

Abstract
The periodic emergence of new infectious agents and the genetic and antigenic evolution of existing agents necessitate the improvement of technology for the rapid development of diagnostic assays. The porcine epidemic diarrhea virus (PEDV) emerged in the United States in 2013, causing severe economic damage to the pork industry. The primary goal of this study was to develop methods to reduce the lead time for serological assay development. An approach involving the computational prediction of diagnostic targets, followed by a rapid synthesis of antigens, was adopted to achieve this objective. To avoid cross-reactivity with other closely related swine coronaviruses, the N protein sequences of PEDV were analyzed to identify sequences unique to PEDV. The potential antigenicity of the identified sequence was predicted computationally using the Jameson-Wolf method. A sequence with a high antigenic index was rapidly synthesized using an in vitro transcription and translation system to yield the diagnostic antigen. The computationally designed enzyme-linked immunosorbent assay (ELISA) was validated using 169 field sera, whose statuses were determined by a PEDV-specific immunofluorescence assay. Comparison of the computationally designed ELISA to a conventionally developed ELISA, using bacterially expressed N protein, and to the immunofluorescence assay showed a high degree of agreement among the three tests (mean kappa statistic, 0.842). The sensitivity and specificity, compared to the conventionally developed assay, were 90.62 and 95.18, respectively. Therefore, the described approach is useful in reducing the development time for serological assays in the face of an infectious disease outbreak.
INTRODUCTION
The number of newly emerging infectious diseases which affect swine has increased rapidly in the past 2 decades. Several economically important diseases, like the porcine reproductive and respiratory disease syndrome virus (PRRSV), porcine circovirus strain 2 (PCV2), and porcine coronaviruses, have emerged to cause severe production losses. In addition, the genetic instability of existing viruses, such as swine influenza viruses (SIV) and rotaviruses, result in the periodic evolution of new variants which may be antigenically distinct and consequently require the periodic updating of diagnostic tests. The possibility of the introduction of foreign animal diseases, such as African swine fever and foot mouth disease, is also a continual threat to the industry (1). A preparedness plan for such diseases would be incomplete without the technology for the rapid development of vaccines and diagnostic tests. Hence, the quick development and availability of diagnostic tests are central to the effective prevention of the initial spread and for the subsequent surveillance of emerging infectious diseases.
Serological diagnostic assays, especially enzyme-linked immunosorbent assays (ELISAs), are widely deployed in veterinary and human medicine. Compared to molecular detection methods, ELISAs have the advantages of being cost-effective and able to detect prior exposure in the absence of active infection. Traditional virus neutralization assays are useful in quantifying protective antibody responses and can be set up relatively quickly, once the virus culture protocols are established. Traditional technology for the development of serological assays for viruses necessitates the production of diagnostic targets by recombinant DNA technology and protein production, a process which is tedious and requires a lead development time of a few months (2), especially in the case of a newly identified agent. Similarly, first-generation methods for developing serological assays involve culturing the agent and purifying it by physical means to use the whole particle as the target antigen. The disadvantage of using the whole complement of viral proteins as the capture antigen is that this approach usually generates a high background and possible cross-reactivity, thus compromising specificity and sensitivity of the assay. Control measures instituted within the first few weeks after the emergence of a new infectious disease are critical for preventing the spread of infection. With the increasing incidence of newly emerging diseases, the availability of technology which can reduce the lead development time for serological assays will be important for the rational institution of control measures in the early stages of an outbreak.
As an example of the above-described scenario, the porcine epidemic diarrhea virus (PEDV), which was previously widely prevalent in Asian and European countries, emerged in the United States in May 2013. Porcine epidemic diarrhea is characterized by acute diarrhea and vomiting in neonatal piglets, with mortality rates of about 90% (3). Within months of the first detection, the virus spread rapidly across the nation, resulting in the culling of roughly 7 million piglets, or 10% of the total population (4). The diagnosis of early cases was complicated by the fact that the diarrhea observed in clinical cases of PEDV is similar to the clinical signs of the transmissible gastroenteritis virus (TGEV) and swine rotaviruses. Early virus isolation and culture and the development of an immunofluorescence assay (IFA) were complicated by the strict trypsin requirements for viral growth. Trypsin is easily inactivated by fetal bovine serum (FBS) used for cell culture. In addition, it may degrade at 37°C and have to be replenished periodically to ensure availability, thus increasing the laboriousness of the assay. The subsequent development of ELISAs based on recombinant nucleocapsid (N) or spike (S) proteins were accomplished some months after the first index case of PEDV was detected (5, 6).
Hence, in this study, we have used PEDV as a model for an emerging infectious disease to develop computationally directed methods which can significantly reduce the lead development time for serological assays, enabling a more rapid response in the face of an outbreak. We also present data to validate the performance of the computationally designed (CD) assay in comparison to an assay developed by conventional methods. Therefore, this study provides proof of concept for the potential advantages of an in silico design for diagnostic antigens and for advancing methods of the rapid development of serological tools.
MATERIALS AND METHODS
Samples for assay development and validation.
A total of 169 field serum samples from PEDV-suspect and -nonsuspect cases which were submitted to the South Dakota State Animal Disease Research and Veterinary Diagnostic Laboratory were used for assay development. The selection of samples was random, and identify of the samples was blinded. All experimentation was carried out in compliance with the institutional policies of the North Dakota State University and South Dakota State University and their institutional biosafety committees. A positive-control serum sample for PEDV was purchased from the National Veterinary Services Laboratory (NVSL) (Ames, IA). Positive-control serum samples for TGEV, porcine respiratory coronavirus (PRCV), and the porcine delta coronavirus (PDCoV), collected from experimentally infected pigs (7–9), were provided by Linda Saif (Ohio State University).
Virus culture.
Vero 76 cells (ATCC CRL 1586) were grown to 70% confluence overnight in Dulbecco's modified Eagle medium (DMEM) (Corning, Manassas, VA, USA) with 10% FBS (Atlanta Biologicals, Flowery Branch, GA, USA) and 100 units/ml of penicillin, and 100 μg/ml of streptomycin (Corning, Manassas, VA, USA). Before infection with PEDV, the cells were washed twice with Hanks' balanced salt solution (GE Healthcare Cell Culture, Logan, UT). Virus (PEDV CO 2013; NVSL, Ames, IA) was added at an index of multiplicity (MOI) of 0.1 in infection media (DMEM) (GE Healthcare Cell Culture, Logan, UT), 10 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (product T1426; Sigma-Aldrich, St. Louis, MO), 7% tryptose phosphate broth (Gibco BRL, Grand Island, NY), 100 units/ml of penicillin, and 100 μg/ml of streptomycin (GE Healthcare Cell Culture, Logan, UT). After incubation in a 5% CO2 incubator at 37°C for 2 h, the supernatant was replaced with fresh infection media. The virus culture was harvested when 80% cytopathic effect was observed, in about 48 h, and stored at −80°C until further use.
Immunofluorescence assay.
Vero cells were seeded in 96-well tissue culture plates (ThermoScientific, Waltham, MA) and grown to confluence over 3 to 4 days. Cells were washed three times with serum-free medium containing 2.5 μg/ml of TPCK-treated trypsin (product T1426; Sigma-Aldrich, St. Louis, MO). Cells were infected with 150 μl of the PEDV virus culture, and the titer was adjusted to 103 to 104 50% tissue culture infective dose in infection media. Twenty-four hours after infection, cells were washed with phosphate-buffered saline with Tween (Boston Bioproducts, Ashland, MA) and fixed with prechilled acetone and methanol (1:1) at 4°C for 1 h. Fixed cells were incubated with test serum samples at a 1:40 for 1 h at 37°C and then with fluorescein isothiocyanate-labeled anti-swine IgG (KPL, Gaithersburg, MD, USA) antibody at a 1:100 dilution at 37°C for 1 h. Positive and negative controls were included in each plate. Finally, the cells were observed with a fluorescence microscope for characteristic cytoplasmic fluorescence. All of the 169 field serum samples used for the assay optimization were tested by the indirect fluorescent antibody and classified as positive or negative.
Bacterial expression and purification of the PEDV nucleocapsid protein.
To develop a conventional ELISA for comparison with the CD N protein (CD-NP) ELISA, a stretch of 283 amino acids comprising the C-terminal end of the PEDV nucleocapside protein (NP) was expressed in Escherichia coli. Genomic RNA from the PEDV culture was extracted by an RNeasy minikit (Qiagen, Valencia, CA, USA), as per the manufacturer's instructions. The iScript cDNA synthesis kit (Bio-Rad, Richmond, CA, USA) was used to generate cDNA, following the manufacturer's instructions. Primers 5′-GCTAGTCGACGGTCCAGATCTCCAAGTAA CAACAGAG-3′ and 5′-TGACCTCGAGATTTCCTGTGTCGAAGATCTCGTTG-3′ were used for amplification of the target DNA. The amplified DNA was directionally cloned into the pET-28a (Novagen, Madison, WI) plasmid for bacterial expression, using the XhoI and SalI enzymes (New England Biologicals, Ipswich, MA). Following the verification of successful cloning by restriction digestion and sequencing, E. coli BL21(DE3) cells (Invitrogen, Carlsbad, CA) were transformed with the plasmids. Transformed cultures were induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (Sigma-Aldrich, St. Louis, MO), for 4 h at 37°C. The soluble protein fraction was extracted with BugBuster protein extraction reagent (EMD Millipore, Billerica, MA) and purified by affinity purification (His-spin protein miniprep purification kit; Zymo Research, Orange, CA), according to the manufacturer's protocols. The purified protein was assessed by a Western blot, as described below.
Computational design of targets for the rapid ELISA.
To eliminate possible cross-reactivity to the other closely related swine coronaviruses, namely, TGEV and PRCV, approximately 50 N protein amino acid sequences for the three viruses were downloaded from GenBank and aligned using Clustal W (MegAlign Pro, LaserGene core suite 9.0; DNAStar, Inc., Madison, WI). Stretches of conserved amino acid sequence, which were unique to the PEDV N protein, were then analyzed for predicted antigenicity using the Protean tool of the LaserGene core suite 9.0 (DNAStar, Inc., Madison, WI). The Jameson-Wolf algorithm (10), which combines secondary structure information with backbone flexibility to predict surface accessibility, was used to determine the predicted antigenic index, with a threshold value of 1.7. The ABCpred B-cell epitope prediction server (11) and the Immune Epitope Database Bepipred B-cell epitope prediction tool were used to predict linear B-cell epitopes in the target region.
Expression of the computationally designed target by in vitro transcription and translation.
Genomic RNA from the PEDV culture was extracted by an RNeasy minikit (Qiagen, Valencia, CA, USA), as per the manufacturer's instructions. The iScript cDNA synthesis kit (Bio-Rad, Richmond, CA, USA) was used to generate cDNA, following the manufacturer's instructions. Primers for amplification of the nucleoprotein target were designed to contain a T7 promoter site and His tag in the forward primer, while the reverse primer included a polyA termination site. The sequences of the primers were 5′-GGATCCTAATACGACTCACTATAGGGAACAGC CACCATGCGTGCAAATTCACGTAGCAG-3′ and 5′-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTAATTGTTATTATTATTGCCTCCTCTGTTCTG-3′ (accession number KF267450, positions 1:26430 to 26473). After PCR amplification of the DNA fragment coding for the unique amino acid sequence in the NP, the TNT T7 quick-coupled transcription/translation system (Promega, Madison, WI, USA) was used to rapidly generate antigen for the ELISA, according to the manufacturer's instructions. Briefly, 1 μg of the purified DNA, 1 μl of 1 mM methionine, and 1 μl of PCR enhancer were added to the reaction mix. The mixture was incubated at 30°C for 90 min. The identity of the expressed products was verified by a Western blot using a commercial anti-His antibody (Invitrogen, Carlsbad, CA), anti-PEDV hyperimmune serum (NVSL, Ames, IA), and anti-swine PRCV,TGEV, and PDCoV sera, as described below.
Western blotting.
The NPs obtained by bacterial expression and in vitro transcription and translation were loaded on 12% or 16% SDS-polyacrylamide gels, respectively. After electrophoresis for 90 min at 80 V, the protein was transferred to a polyvinylidene difluoride (PVDF) membrane (ThermoFisher, Grand Island, NY) by semidry electrotransfer. The blot was developed by standard procedures, with PEDV positive-control swine serum (NVSL, Ames, IA) (1:500), experimentally generated swine anti-TGEV, PRCV, and PDCoV sera (1:500), or a commercial anti-His antibody (Invitrogen, Carlsbad, CA) (1:5,000) as the primary antibody. A 1:1,000 dilution of horseradish peroxidase (HRP)-conjugated anti-swine or anti-mouse antibody (KPL, Gaithersburg, MD) was used for secondary detection. The bands were visualized by staining with 4-chloro-1-naphthol (ThermoFisher, Grand Island, NY), except the band shown in Figure 3A, which was developed with 3,3′,5,5′-tetramethylbenzidine (TMB; KPL, Gaithersburg, MD), according to the manufacturer's instructions.
FIG 3.
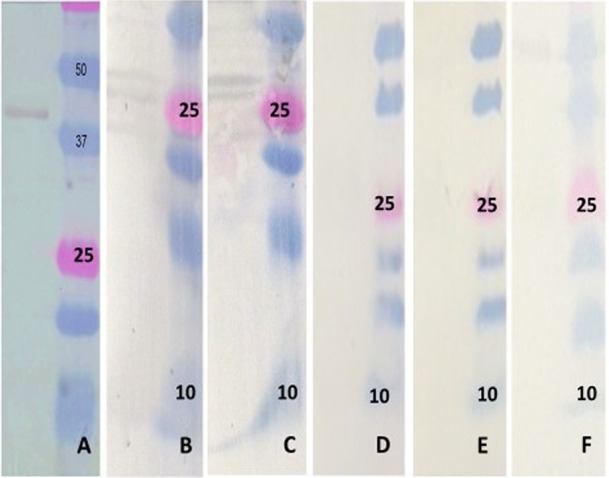
Western blot analysis of the ELISA targets. (A) The bacterially expressed PEDV NP probed with swine anti-PEDV serum, showing a band of approximately 38 kDa (left) and the protein ladder (right). (B) The computationally designed PEDV NP target probed with a commercial anti-His antibody (left) and the protein ladder (right). (C) The computationally designed PEDV NP target probed with swine anti-PEDV serum (left) and the protein ladder (right). (D) The computationally designed PEDV NP target probed with swine anti-TGEV serum (left) and the protein ladder (right). (E) The computationally designed PEDV NP target probed with swine anti-PRCV serum (left) and the protein ladder (right). (F) The computationally designed PEDV NP target probed with swine anti-PDCoV serum (left) and the protein ladder (right).
Optimization of the CD and conventional ELISAs.
The optimal concentration of the in vitro translated nucleoprotein for use as the capture antigen was determined by a checkerboard titration and was approximately 5 μg/ml. The antigen was diluted in coating buffer (0.05 M NaHCO3-Na2CO3 buffer, pH 9.6) and added in 50-μl volumes to each well of the ELISA plate (Costar High Bind Microplate; Corning, Corning, NY) and incubated overnight at room temperature. Plates were washed three times with Tris-buffered saline with Tween 20 (TBST) (Boston Bioproducts, Ashland, MA) and blocked with 100 μl of blocking buffer (4% BSA, 2% sheep serum, and 2% rabbit serum in TBST) for 2 h at 37°C. After three washes with TBST, 50 μl of test sera at a dilution of 1:100 in blocking buffer was added into each well and incubated at 37°C for 2 h. Anti-swine HRP-conjugated IgG (KPL, Gaithersburg, MD) was added at a 1:5,000 dilution to the plate and incubated for 1 h at 37°C. Following three washes with TBST, 50 μl of TMB substrate (KPL, Gaithersburg, MD) was added and incubated in the dark for 5 min. The reaction was stopped with 1 M HCl, and the plates were read at 450 nm with an ELISA reader (BioTek, Winooski, VT).
The conventional ELISA was also optimized essentially as described above, except that general block ELISA blocking buffer (Immunochemistry Technologies, Bloomington, MN) was used for blocking the plates.
All samples were assessed in duplicate in two independent assays for both the computationally designed assay and the conventional ELISA. Positive-control serum samples purchased from NVSL (Ames, IA) as well as serum samples from pigs infected with TGEV, PRCV, PDCoV, PCV2, PRRSV, and SIV were used as controls to assess possible cross-reactivity on both ELISAs. The final values for analysis were calculated as signal-positive sample ratios for each plate. Intraassay variation was assessed by testing 4 samples at 4 different times.
Statistical analysis.
Receiver operating characteristics analysis (12) was used to obtain cutoff values to distinguish between positive and negative samples. The assay sensitivities, specificities, and positive and negative predicted values were calculated at the selected cutoff values. The IFA was used as the gold standard for the conventional ELISA, while the CD-NP ELISA was assessed using either the IFA or the conventional ELISA as the gold standard. Agreement between the dichotomized ELISA and IFA values was assessed using the kappa coefficient association method. All statistical analysis was carried out using MedCalc software (MedCalc, Ostend, Belgium).
RESULTS
Immunofluorescence assay.
Assay validation of the IFA was carried out by a comparison of testing results for total of 100 samples from 5 cases at the South Dakota State University and Iowa State University veterinary diagnostic labs. Complete agreement was obtained for all but one sample. Of the 169 field samples tested by the IFA, 70 were classified as positive and 99, as negative. As expected, the PEDV positive-control serum showed clear cytoplasmic fluorescence typical of PEDV on the IFA, while the negative serum and the TGEV- and PRCV-specific sera did not (Fig. 1).
FIG 1.
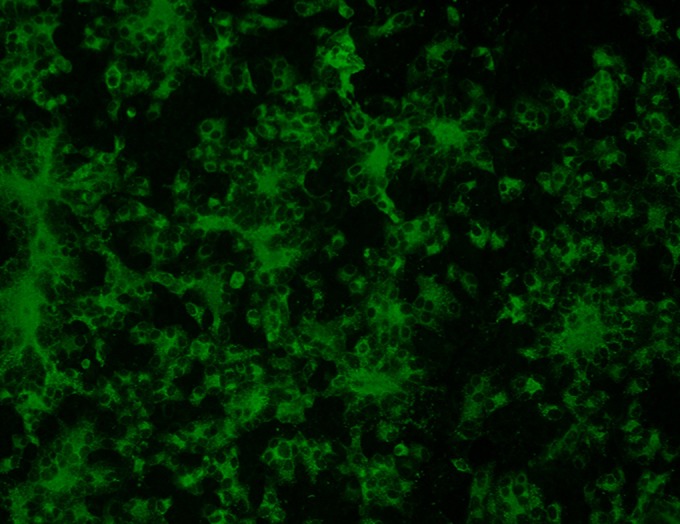
Immunoflorescence assay (IFA) of PEDV-infected cells. Representative image of Vero cells infected with PEDV and stained with a polyclonal swine anti-PEDV antibody. Apple-green cytoplasmic florescence and the formation of syncytia are indicative of viral growth. Uninfected cells did not show any florescence (image not shown).
Amino acid sequence analysis for the identification of unique regions in the PEDV N and S proteins.
Sequences of the S protein from PEDV, TGEV, and PRCV, when aligned with the Clustal W program, showed that an insertion was present from positions 23 to 228. While this sequence was completely absent in PRCV and PDCoV, a corresponding region was present in the TGEV spike protein. The percentage identity between PEDV and TGEV for this unique sequence was only 18%. However, as the S protein is more genetically variable, the nucleoprotein was selected for further analysis and optimization. Sequence alignments of the complete PEDV NP with its counterpart for PRCV and TGEV indicated that a unique fragment of 45 amino acids length at positions 153 to 198 was present in the PEDV NP (Fig. 2). To ensure that the unique, identified region was conserved in a majority of PEDV strains, about 50 PEDV NP sequences, randomly selected from GenBank, were aligned by Clustal W. The alignment showed that the fragment was highly conserved in PEDV. The only changes detected were, in positions 191 to 193, GGN to GNN in 7 sequences and, in positions 204 to 206, SKN to SKS in 5 of the 50 sequences. Therefore, the conserved fragment was likely to provide a wide coverage of PEDV strains when used as a diagnostic target.
FIG 2.

Identification of an unique amino acid sequence in the PEDV N protein. Alignments of representative PEDV, TGEV, PRCV, and PDCoV N proteins show residues 143 to 219 of the N protein. The unique region is indicated in bold letters. The computationally predicted B-cell epitopes are underlined. The lower panel shows the antigenicity analysis of the ELISA targets for the same residues. Solid bars represent turn regions, as identified by Chou-Fasman method; horizontal bars represent coil regions, as identified by Garnier-Robson method; slashed bars represent the Jameson-Wolf antigenicity index. The positive height of the peak represents the predicted antigenicity index at a scale of ±1.7.
Predicted antigenicity of the selected targets.
The unique amino acid sequence of the NP was selected for further computational analysis. The Jameson-Wolf method of antigenicity prediction was used to test whether the selected target had a high probability of being antigenic and, therefore, of eliciting antibody responses in infected animals. The entire stretch of the unique amino acid sequence identified in the nucleoprotein was predicted to be highly immunogenic, with an average index value of 1.7—the highest on the scale. Secondary structure prediction by the Chou-Fasman and Garnier-Robson methods indicated that the sequence was largely composed of turn regions, interspersed with a few coils. The ABCpred prediction server predicted two epitopes with high scores: QGRGASQNRGGNNNN (score, 0.92) and an overlapping epitope QNRGGNNNNNNKSRNQ (score, 0.70). The Bepipred server assigned high scores to all of the residues in the target region. To avoid disruption of possible secondary structure and conformational epitopes, the entire identified unique region of the NP was used for further synthesis and validation as a computationally designed diagnostic target (Fig. 2).
Synthesis of the diagnostic targets.
As expected, the bacterially expressed NP had specific reactivity with anti-PEDV positive-control serum and was detected at a molecular weight of about 38 kDa (Fig. 3A) on the Western blot. Rapid synthesis of the computationally designed NP target by in vitro transcription and translation resulted in the production of protein which was recognized by an anti-His tag monoclonal antibody at the expected molecular weight of approximately 10 kDa (Fig. 3B). Darker bands between 20 and 30 kDa were also detected and could indicate dimerization or trimerization of the 10-kDa product. The pattern of detection with the polyclonal swine PEDV antibody was similar to that of the anti-His antibody (Fig. 3C). No cross-reactivity was detected to TGEV, PRCV, or PDCoV (Fig. 3D to F).
Validation of the conventional and computationally designed ELISAs.
Receiver operator characteristics (ROC) analysis (12) of the conventional ELISA with the IFA as the gold standard generated sensitivity and specificity values of 91.94% and 93.00%, respectively, at a cutoff value of 0.47 (Table 1; Fig. 4). The positive and negative predictive values were 90.5% and 94.1%, respectively.
TABLE 1.
Analysis of the conventional and computationally designed ELISAs
| Assay | Gold standard | Area under the curve (mg · h/liter) | Cutoff valuea | Sensitivity (%) | Specificity (%) | Positive predictive value (%) | Negative predictive value (%) |
|---|---|---|---|---|---|---|---|
| Con-NPb | IFAc | 0.948 | 0.47 | 91.94 | 93.00 | 90.5 | 94.1 |
| CD-NPd | IFA | 0.931 | 0.49 | 89.39 | 88.89 | 85.3 | 91.8 |
| CD-NP | Con-NP | 0.961 | 0.43 | 90.62 | 95.18 | 93.5 | 92.9 |
The signal-to-positive ratio cut-off value distinguishes between positive and negative samples.
Con-NP, conventional NP ELISA.
IFA, immunofluorescence assay.
CD-NP, computationally designed NP ELISA.
FIG 4.

ROC analysis. ROC analysis of the conventional NP ELISA with the IFA as the gold standard, with an AUC value of 0.948 (A); of the computationally designed NP ELISA with the conventional ELISA as the gold standard, with an AUC value of 0.961 (B); and of the computationally designed NP ELISA with the IFA as the gold standard, with an AUC value of 0.931 (C).
When the CD-NP ELISA was compared against the conventional ELISA by ROC analysis, the area under the curve was 0.961, the sensitivity and specificity were 90.62% and 95.18%, respectively, at a signal-positive ratio cutoff value of 0.43, and the positive and negative predictive values were 93.5% and 92.9%, respectively. In comparison with the IFA as the gold standard, the sensitivity and specificity were 89.39% and 88.89%, respectively, at a signal-positive ratio cutoff value of 0.49, while the positive and negative predictive values were 85.3% and 91.8%, respectively (Table 1; Fig. 4).
Assessment of cross-reactivity.
The panel of positive-control sera for other common swine viruses, such as PRRSV, PCV2, SIV, PRCV, TGEV, and PDCoV, showed no evidence of cross-reactivity when tested on the CD-NP ELISA and the conventional NP ELISA.
Agreement between diagnostic assays.
The kappa statistic values for the comparison of the conventional ELISA versus the IFA, the CD-NP ELISA versus the IFA, and the CD-NP ELISA versus the conventional ELISA all ranged from 0.8 to 0.9 (Table 2). For the interpretation of agreement using the kappa statistic, a value of 0.2 to 0.4 is considered fair, 0.4 to 0.6 is moderate, 0.6 to 0.8 is good, and 0.8 to 0.9 is very good (13). Therefore, the agreement between the three tests was very good. When the signal-positive ratios of the individual samples measured by the conventional and CD assays were plotted against each other in a scatter plot, the relationship was linear, with a majority of samples clustering around the trend line (Fig. 5).
TABLE 2.
Agreement between tests as assessed by the kappa coefficient of association
| Test | Conventional NP ELISA statistic (95% confidence interval) | CD-NP ELISA statistic (95% confidence interval) |
|---|---|---|
| IFA | 0.898 (0.825 to 0.972) | 0.810 (0.719 to 0.902) |
| Conventional NP ELISA | Not applicable | 0.818 (0.724 to 0.912) |
FIG 5.
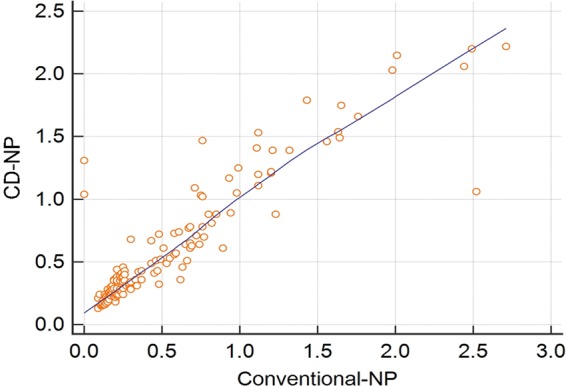
Scatter plot showing the linear relationship between the mean signal-positive ratios of the individual samples when assessed by the conventional or the computationally designed PEDV NP ELISAs. The mean values of the signal-positive ratios of the individual samples for the conventional assay are depicted on the x axis, while the values for the computationally designed assay are plotted on the y axis.
Interassay variation.
An assessment of interassay variation using a panel of 4 samples consisting of either high positive, gray zone, or negative samples on 4 separate assays showed that the interassay variation was minimal. The mean standard deviation of the samples for the conventional ELISA was 0.03, while it was 0.04 for the CD-NP ELISA (Table 3).
TABLE 3.
Assessment of the interassay variation
| Sample | OD value |
|||||
|---|---|---|---|---|---|---|
| CD-NP ELISA |
Conventional NP ELISA |
|||||
| Mean | Median | Standard deviation | Mean | Median | Standard deviation | |
| Sample 1 | 1.15 | 1.14 | 0.06 | 1.07 | 1.07 | 0.06 |
| Sample 2 | 0.75 | 0.76 | 0.05 | 0.80 | 0.82 | 0.09 |
| Sample 3 | 0.46 | 0.46 | 0.02 | 0.44 | 0.44 | 0.04 |
| Sample 4 | 0.14 | 0.14 | 0.01 | 0.25 | 0.23 | 0.01 |
DISCUSSION
The emergence of completely new infectious diseases or the reemergence of existing infectious diseases with altered virulence and antigenicity is a periodic occurrence and a cause of concern in both human and veterinary medicine (14). With the recent changes in animal husbandry practices to favor intensive farming and with the global trade of livestock and animal products, the number of emerging or reemerging infectious diseases affecting swine has increased significantly during the past 20 years (1). Additionally, the introduction of foreign diseases is a consistently looming threat for the nation's food supply. In all of these situations, the rapid and effective detection of disease is central to the control of the spread of infection. However, the majority of diagnostic tests are still produced by conventional methods, which have a relatively long lead time. To address this need, in this study, we have demonstrated that the computational prediction of diagnostic targets followed by the use of rapid synthesis methods can significantly decrease the lead time for serological assay development.
The reliability of the computational method used for the prediction of the antigenicity of the diagnostic targets was verified by the strong reactivity of the synthesized target to sera from PEDV-infected animals. The Jameson-Wolf method is based on the premise that most antigenic sites are located on surface-exposed regions of a protein. The method combines hydropathy, surface probability, and flexibility parameters to generate a surface contour or antigenic index. Previous validation of the algorithm with existing crystal structures showed that it was over 90% accurate (10). While several other online tools for B-cell epitope prediction are available and can be used to predict shorter targets, which can be easily synthesized chemically, their accuracy of prediction is variable, especially in the absence of a crystal structure or experimentally characterized epitopes to verify predictions (15–18). For example, at a sensitivity and specificity of 0.5, the ABCpred server has an accuracy of 65.93%. The higher the score, the greater the probability that the selected sequence is an epitope. High scores of 0.92 and 0.70 were assigned to the two predicted epitopes, and the Bepipred server assigned high probability scores to all of the residues. However, to avoid increasing the lead development time by pursuing detailed linear or conformational epitope mapping experiments, the Jameson-Wolf algorithm was selected as being appropriate for the prediction of surface-exposed antigenic regions. Another advantage of using a longer stretch of peptide sequence is that preservation of the secondary structure could help capture antibodies targeting some conformational epitopes as well as linear epitopes. The accuracy of the prediction was validated by our findings that the predicted target was indeed antigenic. In this study, the time for the optimization of the expression and purification of the recombinant NP took about 3 to 4 months, while the preparation of the in vitro transcribed and translated antigen took less than 2 weeks. However, as algorithms for the prediction of B-cell epitopes are further improved, lead development time may be further shortened by the chemical synthesis of the targets in a cost-effective manner, thus eliminating the need for nucleic acid amplification. However, when a completely new pathogen with no sequence information emerges, the limitation of the described approach is that it cannot be used until the coding sequences for immunogenic proteins are obtained.
Abundant quantities of the NP are produced during infections with coronaviruses, resulting in strong, early antibody responses against this antigen. The spike protein is also highly immunogenic and mediates receptor binding and endosomal fusion. Both the N and S proteins are widely used in the complete or truncated forms for diagnostic test development for human and animal coronaviruses (6, 19). However, both structural and nonstructural proteins are conserved to various degrees within coronaviruses and even between animal and human coronaviruses (20). Indeed, antibodies against TGEV and the feline coronavirus show cross-neutralizing activity (3). Cross-reactivity between the human coronaviruses and human and animal coronaviruses is reported to confound diagnosis and the accuracy of seroprevalence studies which help to identify natural reservoirs for human coronaviruses (21). However, cross-reactivity has not been reported in studies where partial or complete proteins have been used for serological assay development for PEDV (5, 6), except in one study where cross-reactivity was observed between TGEV and PEDV. The reactivity was attributed to a conserved epitope in the N-terminal region of the nucleoprotein (22). Cross-reactivity between PEDV and the PDCoV N proteins was recently reported and attributed to conserved regions in the N protein which could carry B-cell epitopes. The four regions identified included 47-GYW-49, 67-FYYTGTGPRGNLKY-82,194-PKG-197, and 329-EWD-332 (23). In this study, the bacterially expressed NP did not cross-react with the other coronaviruses, probably because the cross-reacting epitopes were excluded in the construct. In addition, selection of the unique amino acid sequence for the CD assay development helped to address possible cross-reactivity issues. Our approach could therefore have a broader application in resolving cross-reactivity issues in coronaviral serological assays.
Several conventionally developed immunoassays using the whole virus (24), or parts of the spike protein, including the S1 domain (5), the N-terminal domain (25), and the S3 domain (26), have been reported in literature (27). Similarly, recombinant N protein-based ELISAs for the detection of PEDV-specific antibodies are reported to have high levels of sensitivity and specificity (6, 28). All studies reported sensitivity and specificity values in the range of 90% to 95%. The computationally designed assay developed in this study was comparable in performance to other published assays, as well as to the IFA (Table 1). As both the N and S protein ELISA results are reported to correlate very well with serum neutralization tests, it is likely that there is additional utility for antibody-binding assays in determining the levels of protection (5, 25).
While there are no accepted gold-standard tests for PEDV, after it emerged in the United States in 2013, the IFA was the first serological tool to be developed and is considered to be a reliable test. While IFAs are generally considered less sensitive than ELISAs, the correlation between ELISAs and IFAs for PEDV, while high, is not perfect. Similarly, others have reported that ELISA-positive samples were negative on the IFA and vice versa (29). The likely reasons for the discrepancy between assays include the possible glycosylation and preferential accessibility of structural proteins in IFAs compared to ELISAs. However, the agreements between the conventional ELISA and IFA and between the CD-ELISA and IFA were high in this study (Table 2), validating the soundness of the analysis.
In conclusion, the approach described in this study supports the hypothesis that lead development time for serological assays for emerging pathogens can be reduced by in silico analysis. It further supports the possible adaptation to the methodology to point-of-care diagnostic kits, to enable the rapid development and deployment of the serological tool in possible emergency situations.
ACKNOWLEDGMENTS
This work was supported by the North Dakota State Board of Agricultural Research (project FARG090242) and the USDA (project ND02425). We thank Linda Saif, Ohio State University, for providing the TGEV, PRCV, and PDCoV control serum samples. We thank the staff and faculty of the NDSU veterinary microbiology department, the NDSU veterinary diagnostic laboratory, and the SDSU veterinary diagnostic laboratory for their assistance.
REFERENCES
- 1.Meng XJ. 2012. Emerging and reemerging swine viruses. Transbound Emerg Dis 59(suppl 1):S85–S102. [DOI] [PubMed] [Google Scholar]
- 2.Hobson-Peters J. 2012. Approaches for the development of rapid serological assays for surveillance and diagnosis of infections caused by zoonotic flaviviruses of the Japanese encephalitis virus serocomplex. J Biomed Biotechnol 2012:379738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horzinek MC, Lutz H, Pedersen NC. 1982. Antigenic relationships among homologous structural polypeptides of porcine, feline, and canine coronaviruses. Infect Immun 37:1148–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jung K, Saif LJ. 2015. Porcine epidemic diarrhea virus infection: etiology, epidemiology, pathogenesis and immunoprophylaxis. Vet J 204:134–143. doi: 10.1016/j.tvjl.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerber PF, Gong Q, Huang YW, Wang C, Holtkamp D, Opriessnig T. 2014. Detection of antibodies against porcine epidemic diarrhea virus in serum and colostrum by indirect ELISA. Vet J 202:33–36. doi: 10.1016/j.tvjl.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okda F, Liu X, Singrey A, Clement T, Nelson J, Christopher-Hennings J, Nelson EA, Lawson S. 2015. Development of an indirect ELISA, blocking ELISA, fluorescent microsphere immunoassay and fluorescent focus neutralization assay for serologic evaluation of exposure to North American strains of porcine epidemic diarrhea virus. BMC Vet Res 11:180. doi: 10.1186/s12917-015-0500-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costantini V, Lewis P, Alsop J, Templeton C, Saif LJ. 2004. Respiratory and fecal shedding of porcine respiratory coronavirus (PRCV) in sentinel weaned pigs and sequence of the partial S-gene of the PRCV isolates. Arch Virol 149:957–974. doi: 10.1007/s00705-003-0245-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu H, Jung K, Vlasova AN, Chepngeno J, Lu Z, Wang Q, Saif LJ. 2015. Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. J Clin Microbiol 53:1537–1548. doi: 10.1128/JCM.00031-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim L, Hayes J, Lewis P, Parwani AV, Chang KO, Saif LJ. 2000. Molecular characterization and pathogenesis of transmissible gastroenteritis coronavirus (TGEV) and porcine respiratory coronavirus (PRCV) field isolates cocirculating in a swine herd. Arch Virol 145:1133–1147. doi: 10.1007/s007050070114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jameson BA, Wolf H. 1988. The antigenic index: a novel algorithm for predicting antigenic determinants. Comput Appl Biosci 4:181–186. [DOI] [PubMed] [Google Scholar]
- 11.Saha S, Raghava GP. 2006. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65:40–48. doi: 10.1002/prot.21078. [DOI] [PubMed] [Google Scholar]
- 12.Greiner M, Pfeiffer D, Smith RD. 2000. Principles and practical application of the receiver-operating characteristic analysis for diagnostic tests. Prev Vet Med 45:23–41. doi: 10.1016/S0167-5877(00)00115-X. [DOI] [PubMed] [Google Scholar]
- 13.Altman DG. 1990. Practical statistics for medical research. Chapman and Hall, London, United Kingdom. [Google Scholar]
- 14.Morens DM, Fauci AS. 2013. Emerg infectious diseases: threats to human health and global stability. PLoS Pathog 9:e1003467. doi: 10.1371/journal.ppat.1003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun P, Ju H, Zhang B, Gu Y, Liu B, Huang Y, Zhang H, Li Y. 2015. Conformational B-cell epitope prediction method based on antigen preprocessing and mimotopes analysis. Biomed Res Int 2015:257030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao J, Kurgan L. 2014. Computational prediction of B-cell epitopes from antigen sequences. Methods Mol Biol 1184:197–215. doi: 10.1007/978-1-4939-1115-8_11. [DOI] [PubMed] [Google Scholar]
- 17.Yao B, Zheng D, Liang S, Zhang C. 2013. Conformational B-cell epitope prediction on antigen protein structures: a review of current algorithms and comparison with common binding site prediction methods. PLoS One 8:e62249. doi: 10.1371/journal.pone.0062249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Constans M, Ssemadaali M, Kolyvushko O, Ramamoorthy S. 2015. Antigenic Determinants of possible vaccine escape by porcine circovirus subtype 2b viruses. Bioinform Biol Insights 9(suppl 2):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suresh MR, Bhatnagar PK, Das D. 2008. Molecular targets for diagnostics and therapeutics of severe acute respiratory syndrome (SARS-CoV). J Pharm Pharm Sci 11:1s–13s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motokawa K, Hohdatsu T, Hashimoto H, Koyama H. 1996. Comparison of the amino acid sequence and phylogenetic analysis of the peplomer, integral membrane and nucleocapsid proteins of feline, canine and porcine coronaviruses. Microbiol Immunol 40:425–433. doi: 10.1111/j.1348-0421.1996.tb01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer B, Drosten C, Muller MA. 2014. Serological assays for emerging coronaviruses: challenges and pitfalls. Virus Res 194:175–183. doi: 10.1016/j.virusres.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin CM, Gao X, Oka T, Vlasova AN, Esseili MA, Wang Q, Saif LJ. 2015. Antigenic relationships among porcine epidemic diarrhea virus and transmissible gastroenteritis virus strains. J Virol 89:3332–3342. doi: 10.1128/JVI.03196-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Y, Zhang Yu, Liang Xueya, Oglesbee Michael, K Steven, Neihaus Andrew, Wang Guiping, Jia Aiqing, Song H, Li Jianrong. 2016. Two-way antigenic cross-reactivity between porcine epidemic diarrhea virus and porcine deltacoronavirus. Vet Microbiol 186:90–96. doi: 10.1016/j.vetmic.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hofmann M, Wyler R. 1990. Enzyme-linked immunosorbent assay for the detection of porcine epidemic diarrhea coronavirus antibodies in swine sera. Vet Microbiol 21:263–273. doi: 10.1016/0378-1135(90)90037-V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Zheng F, Fan B, Muhammad HM, Zou Y, Jiang P. 2015. Development of an indirect ELISA based on a truncated S protein of the porcine epidemic diarrhea virus. Can J Microbiol 61:811–817. doi: 10.1139/cjm-2015-0213. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Chen J, Shi D, Shi H, Zhang X, Yuan J, Jiang S, Feng L. 2016. Immunogenicity and antigenic relationships among spike proteins of porcine epidemic diarrhea virus subtypes G1 and G2. Arch Virol 161:537–547. doi: 10.1007/s00705-015-2694-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knuchel M, Ackermann M, Muller HK, Kihm U. 1992. An ELISA for detection of antibodies against porcine epidemic diarrhea virus (PEDV) based on the specific solubility of the viral surface glycoprotein. Vet Microbiol 32:117–134. doi: 10.1016/0378-1135(92)90100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou XL, Yu LY, Liu J. 2007. Development and evaluation of enzyme-linked immunosorbent assay based on recombinant nucleocapsid protein for detection of porcine epidemic diarrhea (PEDV) antibodies. Vet Microbiol 123:86–92. doi: 10.1016/j.vetmic.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carvajal A, Lanza I, Diego R, Rubio P, Carmenes P. 1995. Evaluation of a blocking ELISA using monoclonal antibodies for the detection of porcine epidemic diarrhea virus and its antibodies. J Vet Diagn Invest 7:60–64. doi: 10.1177/104063879500700109. [DOI] [PubMed] [Google Scholar]