

S. aureus is currently one of the most pervasive multidrug-resistant pathogens and commonly causes nosocomial infections. Clinicians are faced with a dwindling armamentarium to treat infections caused by S. aureus, as resistance develops to current antibiotics. This accentuates the urgent need for antimicrobial drug discovery. In the present study, we characterized the global gene expression profile of S. aureus treated with FADDI-019, a novel synthetic polymyxin analogue. In contrast to the concentration-dependent killing and rapid regrowth in Gram-negative bacteria treated with polymyxin B and colistin, FADDI-019 killed S. aureus progressively without regrowth at 24 h. Notably, FADDI-019 activated several vancomycin resistance genes and significantly downregulated the expression of a number of virulence determinants and enterotoxin genes. A synergistic combination with sulfamethoxazole was predicted by pathway analysis and demonstrated experimentally. This is the first study revealing the transcriptomics of S. aureus treated with a novel synthetic polymyxin analog.
KEYWORDS: Staphylococcus aureus, gene expression, polymyxins
ABSTRACT
Polymyxin B and colistin are exclusively active against Gram-negative pathogens and have been used in the clinic as a last-line therapy. In this study, we investigated the antimicrobial activity of a novel polymyxin, FADDI-019, against Staphylococcus aureus. MIC and time-kill assays were employed to measure the activity of FADDI-019 against S. aureus ATCC 700699. Cell morphology was examined with scanning electron microscopy (SEM), and cell membrane polarity was measured using flow cytometry. Transcriptome changes caused by FADDI-019 treatment were investigated using transcriptome sequencing (RNA-Seq). Pathway analysis was conducted to examine the mechanism of the antibacterial activity of FADDI-019 and to rationally design a synergistic combination. Polymyxin B and colistin were not active against S. aureus strains with MICs of >128 mg/liter; however, FADDI-019 had a MIC of 16 mg/liter. Time-kill assays revealed that no S. aureus regrowth was observed after 24 h at 2× to 4× MIC of FADDI-019. Scanning electron microscopy (SEM) and flow cytometry results indicated that FADDI-019 treatment had no effect on cell morphology but caused membrane depolarization. The vancomycin resistance genes vraRS, as well as the VraRS regulon, were activated by FADDI-019. Virulence determinants controlled by SaeRS and the expression of enterotoxin genes yent2, sei, sem, and seo were significantly downregulated by FADDI-019. Pathway analysis of transcriptomic data was predictive of a synergistic combination comprising FADDI-019 and sulfamethoxazole. Our study is the first to examine the mechanism of the killing of a novel polymyxin against S. aureus. We also show the potential of transcriptomic and pathway analysis as tools to design synergistic antibiotic combinations.
IMPORTANCE S. aureus is currently one of the most pervasive multidrug-resistant pathogens and commonly causes nosocomial infections. Clinicians are faced with a dwindling armamentarium to treat infections caused by S. aureus, as resistance develops to current antibiotics. This accentuates the urgent need for antimicrobial drug discovery. In the present study, we characterized the global gene expression profile of S. aureus treated with FADDI-019, a novel synthetic polymyxin analogue. In contrast to the concentration-dependent killing and rapid regrowth in Gram-negative bacteria treated with polymyxin B and colistin, FADDI-019 killed S. aureus progressively without regrowth at 24 h. Notably, FADDI-019 activated several vancomycin resistance genes and significantly downregulated the expression of a number of virulence determinants and enterotoxin genes. A synergistic combination with sulfamethoxazole was predicted by pathway analysis and demonstrated experimentally. This is the first study revealing the transcriptomics of S. aureus treated with a novel synthetic polymyxin analog.
INTRODUCTION
Polymyxin B and colistin (i.e., polymyxin E) are naturally occurring cationic antimicrobial peptides (CAMPs) that have been used in the clinic for over 50 years to treat multidrug-resistant Gram-negative bacterial infections (1–3). Polymyxins primarily interact with lipid A, which is located in the outer leaflet of the Gram-negative outer membrane (OM) (1, 3, 4). It has been reported that CAMPs interact with cell membranes and form membrane-spanning pores, which results in cell death (5, 6). In addition to causing membrane perturbation, CAMPs can also inhibit intracellular metabolic processes, including biosynthesis of the cell wall, nucleic acids, and proteins (7, 8). In these cases, the cell death may be the result of multiple inhibitory effects. It has been shown that the interaction of polymyxins with lipid A is essential to their antimicrobial activity in Gram-negative bacteria (9). Gram-positive bacteria lack an OM or lipid A, rendering them inherently resistant to polymyxin B and colistin. Interestingly, we have shown that novel polymyxins can have very different activities against both Gram-negative and Gram-positive pathogens (3, 9). Specifically, FADDI-019 is a novel polymyxin B derivative with a d-octylglycine at position 6 and an octanoyl fatty acyl chain at the N terminus, as opposed to polymyxin B1 and colistin A, which have either a d-phenylalanine (polymyxin B1) or d-leucine (colistin A) at position 6 and a 6-S-methlyoctanoyl fatty acyl chain (9). With increased hydrophobicity within its heptapeptide ring, FADDI-019 exhibits increased activity against Gram-positive bacteria, including Staphylococcus aureus (9). More recently, other polymyxin-based synthetic CAMPs have been designed that exhibit antimicrobial activities against Gram-positive bacteria (10, 11). Synthetic CAMPs containing tryptophan, arginine, and N-leucine alterations showed greater activities against Gram-positive bacteria; in addition, these CAMPs showed low cytotoxicities and activities against Gram-negative bacteria, equivalent to those of polymyxin B (10, 12). Gallardo-Godoy et al. generated a library of synthetic polymyxin B derivatives, four of which showed significantly reduced MICs against S. aureus (11). Despite the growing number of synthetic polymyxin-like CAMPs, there is currently no understanding of the transcriptional changes caused by their activity against Gram-positive bacteria.
Many Gram-negative species use well-characterized two-component systems to develop resistance to polymyxin (13, 14). In Salmonella enterica serotype Typhimurium, polymyxins activate the two-component system PhoPQ, causing the activation of the PhoPQ regulon (13, 15). The pmr (polymyxin resistance) locus is part of that regulon, and the activation of the pmr locus results in modifications of lipid A, which in turn serve to decrease the interaction with polymyxins. This mechanism has also been shown in Pseudomonas aeruginosa, Escherichia coli, and Klebsiella pneumoniae (16–20). Due to the inactivity of polymyxin B and colistin against Gram-positive bacteria, the transcriptional response of Gram-positive bacteria to polymyxins has not been investigated. The transcriptional responses to CAMPs other than polymyxins have been reported in S. aureus and Bacillus subtilis (21, 22). Pietiäinen et al. characterized the transcriptional response of S. aureus to three CAMPs, ovisporin-1, temporin L, and dermaseptin K4-S4 (22). Significant activation of the vraSR and vraDE genes, which are involved in vancomycin resistance and cell wall homeostasis, suggested that these CAMPs perturb the cell wall. In B. subtilis, treatment with CAMPs resulted in the activation of the SigW and SigM cytoplasmic sigma factors and the YxdJK and LiaRS two-component systems (21). In scenarios where antimicrobials target multiple components of the bacterial cell, transcriptome analysis is capable of identifying affected pathways (21–24). In the present study, we characterized the response of S. aureus ATCC 700699 treated with a novel synthetic polymyxin, FADDI-019, using phenotypic assays and transcriptomics. Furthermore, we successfully predicted a synergistic combination by targeting a choke point identified using pathway analysis of our transcriptomic data.
RESULTS
Antimicrobial activity of FADDI-019 against S. aureus.
In order to characterize the antimicrobial activities of FADDI-019, the MICs of FADDI-019 were measured against S. aureus ATCC 700699 (Mu50), S. aureus ATCC 700698 (Mu3), P. aeruginosa ATCC 27853, and Acinetobacter baumannii ATCC 19606 (Table 1). The MICs of FADDI-019 against S. aureus strains ATCC 700699 and ATCC 700698 were both 16 mg/liter, consistent with our previous report (9). We also tested polymyxin B and colistin against both S. aureus strains and observed no activity (MICs of >128 mg/liter) for both compounds, confirming that these S. aureus strains are intrinsically resistant to the naturally occurring, clinically available polymyxins. The results in Table 1 show that FADDI-019 had MICs between 1 and 2 mg/liter against Gram-negative bacteria. This level of activity is comparable to those of polymyxin B and colistin against the same strains (Table 1). In summary, FADDI-019 displayed antibacterial activity against S. aureus strains that were resistant to polymyxin B and colistin.
TABLE 1 .
MICs of polymyxin B, colistin, and FADDI-019 against Gram-positive and Gram-negative strains
| Strain | MIC (mg/liter) of: |
||
|---|---|---|---|
| Polymyxin B | Colistin | FADDI-019 | |
| A. baumannii ATCC 19606 | 0.5 | 1 | 1 |
| A. baumannii ATCC 17978 | 2 | 0.5 | 2 |
| P. aeruginosa ATCC 27853 | 2 | 2 | 2 |
| S. aureus ATCC 700698 | >128 | >128 | 16 |
| S. aureus ATCC 700699 | >128 | >128 | 16 |
FADDI-019 killing kinetics against S. aureus and SEM analysis of cell morphology.
Figure 1 shows the killing kinetics of FADDI-019 at three concentrations (i.e., 2×, 4×, and 8× MIC) against S. aureus strain ATCC 700699. Contrary to the typical rapid-killing kinetics of polymyxin B and colistin against Gram-negative bacteria (25–28), FADDI-019 caused slow killing against S. aureus ATCC 700699 at all concentrations tested; interestingly, regrowth was not observed even at 2× MIC. Scanning electron microscopy (SEM) was used to investigate the effects of FADDI-019 treatment on cell morphology and the bacterial cell surface of S. aureus ATCC 700699 (Fig. 2). Treatment with polymyxin B (99.5 mg/liter) was used for comparison, which showed no significant morphological alterations of S. aureus ATCC 700699 cells. Interestingly, compared to the results for the untreated control, no major perturbations of cell morphology were observed in the treated samples at 1× or 4× MIC of FADDI-019, even with significant bacterial killing (Fig. 1). Cell morphology changes were only evident at 8× MIC of FADDI-019 against S. aureus ATCC 700699.
FIG 1 .
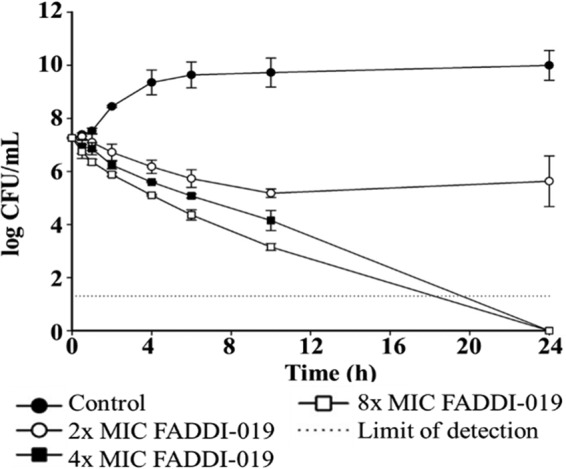
Time-kill kinetics of FADDI-019 against S. aureus ATCC 700699. The error bars show the standard deviations of the results from three independent biological repeats.
FIG 2 .

SEM images of S. aureus ATCC 700699 cells left untreated (A) or treated with polymyxin B (PMB; 99.5 mg/liter) (B), FADDI-019 (1× MIC) (C), FADDI-019 (4× MIC) (D), and FADDI-019 (8× MIC) (E) for 1 h. The white scale bar in each figure represents 500 nm.
Flow cytometry analysis of the membrane polarity during FADDI-019 treatment.
S. aureus cells treated with polymyxin B at 99.5 mg/liter for up to 1 h showed no indication of depolarization relative to the results for the control at time zero (Fig. 3). Interestingly, there was a significant shift in the red fluorescence when cells were treated with FADDI-019 at 4× MIC within 15 min, indicating that treatment with FADDI-019 resulted in membrane depolarization (Fig. 3). In addition, there was no evidence of recovery from membrane depolarization after 60 min of treatment (Fig. 3), which suggests that the depolarization effect of FADDI-019 was permanent.
FIG 3 .

Scatter plots of flow cytometry analysis of S. aureus ATCC 70099 during treatment with polymyxin B and FADDI-019. DiOC2(3) was used to stain the cells, and the fluorescence of each incident (indicated as a dot) in the red and green channels was measured and plotted onto a 2-D scatterplot. Shifting of the scatter in the red fluorescence channel is indicative of membrane depolarization.
Transcriptome profiling and enrichment of S. aureus ATCC 700699 in response to FADDI-019 treatment.
The transcriptome variances of all samples, treatments, and time points were analyzed by principal component analysis (PCA) (Fig. 4A). PCA analysis separated FADDI-019-treated from untreated samples across principal component 1, indicating that treatment with FADDI-019 was responsible for 51.5% of total variance across all samples (Fig. 4A). Samples taken at different time points were separated across principal component 2, which was responsible for 14.9% of the total variance between all samples (Fig. 4A). Individual repeats clustered together, showing the excellent reproducibility of our transcriptomic study (Fig. 4A). Voom and limma linear modeling and Degust (Fig. 4B) were used to enrich for genes that were differentially regulated at 15 min and sustained this response after 60 min of FADDI-019 treatment (false discovery rate [FDR] = 0.05, >2-fold change) (Fig. 4B). After this enrichment, a total of 208 differentially regulated genes were identified, of which 140 were significantly upregulated and 68 significantly downregulated (FDR = 0.05, >2-fold change) (Fig. 4B). The use of a time course allowed enrichment analysis, which limited the list of differentially regulated genes to those involved in a specific response to FADDI-019 (see Table S1 in the supplemental material).
FIG 4 .

(A) PCA results for all data, with individual samples plotted on principal component 1 (horizontal axis) and principal component 2 (vertical axis). (B) Graphical representation of the gene enrichment process using Degust. Individual differentially regulated genes are represented as horizontal red lines. The vertical axes represent the log2 fold change of all genes at each time point in control and treated conditions (indicated above each axis) relative to 15 min of treatment with FADDI-019. Gray boxes indicate the gating cutoffs used to enrich the gene list for genes that are greater than 1 log2 fold differentially expressed throughout the control samples but >1 log2 fold differentially expressed after 60 min.
Full list of differentially regulated genes and their respective expression ratios, expressed in log2, false discovery rate (FDR) values, annotations, and pathways. Download Table S1, PDF file, 0.4 MB (396.2KB, pdf) .
Copyright © 2016 Zhao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Induction of the vancomycin resistance regulon in S. aureus ATCC 700699 by FADDI-019.
Genes involved in similar cellular processes are often regulated in kind (29). We analyzed the enriched gene lists to identify the pathways affected by FADDI-019 treatment. Analysis of genes upregulated by FADDI-019 in S. aureus ATCC 700699 showed a significant increase in the expression of the vancomycin resistance genes. Constituents of the two-component signaling system comprising vraRS were upregulated by FADDI-019 5.04- and 4.90-fold, respectively (Fig. 5A). In order to confirm the activation of the remaining VraSR regulon, we analyzed the expression of the genes known to be under the control of the VraSR two-component system. These included hypothetical proteins SAV1424, SAV1423, SAV1422, SAV1421, and SAV2556, which were upregulated 3.50-, 3.90-, 3.73-, 3.50-, and 4.28-fold, respectively, hypothetical membrane protein SAV1650, which was upregulated 3.54-fold, and a peptidyl-prolyl isomerase, prsA, which was upregulated 3.41-fold (Fig. 5A). These results show that, in addition to the activation of VraSR, genes in the VraSR regulon were also upregulated during FADDI-019 treatment.
FIG 5 .

(A to I) Heat map representation of gene expression relative to that in the control at time zero. Gene numbers and annotations are displayed adjacent to each map. Red-to-blue colors represent the log2 fold change, ranging from 6(log2)-fold upregulated (red) through no change (white) to −6(log2)-fold change (blue). Each map was generated using data from three biological repeats, and all genes represented have a >2-fold change in expression after 15 min of treatment relative to their expression in control samples.
Activation of genes involved in the metabolism of energy, folate, and amino acids by FADDI-019.
FADDI-019-treated S. aureus ATCC 700699 showed an increase in the expression of genes involved in basic metabolic processes, including peptide import, metabolism of folate and sugar, and amino acid biosynthesis. The phosphotransferase system genes pstSACB, which code for proteins involved in sugar import, were upregulated 2.08-, 2.88-, 2.88-, and 3.57-fold, respectively (Fig. 5B). In addition, the galactose metabolism genes lacCDEF and the malA gene were upregulated 2.37-, 2.18-, 2.27-, 2.09-, and 2.79-fold, correspondingly (Fig. 5C). Genes involved in folate biosynthesis, fhs and folD, were also upregulated 2.14- and 2.47-fold, respectively (Fig. 5D). The most-upregulated genes during FADDI-019 treatment were those involved in amino acid biosynthesis, and the levels of gene expression ranged from 13.12- to 23.07-fold higher than in the control (Fig. 5E). In addition to amino acid biosynthesis, peptide import genes were also significantly upregulated. The oppABCDF locus encodes a peptide import system, and genes in this operon were 3.11- to 3.31-fold upregulated in FADDI-019-treated cells relative to their expression in the control (Fig. 5F).
Repression of key virulence factors in S. aureus ATCC 700699 treated with FADDI-019.
Of the 208 genes that were differentially expressed during the FADDI-019 treatment, 68 were downregulated. The main pathways repressed by FADDI-019 treatment are involved in virulence. The saeSR genes, which code for the SaeSR two-component system, were downregulated −3.89-fold and −4.03-fold, respectively (Fig. 5G). The SaeSR two-component system is responsible for the activation of numerous S. aureus virulence genes, including coa, sbi, fnbB, saeQ, and saeP (Fig. 5G) (30–35). The coa, sbi, and fnbB genes, which code for host cell-associated proteins, were downregulated −4.49-fold, −6.83-fold, and −2.58-fold, correspondingly (Fig. 5G). The saeQ and saeP genes, which encode regulatory proteins that increase the phosphatase activity of SaeS, were repressed −4.19-fold and −8.88-fold, respectively (Fig. 5G). In addition to the repression of virulence pathways, FADDI-019 treatment also had significant effects on known pathogenicity determinants in S. aureus. Four enterotoxin genes, yent2, sei, sem, and seo, which encode proteins that are the causative agents of toxic shock, were downregulated −2.09-fold, −2.67-fold, −2.19-fold, and −2.94-fold, respectively (Fig. 5H) (36). The import of polyamines has been linked to a number of processes, including signaling and infection (37, 38). The putrescine/spermidine importer potABCD was repressed by FADDI-019 treatment. The potABCD genes were significantly downregulated, by −2.85-fold, −2.96-fold, −2.80-fold, and −2.56-fold, respectively, relative to their expression in the untreated control (Fig. 5I).
Identification of a folate metabolism chokepoint.
We used a systems biology approach to identify metabolic chokepoints based on the transcriptomic response to FADDI-019 treatment. Pathway analysis using the transcriptome data identified that each amino acid biosynthesis system was overexpressed (Fig. 6A). Importantly, the folate metabolic pathways were also overexpressed (Fig. 6B). Tetrahydrofolate is a key metabolite for amino acid biosynthesis (39); our pathway analysis indicated that amino acid metabolism is dependent on folate metabolism and that the tetrahydrofolate synthesis pathway is a suitable chokepoint to inhibit both processes. Sulfamethoxazole is an antimicrobial that inhibits the tetrahydrofolate metabolic pathway (Fig. 5C) (40–42). We measured the MICs of sulfamethoxazole in combination with FADDI-019 against S. aureus. Table 2 shows the MICs of FADDI-019 and sulfamethoxazole against S. aureus ATCC 700699. Sulfamethoxazole alone had a MIC greater than 128 mg/liter, indicating that it had no antimicrobial activity against S. aureus ATCC 700699. In combination, 16 mg/liter sulfamethoxazole decreased the FADDI-019 MIC substantially, to 4 mg/liter, against S. aureus ATCC 700699.
FIG 6 .

(A) Metabolic pathways of the amino acid biosynthesis affected by FADDI-019 treatment. (B) Tetrahydrofolate pathway affected by FADDI-019 treatment. (C) Tetrahydrofolate biosynthesis pathway targeted by sulfamethoxazole and trimethoprim. Arrows indicate reactions catalyzed by known enzymes in S. aureus 700698, and red arrows indicate reactions catalyzed by enzymes with >2-fold increased expression.
TABLE 2 .
MICs of FADDI-019 and sulfamethoxazole against Gram-positive strains
| Strain | MIC (mg/liter) of: |
||
|---|---|---|---|
| FADDI-019 | SMXa | FADDI-019 + SMX | |
| S. aureus ATCC 700698 | 16 | >128 | 4/16 |
| S. aureus ATCC 700699 | 16 | >128 | 4/16 |
SMX, sulfamethoxazole.
DISCUSSION
Polymyxins have been used as a last-line therapeutic option against Gram-negative pathogens (43–45). However, as their mode of action requires the initial binding to lipid A in the outer membrane, polymyxin B and colistin are inactive against Gram-positive bacteria (3, 46). Furthermore, resistance to polymyxins is frequently acquired in vitro within 24 h of treatment by loss or modification of lipid A (16, 17, 47–50). Previously, we have shown that a novel polymyxin, FADDI-019, has antimicrobial activity against Gram-positive bacteria (3, 9). FADDI-019 was designed with a major alteration from the clinically available polymyxin B and colistin. FADDI-019 has a d-octylglycine at position 6, whereas polymyxin B and colistin have d-phenylalanine and d-leucine residues, respectively, at this location (3, 9). Hence, the hydrophobicity of FADDI-019 at position 6 is greater than that of polymyxin B or colistin. The predicted effect of this difference is to increase the hydrophobic reach of the molecule at position 6. Previously, we have shown that both polar and hydrophobic residues are important for the action of polymyxins against Gram-negative bacteria (3). The activity of FADDI-019 against Gram-positive bacteria indicated that increased hydrophobicity is crucial for the antimicrobial activity of the core polymyxin structure against Gram-positive bacteria. It is evident that FADDI-019 has a wider antimicrobial spectrum than do polymyxin B and colistin (3). Time-kill assays revealed that no regrowth of S. aureus ATCC 700699 was observed after treatment, which is in contrast to the killing of polymyxins against Gram-negative bacteria, where regrowth of polymyxin-resistant variants is frequently observed even within 12 h (26, 27). In addition to the aforementioned properties of FADDI-019, we have previously reported that, in rodent models, FADDI-019 has a tolerability equivalent to that of colistin and polymyxin B after intravenous (0.75 mg/kg of body weight) and subcutaneous (40 mg/kg) administration (3). FADDI-019 also lacks hemolytic activity, similar to polymyxin B and colistin. However, it should be noted that the MIC of FADDI-019 against S. aureus is between 4- and 8-fold higher than that of polymyxin B or colistin against Gram-negative bacteria (26, 51) and that this may limit the use of FADDI-019 as a therapeutic.
The analysis of cell morphology by SEM after treatment revealed that cell blebbing was not induced by FADDI-019 at 1× or 4× MIC, although morphology changes were evident at 8× MIC. Membrane blebbing is a common feature of polymyxin-treated Gram-negative bacteria (52, 53), so the absence of membrane blebbing is suggestive of an alternative killing mechanism. To further investigate the effect of FADDI-019 treatment, we examined the membrane polarity of S. aureus ATCC 700699 cells by flow cytometry. FADDI-019 depolarized the cell membrane within 15 min, indicating that, despite the lack of membrane blebbing, FADDI-019 interacted with the S. aureus cell membrane. Our results show that the antimicrobial mechanism of FADDI-019 against S. aureus is different from previously characterized polymyxin mechanisms against Gram-negative pathogens (5). Furthermore, the lack of lipid A, absence of membrane blebbing, and extensive membrane depolarization are suggestive of an alternative target of FADDI-019.
Analysis of transcriptomic changes caused by FADDI-019 treatment showed that a number of major pathways associated with vancomycin resistance were upregulated. The vraSR two-component system, which was originally characterized for its role in vancomycin resistance, is responsible for coordinating the response to antimicrobials that target the cell wall (54). Transcriptomic studies that assayed the effects of ovisporin-1, temporin L, and dermaseptin K4-S4 against S. aureus ATCC 25904 have been reported previously (21, 22). Unlike the cyclic FADDI-019, ovisporin-1, temporin L, and dermaseptin K4-S4 are all linear alpha-helical CAMPs. The upregulation of vancomycin resistance genes was also observed with alpha-helical CAMP treatments against S. aureus (22), which suggests that cell wall homeostasis has a key role in the response to all CAMPs that show antimicrobial activity against Gram-positive bacteria. Amino acid biosynthesis genes and peptide import systems were also upregulated in response to alpha-helical CAMP treatments (21). The predominant amino acid biosynthesis pathway upregulated by FADDI-019 treatment was the leucine, isoleucine, and valine superpathway. Components of other amino acid pathways (e.g., glutamine, methionine, serine, and tryptophan) were also upregulated (Fig. 5E). In addition to amino acid biosynthesis, the OppABCDF machinery, which imports tripeptides from the environment (55), was upregulated in response to FADDI-019. The OppABCDF machinery is important for quorum sensing and nutrition (55). Opp transporters have been shown to supply bacteria with exogenous peptides that serve as amino acid resources. The activation of the Opp transport system is therefore synonymous with amino acid production. The role of amino acid biosynthesis and peptide import in the response to CAMPs is currently not well understood. However, in response to FADDI-019 treatment, we observed the strongest activation of gene expression, compared to all other differentially expressed genes, in amino acid biosynthesis genes. Taken together, we propose that the increase in vancomycin resistance gene expression and cell wall metabolism drives the need for increased amino acid production and import of tripeptides.
FADDI-019 also significantly repressed genes involved in the virulence of S. aureus. The repression of saeRS and the SaeRS-regulated virulence factor sbi by other, alpha-helical CAMPs, i.e., ovisporin-1, temporin L, and dermaseptin K4-S4, has been shown previously (21, 22). However, other known SaeRS-regulated genes (e.g., coa, fnbB, and saeQP), which were similarly repressed in our study, were not repressed by those alpha-helical CAMPs in S. aureus ATCC 25904 (21, 22). These differences are most likely due to differences in the mode of action between FADDI-019 and alpha-helical CAMPs and/or biological variability between S. aureus strains. In addition to the repression of SaeRS-regulated genes, four enterotoxin genes were also dramatically repressed by FADDI-019 treatment. Interestingly, this response is also specific to FADDI-019 and has not been reported for other CAMPs. Our data suggest that, in addition to killing S. aureus ATCC 700699, it is very likely that FADDI-019 reduced the virulence of the infecting bacterial cells as a secondary effect. When making comparisons between strains, it is important to note that the strain used in this study is a vancomycin-intermediate S. aureus (VISA), whereas S. aureus ATCC 25904 is vancomycin susceptible. VISA strains have been shown to have mutations in regulatory genes, including vraSR, which alter the transcription profile and result in intermediate vancomycin resistance (56, 57). The SaeSR two-component system is not known to be differentially regulated in different VISA strains (56).
Our transcriptomic analysis of S. aureus ATCC 700699 during FADDI-019 exposure demonstrated that a large number of essential metabolism processes, including folate biosynthesis, were upregulated (Fig. 5D). Some of these changes have been previously observed with alpha-helical CAMPs (22); however, the upregulation of folate biosynthesis genes was not reported with alpha-helical-CAMP treatment in that study. We hypothesized that these perturbations are important for S. aureus ATCC 700699 during FADDI-019 treatment. Pathway analysis (Fig. 5B) revealed that tetrahydrofolate production would be a suitable chokepoint target for a synergistic combination with FADDI-019. Indeed, as a test of this hypothesis, we subsequently demonstrated that FADDI-019 and sulfamethoxazole in combination were synergistic, with substantial reductions in the MICs of both compounds (Table 2). Our systems approach, exemplified here, holds much promise for identifying novel approaches to identify rational antimicrobial combinations in the future.
In conclusion, in an era of burgeoning multidrug resistance and diminishing therapeutic options, it is crucial that every effort must be made to discover antibacterial compounds against problematic pathogens. This is the first study reporting the transcriptomics of an unexpected antibacterial activity of a novel synthetic polymyxin analog against Gram-positive S. aureus. Importantly, our systems approach using transcriptomics has shown that pathway analysis of responses to drugs has a significant potential in predicting synergistic antibiotic combinations.
MATERIALS AND METHODS
Bacterial strains, culturing conditions, and susceptibility tests.
S. aureus ATCC 700699 (Mu50, VISA), S. aureus ATCC 700698 (Mu3, heterogeneous VISA), A. baumannii ATCC 17978, A. baumannii ATCC 19606, and P. aeruginosa ATCC 27853 were grown at 37°C on Mueller-Hinton (MH) agar plates or in cation-adjusted Mueller-Hinton broth (CAMHB) (Ca2+ at 22.5 mg/liter and Mg2+ at 11.25 mg/liter). No antibiotic selection was used unless stated otherwise. The MICs of FADDI-019, colistin (Sigma-Aldrich, Castle Hill, Australia), polymyxin B (Sigma-Aldrich), and sulfamethoxazole (Sigma-Aldrich) were determined by broth microdilution in CAMHB according to the Clinical and Laboratory Standards Institute protocol (n = 3) (63). The synergistic effect by FADDI-019 with sulfamethoxazole was examined using a checkerboard method (see Table S2 in the supplemental material) (58).
Checkerboard analysis of sulfamethoxazole and FADDI-019 combination therapy. Download Table S2, PDF file, 0.04 MB (38.9KB, pdf) .
Copyright © 2016 Zhao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Time-kill studies.
The time-kill kinetics of FADDI-019 against S. aureus ATCC 700699 was examined in three replicates at 2×, 4×, and 8× MIC with a log-phase broth culture (optical density at 600 nm [OD600] of 0.4, 107 CFU/ml) (59). Viable bacteria were enumerated on nutrient agar plates from samples collected at 0, 0.5, 1, 2, 4, 6, 10, and 24 h after treatment. Colonies were counted following overnight incubation at 37°C, and the lower limit of detection was 20 CFU/ml.
Preparation of cells for SEM.
Cultures of S. aureus ATCC 700699 were incubated at 37°C with aeration until the OD600 reached 0.4. FADDI-019 was added to a final concentration of 1×, 4×, or 8× MIC, and cultures were incubated for an hour at 37°C. An untreated sample and a sample treated with 99.5 mg/liter polymyxin B were used as controls. After the incubation, cells were pelleted by centrifugation at 5,000 × g for 10 min. Cells were fixed by the addition of 2.5% glutaraldehyde for 1 min and then washed three times using phosphate-buffered saline (PBS) (pH 7.4). The bacterial cultures were air dried onto polyethylenimine-coated coverslips and immersed for an hour in 2.5% glutaraldehyde in PBS. The slides were then washed with PBS three times, and dehydration was done using 10% increments of ethanol in water from 0 to 100% for 10 min at each step. The coverslips were dried using a Balzers critical point dryer (Balzers, Liechtenstein, Germany) prior to mounting on 20-mm aluminum stubs with double-sided carbon tabs. The cells were coated with gold and imaged with a Philips XL30 field-emission scanning electron microscope (SEM; Philips, Eindhoven, Holland) at the University of Melbourne (Victoria, Australia).
Flow cytometry.
Cultures (10 ml) of S. aureus ATCC 700699 were incubated at 37°C until the OD600 reached 0.4 and then treated with either polymyxin B (99.5 mg/liter) or FADDI-019 at 4× MIC. Samples (300 µl) were taken from each culture at 0, 15, and 60 min after treatment and incubated for 10 min with DiOC2(3) (3,3'-diethyloxacarbocyanine iodide; Thermo Fisher, Victoria, Australia). Samples (300 µl) were analyzed by flow cytometry (NovoCyte; ACEA Biosciences, Inc.) with the laser set to emit at 488 nm, and fluorescence was measured in the red and green channels. All incidents were plotted on a two-dimensional (2-D) scatterplot using their respective fluorescence intensities in each channel.
RNA-Seq transcriptomics.
S. aureus ATCC 700699 was grown to an OD600 of 0.4 from an initial absorbance of 0.005, and FADDI-019 was added to a final concentration of 4× MIC. Samples were collected at 0, 15, and 60 min posttreatment and preserved with RNAprotect (Qiagen, United States) following the manufacturer’s instructions. Cells were pelleted by centrifugation at 5,000 × g for 10 min at 4°C. RNA was isolated using an RNeasy minikit (Qiagen) in accordance with the manufacturer’s instructions, with the following additions. Cell pellets were homogenized in 1 ml Tris-buffered saline (TBS) (20 mM Tris, pH 7.5) containing 0.4 mg of lysostaphin and incubated at 37°C for 15 min. Subsequently, 20 mg of lysozyme in TE buffer (20 mM Tris, pH 7.5, 2 mM EDTA, pH 7.8) was added and the sample was incubated at 25°C for 10 min. Control samples were collected from an antibiotic-free culture, and each experiment was repeated three times. RNA sequencing was conducted by the MHTP High-Throughput Sequencing Facility at the Hudson Institute of Medical Research (Clayton, Victoria, Australia).
Bioinformatic analysis.
RNA sequence reads were independently aligned with the genome sequence of S. aureus ATCC 700699 (NCBI accession number NC_002758.2) using Subread (Victorian Bioinformatics Consortium, http://www.vicbioinformatics.com/software.shtml) (60, 61). The RNA sequence data from biological replicates were analyzed using the voom and limma linear modeling methods via the Degust interactive Web-based RNA-seq visualization software (http://www.vicbioinformatics.com/degust/). Differentially expressed genes were defined as those with a change in expression of >2-fold and a corresponding false discovery rate (FDR) of <0.05. The gene ontology terms and pathways were annotated via BioCyc and KEGG. Principal component analysis was performed using the R statistical computing package (62). For pathway analysis, genes shown to be differentially expressed at 15 min after treatment relative to their expression in the control were mapped onto the metabolic network of S. aureus ATCC 700699 (biocyc.org). Pathways containing multiple differentially expressed genes were selected for chokepoint analysis (biocyc.org).
ACKNOWLEDGMENTS
We are grateful to Simon Crawford (University of Melbourne, Parkville) for his technical support regarding SEM experiments. We are also grateful to Trevor Wilson (Monash Health Translational Precinct, Clayton, Australia) for library preparation and RNA sequencing.
J.L., T.V., R.L.N., P.E.T., and K.D.R. are supported by a research grant from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI098771), and J.L. and T.V. are also supported by R01 AI111965. J.L. is an Australian NHMRC Senior Research Fellow. T.V. is an Australian NHMRC Industry Career Development Research Fellow.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
REFERENCES
- 1.Hancock RE. 1997. Peptide antibiotics. Lancet 349:418–422. doi: 10.1016/S0140-6736(97)80051-7. [DOI] [PubMed] [Google Scholar]
- 2.Hancock RE, Lehrer R. 1998. Cationic peptides: a new source of antibiotics. Trends Biotechnol 16:82–88. doi: 10.1016/S0167-7799(97)01156-6. [DOI] [PubMed] [Google Scholar]
- 3.Velkov T, Roberts KD, Nation RL, Thompson PE, Li J. 2013. Pharmacology of polymyxins: new insights into an “old” class of antibiotics. Future Microbiol 8:711–724. doi: 10.2217/fmb.13.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azad MA, Huang JX, Cooper MA, Roberts KD, Thompson PE, Nation RL, Li J, Velkov T. 2012. Structure-activity relationships for the binding of polymyxins with human alpha-1-acid glycoprotein. Biochem Pharmacol 84:278–291. doi: 10.1016/j.bcp.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Powers JP, Hancock RE. 2003. The relationship between peptide structure and antibacterial activity. Peptides 24:1681–1691. doi: 10.1016/j.peptides.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 6.Meredith JJ, Dufour A, Bruch MD. 2009. Comparison of the structure and dynamics of the antibiotic peptide polymyxin B and the inactive nonapeptide in aqueous trifluoroethanol by NMR spectroscopy. J Phys Chem B 113:544–551. doi: 10.1021/jp808379x. [DOI] [PubMed] [Google Scholar]
- 7.Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 8.Hancock RE, Diamond G. 2000. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol 8:402–410. doi: 10.1016/S0966-842X(00)01823-0. [DOI] [PubMed] [Google Scholar]
- 9.Velkov T, Roberts KD, Nation RL, Wang J, Thompson PE, Li J. 2014. Teaching “old” polymyxins new tricks: new-generation lipopeptides targeting gram-negative “superbugs.” ACS Chem Biol 9:1172–1177. doi: 10.1021/cb500080r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabanal F, Grau-Campistany A, Vila-Farrés X, Gonzalez-Linares J, Borràs M, Vila J, Manresa A, Cajal Y. 2015. A bioinspired peptide scaffold with high antibiotic activity and low in vivo toxicity. Sci Rep 5:10558. doi: 10.1038/srep10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallardo-Godoy A, Muldoon C, Becker B, Elliott AG, Lash LH, Huang JX, Butler MS, Pelingon R, Kavanagh AM, Ramu S, Phetsang W, Blaskovich MA, Cooper MA. 2016. Activity and predicted nephrotoxicity of synthetic antibiotics based on polymyxin B. J Med Chem 59:1068–1077. doi: 10.1021/acs.jmedchem.5b01593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grau-Campistany A, Manresa Á, Pujol M, Rabanal F, Cajal Y. 2016. Tryptophan-containing lipopeptide antibiotics derived from polymyxin B with activity against Gram positive and Gram negative bacteria. Biochim Biophys Acta 1858:333–343. doi: 10.1016/j.bbamem.2015.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Macfarlane EL, Kwasnicka A, Hancock RE. 2000. Role of Pseudomonas aeruginosa PhoP-PhoQ in resistance to antimicrobial cationic peptides and aminoglycosides. Microbiology 146:2543–2554. doi: 10.1099/00221287-146-10-2543. [DOI] [PubMed] [Google Scholar]
- 14.Dalebroux ZD, Matamouros S, Whittington D, Bishop RE, Miller SI. 2014. PhoPQ regulates acidic glycerophospholipid content of the Salmonella Typhimurium outer membrane. Proc Natl Acad Sci U S A 111:1963–1968. doi: 10.1073/pnas.1316901111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Llobet E, Campos MA, Giménez P, Moranta D, Bengoechea JA. 2011. Analysis of the networks controlling the antimicrobial-peptide-dependent induction of Klebsiella pneumoniae virulence factors. Infect Immun 79:3718–3732. doi: 10.1128/IAI.05226-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee H, Hsu FF, Turk J, Groisman EA. 2004. The PmrA-regulated pmrC gene mediates phosphoethanolamine modification of lipid A and polymyxin resistance in Salmonella enterica. J Bacteriol 186:4124–4133. doi: 10.1128/JB.186.13.4124-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arroyo LA, Herrera CM, Fernandez L, Hankins JV, Trent MS, Hancock RE. 2011. The pmrCAB operon mediates polymyxin resistance in Acinetobacter baumannii ATCC 17978 and clinical isolates through phosphoethanolamine modification of lipid A. Antimicrob Agents Chemother 55:3743–3751. doi: 10.1128/AAC.00256-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng HY, Chen YF, Peng HL. 2010. Molecular characterization of the PhoPQ-PmrD-PmrAB mediated pathway regulating polymyxin B resistance in Klebsiella pneumoniae CG43. J Biomed Sci 17:60. doi: 10.1186/1423-0127-17-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SY, Choi HJ, Ko KS. 2014. Differential expression of two-component systems, pmrAB and phoPQ, with different growth phases of Klebsiella pneumoniae in the presence or absence of colistin. Curr Microbiol 69:37–41. doi: 10.1007/s00284-014-0549-0. [DOI] [PubMed] [Google Scholar]
- 20.Helander IM, Kato Y, Kilpeläinen I, Kostiainen R, Lindner B, Nummila K, Sugiyama T, Yokochi T. 1996. Characterization of lipopolysaccharides of polymyxin-resistant and polymyxin-sensitive Klebsiella pneumoniae O3. Eur J Biochem 237:272–278. doi: 10.1111/j.1432-1033.1996.0272n.x. [DOI] [PubMed] [Google Scholar]
- 21.Pietiäinen M, Gardemeister M, Mecklin M, Leskelä S, Sarvas M, Kontinen VP. 2005. Cationic antimicrobial peptides elicit a complex stress response in Bacillus subtilis that involves ECF-type sigma factors and two-component signal transduction systems. Microbiology 151:1577–1592. doi: 10.1099/mic.0.27761-0. [DOI] [PubMed] [Google Scholar]
- 22.Pietiäinen M, François P, Hyyryläinen HL, Tangomo M, Sass V, Sahl HG, Schrenzel J, Kontinen VP. 2009. Transcriptome analysis of the responses of Staphylococcus aureus to antimicrobial peptides and characterization of the roles of vraDE and vraSR in antimicrobial resistance. BMC Genomics 10:429. doi: 10.1186/1471-2164-10-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henry R, Vithanage N, Harrison P, Seemann T, Coutts S, Moffatt JH, Nation RL, Li J, Harper M, Adler B, Boyce JD. 2012. Colistin-resistant, lipopolysaccharide-deficient Acinetobacter baumannii responds to lipopolysaccharide loss through increased expression of genes involved in the synthesis and transport of lipoproteins, phospholipids, and poly-beta-1,6-N-acetylglucosamine. Antimicrob Agents Chemother 56:59–69. doi: 10.1128/AAC.05191-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henry R, Crane B, Powell D, Deveson Lucas D, Li Z, Aranda J, Harrison P, Nation RL, Adler B, Harper M, Boyce JD, Li J. 2015. The transcriptomic response of Acinetobacter baumannii to colistin and doripenem alone and in combination in an in vitro pharmacokinetics/pharmacodynamics model. J Antimicrob Chemother 70:1303–1313. doi: 10.1093/jac/dku536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Turnidge J, Milne R, Nation RL, Coulthard K. 2001. In vitro pharmacodynamic properties of colistin and colistin methanesulfonate against Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob Agents Chemother 45:781–785. doi: 10.1128/AAC.45.3.781-785.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poudyal A, Howden BP, Bell JM, Gao W, Owen RJ, Turnidge JD, Nation RL, Li J. 2008. In vitro pharmacodynamics of colistin against multidrug-resistant Klebsiella pneumoniae. J Antimicrob Chemother 62:1311–1318. doi: 10.1093/jac/dkn425. [DOI] [PubMed] [Google Scholar]
- 27.Tan CH, Li J, Nation RL. 2007. Activity of colistin against heteroresistant Acinetobacter baumannii and emergence of resistance in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 51:3413–3415. doi: 10.1128/AAC.01571-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Rayner CR, Nation RL, Owen RJ, Spelman D, Tan KE, Liolios L. 2006. Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 50:2946–2950. doi: 10.1128/AAC.00103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jothi R, Balaji S, Wuster A, Grochow JA, Gsponer J, Przytycka TM, Aravind L, Babu MM. 2009. Genomic analysis reveals a tight link between transcription factor dynamics and regulatory network architecture. Mol Syst Biol 5:294. doi: 10.1038/msb.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuroda H, Kuroda M, Cui L, Hiramatsu K. 2007. Subinhibitory concentrations of beta-lactam induce haemolytic activity in Staphylococcus aureus through the SaeRS two-component system. FEMS Microbiol Lett 268:98–105. doi: 10.1111/j.1574-6968.2006.00568.x. [DOI] [PubMed] [Google Scholar]
- 31.Rogasch K, Rühmling V, Pané-Farré J, Höper D, Weinberg C, Fuchs S, Schmudde M, Bröker BM, Wolz C, Hecker M, Engelmann S. 2006. Influence of the two-component system SaeRS on global gene expression in two different Staphylococcus aureus strains. J Bacteriol 188:7742–7758. doi: 10.1128/JB.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeong DW, Cho H, Jones MB, Shatzkes K, Sun F, Ji Q, Liu Q, Peterson SN, He C, Bae T. 2012. The auxiliary protein complex SaePQ activates the phosphatase activity of sensor kinase SaeS in the SaeRS two-component system of Staphylococcus aureus. Mol Microbiol 86:331–348. doi: 10.1111/j.1365-2958.2012.08198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haupt K, Reuter M, van den Elsen J, Burman J, Hälbich S, Richter J, Skerka C, Zipfel PF. 2008. The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement factor H and C3b. PLoS Pathog 4:e1000250. doi: 10.1371/journal.ppat.1000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arciola CR, Campoccia D, Gamberini S, Baldassarri L, Montanaro L. 2005. Prevalence of cna, fnbA and fnbB adhesin genes among Staphylococcus aureus isolates from orthopedic infections associated to different types of implant. FEMS Microbiol Lett 246:81–86. doi: 10.1016/j.femsle.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 35.Lou Q, Zhu T, Hu J, Ben H, Yang J, Yu F, Liu J, Wu Y, Fischer A, Francois P, Schrenzel J, Qu D. 2011. Role of the SaeRS two-component regulatory system in Staphylococcus epidermidis autolysis and biofilm formation. BMC Microbiol 11:146. doi: 10.1186/1471-2180-11-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collery MM, Smyth CJ. 2007. Rapid differentiation of Staphylococcus aureus isolates harbouring egc loci with pseudogenes ψent1 and ψent2 and the selu or seluv gene using PCR-RFLP. J Med Microbiol 56:208–216. doi: 10.1099/jmm.0.46948-0. [DOI] [PubMed] [Google Scholar]
- 37.Jelsbak L, Thomsen LE, Wallrodt I, Jensen PR, Olsen JE. 2012. Polyamines are required for virulence in Salmonella enterica serovar Typhimurium. PLoS One 7:e36149. doi: 10.1371/journal.pone.0036149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soksawatmaekhin W, Kuraishi A, Sakata K, Kashiwagi K, Igarashi K. 2004. Excretion and uptake of cadaverine by CadB and its physiological functions in Escherichia coli. Mol Microbiol 51:1401–1412. doi: 10.1046/j.1365-2958.2003.03913.x. [DOI] [PubMed] [Google Scholar]
- 39.Umbarger HE. 1978. Amino acid biosynthesis and its regulation. Annu Rev Biochem 47:533–606. doi: 10.1146/annurev.bi.47.070178.002533. [DOI] [PubMed] [Google Scholar]
- 40.Wüst J, Wilkins TD. 1978. Susceptibility of anaerobic bacteria to sulfamethoxazole/trimethoprim and routine susceptibility testing. Antimicrob Agents Chemother 14:384–390. doi: 10.1128/AAC.14.3.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evans ME, Feola DJ, Rapp RP. 1999. Polymyxin B sulfate and colistin: old antibiotics for emerging multiresistant gram-negative bacteria. Ann Pharmacother 33:960–967. doi: 10.1345/aph.18426. [DOI] [PubMed] [Google Scholar]
- 42.Finegold SM, Davis A, Ziment I, Jacobs I. 1969. Chemotherapy guide. Calif Med 111:362. [PMC free article] [PubMed] [Google Scholar]
- 43.Zavascki AP, Goldani LZ, Li J, Nation RL. 2007. Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J Antimicrob Chemother 60:1206–1215. doi: 10.1093/jac/dkm357. [DOI] [PubMed] [Google Scholar]
- 44.Bergen PJ, Bulman ZP, Saju S, Bulitta JB, Landersdorfer C, Forrest A, Li J, Nation RL, Tsuji BT. 2015. Polymyxin combinations: pharmacokinetics and pharmacodynamics for rationale use. Pharmacotherapy 35:34–42. doi: 10.1002/phar.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nation RL, Li J, Cars O, Couet W, Dudley MN, Kaye KS, Mouton JW, Paterson DL, Tam VH, Theuretzbacher U, Tsuji BT, Turnidge JD. 2015. Framework for optimisation of the clinical use of colistin and polymyxin B: the Prato polymyxin consensus. Lancet Infect Dis 15:225–234. doi: 10.1016/S1473-3099(14)70850-3. [DOI] [PubMed] [Google Scholar]
- 46.Mulcahy H, Charron-Mazenod L, Lewenza S. 2008. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog 4:e1000213. doi: 10.1371/journal.ppat.1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moffatt JH, Harper M, Adler B, Nation RL, Li J, Boyce JD. 2011. Insertion sequence ISAba11 is involved in colistin resistance and loss of lipopolysaccharide in Acinetobacter baumannii. Antimicrob Agents Chemother 55:3022–3024. doi: 10.1128/AAC.01732-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou Z, Ribeiro AA, Lin S, Cotter RJ, Miller SI, Raetz CR. 2001. Lipid A modifications in polymyxin-resistant Salmonella typhimurium: PMRA-dependent 4-amino-4-deoxy-L-arabinose, and phosphoethanolamine incorporation. J Biol Chem 276:43111–43121. doi: 10.1074/jbc.M106960200. [DOI] [PubMed] [Google Scholar]
- 49.Beceiro A, Llobet E, Aranda J, Bengoechea JA, Doumith M, Hornsey M, Dhanji H, Chart H, Bou G, Livermore DM, Woodford N. 2011. Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system Antimicrob Agents Chemother 55:3370–3379. doi: 10.1128/AAC.00079-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moffatt JH, Harper M, Mansell A, Crane B, Fitzsimons TC, Nation RL, Li J, Adler B, Boyce JD. 2013. Lipopolysaccharide-deficient Acinetobacter baumannii shows altered signaling through host Toll-like receptors and increased susceptibility to the host antimicrobial peptide LL-37. Infect Immun 81:684–689. doi: 10.1128/IAI.01362-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Owen RJ, Li J, Nation RL, Spelman D. 2007. In vitro pharmacodynamics of colistin against Acinetobacter baumannii clinical isolates. J Antimicrob Chemother 59:473–477. doi: 10.1093/jac/dkl512. [DOI] [PubMed] [Google Scholar]
- 52.Abdul Rahim N, Cheah SE, Johnson MD, Yu H, Sidjabat HE, Boyce J, Butler MS, Cooper MA, Fu J, Paterson DL, Nation RL, Bergen PJ, Velkov T, Li J. 2015. Synergistic killing of NDM-producing MDR Klebsiella pneumoniae by two “old” antibiotics—polymyxin B and chloramphenicol. J Antimicrob Chemother 70:2589–2597. doi: 10.1093/jac/dkv135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hartmann M, Berditsch M, Hawecker J, Ardakani MF, Gerthsen D, Ulrich AS. 2010. Damage of the bacterial cell envelope by antimicrobial peptides gramicidin S and PGLa as revealed by transmission and scanning electron microscopy. Antimicrob Agents Chemother 54:3132–3142. doi: 10.1128/AAC.00124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belcheva A, Golemi-Kotra D. 2008. A close-up view of the VraSR two-component system. A mediator of Staphylococcus aureus response to cell wall damage. J Biol Chem 283:12354–12364. doi: 10.1074/jbc.M710010200. [DOI] [PubMed] [Google Scholar]
- 55.Gardan R, Besset C, Guillot A, Gitton C, Monnet V. 2009. The oligopeptide transport system is essential for the development of natural competence in streptococcus thermophilus strain LMD-9. J Bacteriol 191:4647–4655. doi: 10.1128/JB.00257-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Howden BP, Peleg AY, Stinear TP. 2014. The evolution of vancomycin intermediate Staphylococcus aureus (VISA) and heterogenous-VISA. Infect Genet Evol 21:575–582. doi: 10.1016/j.meegid.2013.03.047. [DOI] [PubMed] [Google Scholar]
- 57.Sieradzki K, Leski T, Dick J, Borio L, Tomasz A. 2003. Evolution of a vancomycin-intermediate Staphylococcus aureus strain in vivo: multiple changes in the antibiotic resistance phenotypes of a single lineage of methicillin-resistant S. aureus under the impact of antibiotics administered for chemotherapy. J Clin Microbiol 41:1687–1693. doi: 10.1128/JCM.41.4.1687-1693.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sopirala MM, Mangino JE, Gebreyes WA, Biller B, Bannerman T, Balada-Llasat JM, Pancholi P. 2010. Synergy testing by Etest, microdilution checkerboard, and time-kill methods for pan-drug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 54:4678–4683. doi: 10.1128/AAC.00497-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bergen PJ, Forrest A, Bulitta JB, Tsuji BT, Sidjabat HE, Paterson DL, Li J, Nation RL. 2011. Clinically relevant plasma concentrations of colistin in combination with imipenem enhance pharmacodynamic activity against multidrug-resistant Pseudomonas aeruginosa at multiple inocula. Antimicrob Agents Chemother 55:5134–5142. doi: 10.1128/AAC.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Law CW, Chen Y, Shi W, Smyth GK. 2014. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin-Labat ML, Smyth GK, Ritchie ME. 2015. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res 43:e97. doi: 10.1093/nar/gkv412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jombart T. 2008. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 63.Clinical and Laboratory Standards Institute. 2015. Performance standards for antimicrobial susceptibility testing; twenty-fifth informational supplement. Approved standard M100-S25. CLSI, Wayne, PA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full list of differentially regulated genes and their respective expression ratios, expressed in log2, false discovery rate (FDR) values, annotations, and pathways. Download Table S1, PDF file, 0.4 MB (396.2KB, pdf) .
Copyright © 2016 Zhao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Checkerboard analysis of sulfamethoxazole and FADDI-019 combination therapy. Download Table S2, PDF file, 0.04 MB (38.9KB, pdf) .
Copyright © 2016 Zhao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.