvest differentiated progenitor cells at day 12:
-
Treat 12-day differentiated cells with trypLE (0.5 ml/well to 6-well plate) at 37 °C in the incubator for 4 min.Insufficient incubation with TrypLE (cells are still attached to the plate) makes it hard to detach all cells from the plate. Excessive incubation with TrypLE (e.g. > 10 min) may cause low viability and dramatic cell loss after switching into N2 medium supplemented with B27 and DAPT.
- Add 3 ml N2 medium to each well and use 5ml pipet to detach all cells by pipetting up and down.
- Combine all cells from each cell line into a 15 ml tube.
- Spin down cells at 800 rpm for 4 min at room temperature.
- Aspirate the supernatant and wash cell pellet again with N2 medium.
- Resuspend cell pellet in 2–3 ml N2 medium plus B27 and 10 μM Rock inhibitor for cells collected from 2–3 wells.
- Use P1000 Gilson pipet to disaggregate cell clusters completely by gently pipeting up and down a minimum of 10 times.
- Use P1000 Gilson pipet to pass cell suspension through falcon tube with cell-strainer cap to get rid of cell clusters.
- Mix 10 μl cell suspension with 10 μl Trypan blue and add 10 μl cell mixture to cell counting chamber slide.
-
Count cells and save the number for the next step.Note: These day12 neuron progenitors can be cryopreserved and stored in liquid nitrogen for a long time period (see Support protocol 1).
-
- Set up neuron differentiation:
- Preparing poly-L-ornithine and laminin coated plates: 2 days before setting up the neuron differentiation experiment, add 0.002% (w/v) poly-L-ornithine (dilute 0.01% poly-L-ornithine solution 5× with distilled water) to cover the bottom of each well and incubate at 37°C for overnight. On the second day, aspirate poly-L-ornithine. Wash the plates with sterile distilled water for 4–5 times. Then add 4 μg /ml laminin (prepare in distilled H2O) to each well and incubate the plates at 37°C for overnight. Aspirate laminin solution, the plates are ready to use.
- After cell counting, calculate and apply indicated number of neuron progenitors with N2 medium plus B27 and 10 μM Rock inhibitor to the poly-L-ornithine and laminin coated plate: 150,000 cells /well for 24-well plate, 300,000 cells/well for 12-well plate, 1 million cells/well for 6-well plate.
- Incubate the plates at 37°C, 5% CO2 in the incubator for 2 to 4 hrs.
-
When cells completely attach to the plate, switch cells into N2 medium plus B27 and 10 μM DAPT (Figure 3B).To reduce cell loss, make sure that cells are attached before switching to the new medium. This step is critical for minimizing the number of non-neuronal cells in the culture.
-
Days 13–16 (Figure 3B): Keep cells in N2 medium plus B27 and 10 μM DAPT. Perform medium change daily. There are 50%–70% cell death happened after 4 days of DAPT treatment.
If there is excessive cell debris in the suspension, changing medium daily will help to remove dead cells and improve survival of differentiated neurons.
On day 17, switch cells into N2 medium supplemented with B27, 20 ng/ml BDNF and keep them in this neuron differentiation medium for another 2 to 3 weeks. Change medium every two days until observing fully differentiated neurons. Neurite formation can be seen since day 14, as shown in Figure 3B.
Support Protocol 1. Cryopreservation of Day 12 neuron progenitors
Day 12 neuron progenitors can be frozen down for long-term storage (Basic protocol 3–1), which greatly shortens the time of the differentiation procedure and enables the sharing of these cells with others. Here we first freeze down progenitor cells in freezing medium in Mr. Frosty (filled with isopropanol) at −80 °C for overnight. Then we transfer the frozen vials into liquid nitrogen for long-term storage.
Materials
2 ml Cryo tubes
Cell freezing medium
Mr. Frosty container
−80C freezer
Liquid nitrogen tank
After counting, spin down neuron progenitors at 800 rpm for 4min at room temperature.
Aspirate supernatant. For 5 million cells, add 1.5ml cell freezing medium and transfer to 2ml cryo tube.
Put the cryo tubes in freezing container and store at −80C freezer for overnight.
-
Transfer the frozen cells to liquid nitrogen tank for long-term storage.
These hES/iPS cells-derived hypothalamic progenitors can be cryopreserved for long-term storage (>11 months). The viability of the frozen progenitors after thawing is around 95%. These thawed cells performed identically to non-frozen cells as described previously (Wang et al., 2015).
Support Protocol 2. Thawing frozen Day 12 neuron progenitors
Here we describe techniques for thawing these cells without affecting their viability and differentiation efficiency. Rock inhibitor is used to improve the survival of neuron progenitors after thawing.
Materials
N2 medium
B27
Y-27632 (Rock inhibitor)
Poly-L-ornithine/laminin
Sterile distilled water
6-well/12-well/24-well/4-well cultured plates (Thermoscientific)
15ml falcon tube
Centrifuge
Prepare Poly-L-Ornithine and laminin coated plates as mentioned previously, (Basic Protocol 3, step 2a). 2 hours later, the plates are ready to use.
Thaw frozen 12 days neuron progenitors in water bath.
Transfer all cells into 15 ml falcon tube containing 5 ml N2 medium.
Spin down cells at 800 rpm for 4 min, wash again with 3 ml N2 medium.
Spin down cells at 800 rpm for 4 min. Aspirate supernatant, add 2–3 ml N2 medium plus B27 and 10 μM Rock inhibitor to resuspend cell pellet.
Count and apply desired number of progenitor cells to each well after aspirating laminin for further neuron differentiation.
Reagents and Solutions
The supplier information for all reagents and solutions below are provided in Appendix Table 1 and Appendix Table 2. The information of antibodies is described earlier (Wang et al., 2015).
FGF-basic 0.1 mg/ml stock
Centrifuge briefly and then reconstitute 100 μg bFGF with 1ml distilled water, to yield a stock solution of 0.1 mg/ml of bFGF. Make 65 μl aliquots and store at −80 °C. Stock solutions are stable for 6 months at −80°C.
SB 431542 10 mM stock
Reconstitute 10 mg SB 431542 with 2.6 ml of dimethyl sulfoxide (DMSO). Filter reconstituted SB solution with Millex filter unit (0.22 μm pore size, Millipore). Then make 50 μl aliquots and store at −20 °C. Stock solutions are stable for 6 months at −20°C.
LDN-193189 2.5 mM stock
Reconstitute 10 mg LDN-193189 with 9.04 ml of DMSO. Filter reconstituted LDN solution with Millex filter unit. Then make 50 μl aliquots and store at −20 °C. Stock solutions are stable for 6 months at −20°C.
Purmorphamine 5 mM stock
Reconstitute 5 mg purmorphamine with 1.92 ml of DMSO. Filter reconstituted purmorphamine solution with Millex filter unit. Then make 20 μl aliquots and store at −20 °C. Stock solutions are stable for 6 months at −20°C.
SHH 100 μg/ml stock
Reconstitute 500 μg with 5 ml PBS plus 0.1% BSA (w/v) to make 100 μg/ml solution. Filter reconstituted SHH solution with Millex filter unit. Then make 50 μl aliquots and store at −80 °C. Stock solutions are stable for 3 months at −80°C.
DAPT 10 mM stock
Reconstitute 10 mg DAPT by adding 2.32 ml of DMSO to the entire vial. Filter reconstituted DAPT solution with Millex filter unit. Then make 50 μl aliquots and store at −20 °C. Stock solutions are stable for 6 months at −20°C.
Ascorbic Acid 0.4 N stock
Dissolve 0.7 g L-Ascorbic acid in 10 ml H2O to make 0.4 N stock solution. Filter reconstituted ascorbic acid solution with Millex filter unit. Make 275 ul aliquots and store at −20 °C. Stock solutions are stable for 6 months at −20°C.
BDNF 40 μg/ml stock
Reconstitute 250 μg BDNF with PBS plus 0.1% BSA to make 40 ug/ml stock solution. Filter with Millex filter unit. Make 25 μl aliquots and store at −80 °C. Stock solutions are stable for 3 months at −80°C.
hES medium
500 ml knockout DMEM
90 ml knockout serum
6.5 ml GlutaMAX
6.5 ml NEAA
6.5 ml penicillin/streptomycin
0.65 ml 2–mercaptoethanol
10 ng/ml bFGF
KSR medium
500 ml knockout DMEM
90 ml knockout serum
6.5 ml GlutaMAX
6.5 ml NEAA
6.5 ml penicillin/streptomycin
0.65 ml β–mercaptoethanol
N2 medium
500 ml DMEM/F12
5.5 ml GlutaMAX
5.5 ml NEAA
5.5 ml penicillin/streptomycin
5.5 ml N2 supplement
5.5 ml 16% (w/v%) D-Glucose
0.2 mM Ascorbic Acid
Cell freezing medium
108 ml FBS
72 ml DMEM
20 ml DMSO
COMMENTARY
Background Information
Hypothalamic-like neurons have been created from mouse embryonic stem cells in a growth factor-free 3D culture system (Wataya et al., 2008), but the generation of human hypothalamic neurons has been more challenging (Merkle et al., 2015; Wataya et al., 2008). Here we describe a protocol for efficient generation of human hypothalamic neurons based on the limited knowledge of hypothalamic development.
Though the precise regulation of hypothalamic development is poorly understood, a growing number of transcription factors and other molecular signals have been identified. Inhibition of TGFβ and BMP signaling in ES cells promotes efficient conversion of hPS cells into Tuj1+ and MAP2+ neurons (Chambers et al., 2009). The hypothalamus develops from the ventral diencephalon where NKX2.1 is highly expressed and required for its development (Maroof et al., 2013). Activation of SHH signaling induces the expression of NKX2.1 in differentiated ES cells in a dose-dependent manner (Liu et al., 2013). Moreover, SHH and Wnt signals regulate the dorsal-ventral specification in neural tube patterning, in which SHH induces ventral neuron development and Wnt induces dorsal neuron development (Marcelle et al., 1997). Inhibiting Notch cofactor in Nkx2.1+ cells increases the number of ARC neurons in mice while expressing constitutively active Notch has the opposite effect (Aujla et al., 2013). The protocol presented here was created and modified based on these previous studies, and we have demonstrated high efficiency of generating hypothalamic ARC-like neurons from hES and iPS cells (Wang et al., 2015). Within 30 days, we can achieve over 90% efficiency in generating hypothalamic ARC-like cell types from hES/iPS cells. Further functional studies suggest these stem cell-derived human hypothalamic cells can produce and secrete neuropeptides, and respond appropriately to metabolic signals such as insulin and leptin. Taken together, the hypothalamic neurons generated with this protocol are suitable for investigating the molecular physiology of hypothalamic processes affecting body weight homeostasis.
Critical Parameters
Stem Cell Quality
Depending on the method of stem cell generation, stem cells can be differentiation resistant, and retain Oct4 expression(Koyanagi-Aoi et al., 2013). Basic quality control of stem cell lines, including ability to silence Oct4 expression is required. Furthermore, different hES/iPS cell lines can have different propensity in the differentiation to specific cell types (Bock et al., 2011). Though we find that most cell lines can be used for hypothalamic differentiation, the use of a stem cell line with particularly efficient differentiation to a neuronal lineage will facilitate studies. Our laboratory is able to provide stem cell lines tested for hypothalamic differentiation. These cell lines can be requested under https://www.cellhub.org/cell-line-information.
Cell Density
Cell density at the time point when neuron differentiation is started is also critical in this protocol. Low cell density may lead to inefficient generation of NKX2.1 neuron progenitors and contamination of undifferentiated stem cells at day 12 that will take over the whole well in later neuron culture. If cells remain 100% confluent for too long before initiating differentiation, it will also affect the differentiation efficiency and cause increased cell death during and after the first 12 days of differentiation. Ideally, start the neuron differentiation immediately once cells reach 95%–100% confluency on matrigel plate.
Fresh differentiation factors
Improper preparation or storage of chemicals and morphogens may cause failed differentiation.
Troubleshooting
Although some troubleshooting has been incorporated into the protocols and mentioned above, here are some additional issues and potential solutions.
No neuron formation after seeding day 12 cells on PO/LA plate
There are many potential causes, as mentioned above, such as some hES/iPS cell lines with low neuron differentiation propensity, low cell density at the start neuron differentiation, inactive reagents. One approach is to include the NKX2.1 GFP/W-hES line or control iPS cell line as a positive control in the experiment to track the differentiation procedure dynamically based on the GFP expression.
A few undifferentiated stem cells spread over the well in late neuron differentiation
Contamination with undifferentiated stem cell in neuron cultures also indicates low efficiency of neuron induction during the first 12 days. These stem cells can survive and proliferate in the N2 medium plus B27 and BDNF, which will further expand and cover the whole dish in culture. Low cell density when initiating neuron differentiation, or inefficient differentiation (inactive reagents) can lead to this issue. Increasing cell density (95–100%) before start neuron differentiation and/or using freshly prepared reagents will help to solve this problem.
Excessive cell debris exist in neuron cultures
After seeding neuron progenitors on poly-L-ornithine and laminin coated plates, treatment with DAPT will not only promote further neuron differentiation but also induce death of neuronal stem cells. Therefore change medium daily from day 13 to day 16 to remove as many dead cells as possible. High cell density when set up neuron differentiation on poly-L-ornithine/laminin coated plates may also lead to more cell death in later neuron cultures. Excessive cell death after DAPT treatment may be noted. An approach is to minimize the duration of DAPT treatment and switch neurons into N2 medium with B27 and BDNF right away.
Dying of neurons at 40 or more days
For neurons cultured beyond 40 days in N2 medium plus B27 and BDNF, surfaces of neuronal cell bodies and neurites are no longer smooth. These neurons are dying gradually. This is the common problem for in vitro monolayer-cultured neurons. To reduce this problem, co-culture with mouse astrocytes should be tried to prolong cell culture. Primary mouse cortical astrocytes were prepared as previous described (Albuquerque et al., 2009). Instead of plating 12 days hypothalamic neuron progenitors directly onto poly-L-ornithine and laminin coated plate, first add isolated mouse astrocytes onto poly-L-lysine coated plate and add neuron precursors on top once the mouse astrocyte culture reaches 100% confluence and stop dividing. The subsequent differentiation procedure is the same as the monolayer culture. In the presence of mouse astrocytes, these neurons can be cultured in vitro for at least 53 days.
Anticipated Results
Over 80% of cells will be NKX2.1+ progenitors by day 12 (Figure 2C). From each well of 6-well plate, around 4–6 million progenitors can be obtained. After 24 days, more than 90% of live cells become hypothalamic ARC-like neurons. Among them, around 20–40% cells will be POMC+ neurons, 40–60% cells will be NPY+ neurons, 10–30% cells will be somatostatin+ neurons (Figure 3C). At day 24, most POMC neurons are also NPY positive. As time proceeds, the percentage of dual POMC+NPY+ neurons decreases while the percentage of POMC monopeptide expressing cell increases, reflecting the maturation of these neurons (Wang et al., 2015). These neurons can produce and secrete large amount of POMC-derived neuropeptides and respond to metabolic signals.
Time Consideration
It takes an average of 12 days to make hypothalamic progenitors from hES/iPS cells. Another 2–4 weeks are required to obtain differentiated hypothalamic ARC-like neurons. Because the neuron progenitors can be frozen down at day 12, it will only take 2 weeks to generate hypothalamic neurons when starting from the neuron progenitor stage. This greatly shortens the time to make functional hypothalamic neurons.
Acknowledgments
This work is supported by RO1 DK52431-20, the Rudin Foundation, the Foundation for Prader-Willi Research, the Diabetes (P30 DK63608-12) and Obesity (P30 DK26687-33) Research Center at Columbia University, a NYSTEM IIRP award #C026184, the Berrie Foundation Program in Cellular Therapies of Diabetes, and the New York Stem Cell Foundation.
Appendix Table 1.
Chemicals.
| Name | Catalog No. | Supplier | Storage condition for stock solution |
|---|---|---|---|
| DMSO | D2650 | Sigma | Room Temperature |
| SB 431542 | 04-0010-10 | Stemgent | −20 °C |
| LDN-193189 | S2618 | Selleckchem | −20 °C |
| Y-27632 | S1049 | Selleckchem | −20 °C |
| DAPT | 2634 | TOCRIS | −20 °C |
| Purmorphamine | 04-0009 | Stemgent | −20 °C |
| SHH | 1845-SH | R&D | −80 °C |
| BDNF | 248-BD | R&D | −80 °C |
| D-Glucose | G8270 | Sigma | 4 °C |
| DPBS | 14190-250 | ThermoFisher | Room Temperature |
| Distilled water | 15230-204 | ThermoFisher | Room Temperature |
| Ascorbic Acid | A-5960 | Sigma | −20 °C |
| 7.5% BSA | 15260-037 | ThermoFisher | 4 °C |
| Poly-L-ornithine | P4957 | Sigma | 4 °C |
| Natural mouse laminin | 23017-015 | ThermoFisher | −80 °C |
Appendix Table 2.
Tissue culture reagents
| Name | Catalog No. | Supplier | Store condition for stock solution |
|---|---|---|---|
| DMEM | 11965-092 | Thermofisher | 4 °C |
| DMEM/F12 | 11320-082 | Thermofisher | 4 °C |
| Fetal Bovine Serum | 16000-044 | Thermofisher | −80 °C |
| Knockout DMEM | A12861-01 | Thermofisher | 4 °C |
| Knockout Serum Replacement | 10828-028 | Thermofisher | −80 °C |
| mTeSR1 | 05850 | Stemcell Technologies | −20 °C |
| Matrigel | 354277 | BD Biosciences | −80 °C |
| FGF-basic | PHG-0021 | Thermofisher | −80 °C |
| Penicillin-Streptomycin (10,000 U/mL) | 15140-163 | Thermofisher | 4 °C |
| NEAA | 11140-050 | Thermofisher | 4 °C |
| Glutamax | 35050-061 | Thermofisher | 4 °C |
| 2-mecaptoethanol | 21985-023 | Thermofisher | 4 °C |
| B27 | 12587-010 | Thermofisher | −20 °C |
| N2-supplement | 17502-048 | Thermofisher | −20 °C |
| TrypLE Express Enzyme | 12605-036 | Thermofisher | Room Temperature |
| Trypan Blue Stain (0.4%) | T10282 | Thermofisher | Room Temperature |
| 0.1% Gelatin | 07903 | Stemcell Technologies | Room Temperature |
Literature Cited
- Albuquerque C, Joseph DJ, Choudhury P, MacDermott AB. Dissection, plating, and maintenance of cortical astrocyte cultures. Cold Spring Harb Protoc. 2009;2009 doi: 10.1101/pdb.prot5273. pdb prot5273. [DOI] [PubMed] [Google Scholar]
- Aujla PK, Naratadam GT, Xu L, Raetzman LT. Notch/Rbpjkappa signaling regulates progenitor maintenance and differentiation of hypothalamic arcuate neurons. Development. 2013;140:3511–3521. doi: 10.1242/dev.098681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsh GS, Schwartz MW. Genetic approaches to studying energy balance: perception and integration. Nature reviews Genetics. 2002;3:589–600. doi: 10.1038/nrg862. [DOI] [PubMed] [Google Scholar]
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature biotechnology. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Qi Y, Mica Y, Lee G, Zhang XJ, Niu L, Bilsland J, Cao L, Stevens E, Whiting P, et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nature biotechnology. 2012;30:715–720. doi: 10.1038/nbt.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford TQ, Roelink H. The notch response inhibitor DAPT enhances neuronal differentiation in embryonic stem cell-derived embryoid bodies independently of sonic hedgehog signaling. Developmental dynamics: an official publication of the American Association of Anatomists. 2007;236:886–892. doi: 10.1002/dvdy.21083. [DOI] [PubMed] [Google Scholar]
- Goulburn AL, Alden D, Davis RP, Micallef SJ, Ng ES, Yu QC, Lim SM, Soh CL, Elliott DA, Hatzistavrou T, et al. A targeted NKX2.1 human embryonic stem cell reporter line enables identification of human basal forebrain derivatives. Stem cells. 2011;29:462–473. doi: 10.1002/stem.587. [DOI] [PubMed] [Google Scholar]
- Kime C, Rand TA, Ivey KN, Srivastava D, Yamanaka S, Tomoda K. Practical Integration-Free Episomal Methods for Generating Human Induced Pluripotent Stem Cells. Current protocols in human genetics/editorial board, Jonathan L Haines [et al] 2015;87:21 22 21–21 22 21. doi: 10.1002/0471142905.hg2102s87. [DOI] [PubMed] [Google Scholar]
- Koyanagi-Aoi M, Ohnuki M, Takahashi K, Okita K, Noma H, Sawamura Y, Teramoto I, Narita M, Sato Y, Ichisaka T, et al. Differentiation-defective phenotypes revealed by large-scale analyses of human pluripotent stem cells. Proc Natl Acad Sci U S A. 2013;110:20569–20574. doi: 10.1073/pnas.1319061110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Weick JP, Liu H, Krencik R, Zhang X, Ma L, Zhou GM, Ayala M, Zhang SC. Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nature biotechnology. 2013;31:440–447. doi: 10.1038/nbt.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallon BS, Park KY, Chen KG, Hamilton RS, McKay RD. Toward xeno-free culture of human embryonic stem cells. Int J Biochem Cell Biol. 2006;38:1063–1075. doi: 10.1016/j.biocel.2005.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelle C, Stark MR, Bronner-Fraser M. Coordinate actions of BMPs, Wnts, Shh and noggin mediate patterning of the dorsal somite. Development. 1997;124:3955–3963. doi: 10.1242/dev.124.20.3955. [DOI] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying SW, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell stem cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkle FT, Maroof A, Wataya T, Sasai Y, Studer L, Eggan K, Schier AF. Generation of neuropeptidergic hypothalamic neurons from human pluripotent stem cells. Development. 2015;142:633–643. doi: 10.1242/dev.117978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BR, Hartman BH, Georgi SA, Lan MS, Reh TA. Transient inactivation of Notch signaling synchronizes differentiation of neural progenitor cells. Developmental biology. 2007;304:479–498. doi: 10.1016/j.ydbio.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- van der Klaauw AA, Farooqi IS. The hunger genes: pathways to obesity. Cell. 2015;161:119–132. doi: 10.1016/j.cell.2015.03.008. [DOI] [PubMed] [Google Scholar]
- Wang L, Meece K, Williams DJ, Lo KA, Zimmer M, Heinrich G, Martin Carli J, Leduc CA, Sun L, Zeltser LM, et al. Differentiation of hypothalamic-like neurons from human pluripotent stem cells. The Journal of clinical investigation. 2015 doi: 10.1172/JCI79220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wataya T, Ando S, Muguruma K, Ikeda H, Watanabe K, Eiraku M, Kawada M, Takahashi J, Hashimoto N, Sasai Y. Minimization of exogenous signals in ES cell culture induces rostral hypothalamic differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11796–11801. doi: 10.1073/pnas.0803078105. [DOI] [PMC free article] [PubMed] [Google Scholar]