

Abstract
Background
Coxsackievirus A16 (CV-A16), a major etiopathologic cause of pediatric hand, foot, and mouth disease (HFMD) worldwide, has been reported to have caused several fatalities. Revealing the evolutionary and epidemiologic dynamics of CV-A16 across time and space is central to understanding its outbreak potential.
Methods
In this study, we isolated six CV-A16 strains in China’s Jilin province and construct a maximum clade credibility (MCC) tree for CV-A16 VP1 gene by the Bayesian Markov Chain Monte Carlo method using 708 strains from GenBank with epidemiological information. The evolution characteristics of CV-A16 VP1 gene was also analysed dynamicly through Bayesian skyline plot.
Results
All CV-A16 strains identified could be classified into five major genogroups, denoted by GI–GV. GIV and GV have co-circulated in China since 2007, and the CV-A16 epidemic strain isolated in the Jilin province, China, can be classified as GIV-3. The CV-A16 genogroups circulating recently in China have the same ancestor since 2007. The genetic diversity of the CV-A16 VP1 gene shows a continuous increase since the mid-1990s, with sharp increases in genetic diversity in 1997 and 2007 and reached peak in 2007. Very low genetic diversity existed after 2010. The CV-A16 VP1 gene evolutionary rate was 6.656E-3 substitutions per site per year.
Conclusions
We predicted the dynamic phylogenetic trends, which indicate outbreak trends of CV-A16, and provide theoretical foundations for clinical prevention and treatment of HFMD which caused by a CV-A16.
Electronic supplementary material
The online version of this article (doi:10.1186/s12985-016-0578-3) contains supplementary material, which is available to authorized users.
Keywords: Coxsackievirus A16, HFMD, Molecular evolution, Genetic diversity, Bayesian method
Background
In the pediatric population, hand, foot, and mouth disease (HFMD) is a common self-limiting condition caused by various serotypes of the enterovirus A species that is typically characterized by fever, pharyngalgia, malaise, erythema and herpetic lesions on hands and feet, as well as exanthema on oral mucosa and tongue [1]. A minority of patients develop severe neurologic complications such as acute flaccid paralysis, encephalitis, pulmonary edema, and myocarditis, with fatal outcomes in some severely afflicted patients [2, 3]. Over the last few decades, HFMD has been a very common pediatric infection in the Asia–Pacific region, with sporadic outbreaks reported in Europe and North America. In mainland China, HFMD has been listed as a notifiable disease since 2008 [4–6].
Coxsackievirus A16 (CV-A16) and enterovirus 71 (EV-A71) were major pathogens of HFMD in the past few decades, with EV-A71 more frequently associated with neurologic diseases [7]. Vaccine trials for the first EV-A71 vaccine have reached phase 3 clinical testing in China, with an estimated vaccine protection rate of 90 % against clinical EV-A71 infection-associated HFMD and 80.4 % against other EV-A71-associated diseases [3]. The other major pathogen of HFMD, CV-A16 has had epidemic presence in China and abroad for many years, but generating much lower social concern compared to EV71.
The first CV-A16 was identified in 1951 in South Africa, although there were no reports of an HFMD epidemic at the time [8]. New Zealand reported the first case of HFMD in the world in 1957 [9]. The relationship between CV-A16 and HFMD was confirmed by Atsop in 1959, and officially coined the term “Hand, Foot and Mouth Disease” based on clinical symptoms [10]. Although it is the first-identified HFMD virus, there is no specific antiviral treatment for CV-A16, which thus deserves more research attention. Recent years have seen extensive research efforts toward phylogenetic analysis of the HFMD pathogen based on bioinformatics in order to understand correlations between virus genogroup changes and disease epidemic trends [8, 11, 12]. At present, most extant phylogenetic trees are constructed based on neighbor-joining (NJ) distance, maximum parsimony (MP), and maximum likelihood (ML) [8, 13]. In reality, however, the difference between sequences does not completely represent the evolutionary distance, with large potential for errors [14]. The posterior probability is derived based on the Markov chain by Bayesian statistics, which allow researchers to use prior knowledge for guiding the construction of phylogenetic trees and to infer the maximum posteriori probability for estimating the most likely phylogenetic tree [15]. Moreover, the Bayesian method uses the posterior probability to visually represent phylogenetic relationships, thereby eliminating the need for bootstrapping [15, 16], and is widely used to construct the phylogenetic trees of swine-origin influenza A (H1N1) virus, measles viruses (MV), and EV-A71 and for accurate judgments of the relationship between genetic diversity (g) and epidemics of associated diseases [16–18].
This study was undertaken to investigate evolutionary and epidemiological dynamics of HFMD, with particular focus on CV-A16 genetic history and dynamics, within and between countries where this disease is endemic to facilitate prediction of its emergence in new locations as well as to provide the basis for an effective public health response framework. Bayesian analyses were performed to construct maximum clade credibility (MCC) tree for all sequenced and downloaded sequences, and global population dynamics of CV-A16 over the previous 30 years were reconstructed to examine temporal trends in genetic diversity and association with major epidemics.
Methods
Sample collection
A total of 52 specimens, as well as information on patient demographics, clinical symptoms, and complications were obtained from 2012 to 2013 at Disease Control and Prevention Center of Changchun, China. Stool specimens were processed as described previously for subsequent RNA extraction [19]. Specimens of other types were used directly for viral RNA extraction. The strains used for sequencing were amplified and isolated in RD (rhabdomyosarcoma) cells as previously described [20].
RNA extraction, RT-PCR, and sequencing
Viral RNA was extracted using QIAamp Viral RNA MiniKit (QIAGEN, USA). TaKaRa RNA PCRTM Kit (AMV) was used to do the amplification fragment according to previous reports. The real-time PCR (RT-PCR) was firstly performed to detect the presence of the common (universal) sequence of enterovirus (EV-F, EV-R), and the specific sequences of EV-A71 (EV71S-F, EV71S-R) and CV-A16 (CV-A16S-F, CV-A16S-R). Then using primers (CV-A16VP1-F and CV-A16VP1-R) amplified CV-A16 VP1 whole sequence. Primers and reaction conditions was shown in Additional file 1: Table S1. All results of RT-PCR products were analyzed by 1 % agarose gel electrophoresis [21]. A part of CV-A16 VP1 sequences have been submitted to GenBank (Accession no. KT000389-KT000394).
Sequence collection and phylogenetic analyses
For phylogenetic analysis, a total of 759 CV-A16 VP1 gene sequences before June 2013 were downloaded from GenBank. We retrieved 706 sequences which known collection dates and isolate country for analysis. The accession numbers and specific information of the sequences was listed in Additional file 2: Table S2. These nucleotide sequences were isolated mainly from large HFMD outbreaks and sporadic cases that occurred globally over 1981–2013. Combined with the six sequences isolated from our laboratory, a total of 708 sequences were used in phylogenetic analysis.
Alignment processing and recombination detection
The complete VP1 sequence alignment of the CV-A16 strains was conducted with the Clustal W program in MEGA 6.0. Excess sequence was cut off, and FASTA format that can be used in the BEAST 1.8.2 was exported. Then we use the SEAL (sequence simulation and alignment evaluation software, http://tree.bio.ed.ac.uk/software/seal/) software to edit the nucleotide sequence. RDP3 Restructuring Package was used to detect the recombination of all CV-A16 sequence [22]. Then DAMBE was used for the saturation monitoring, if ISS < ISS.c and p = 0.0000 (extremely significant), then these sequences were unsaturated and suitable for construction of phylogenetic tree [23]. Finally we calculated the best alternative model With JModeltest [24]. The calculation was done after the selection of all the four kinds of patterns and then the statistical of AIC value. The smaller of the AIC value, the better fitting for the model with the data. Then, usually we choose the model with smallest AIC value for the construction of phylogenetic trees.
Bayesian Markov Chain Monte Carlo evolutionary analysis
Bayesian Markov chain Monte Carlo (MCMC) methods were used to construct a maximum clade credibility tree (MCC) using BEASTv1.8.2 (http://beast-mcmc.googlecode.com/files/BEASTv1.8.2.tgz). Tracer v1.6 (http://beast.bio.ed.ac.uk/Tracer) was used to output analysis of sampling data, and then the Tree Annotator program was employed to output the results of MCC tree model. In the end the MCC molecular evolutionary tree graph was illustrated with FigTree1.3 (http://tree.bio.ed.ac.uk/). At the same time, Bayesian skyline plot analyses was used to reconstruct the population history of CV-A16 by measuring the dynamics of VP1 gene genetic diversity over time with 160 typical CV-A16 VP1 (Additional file 3: Figure S1).
JModeltest result revealed that HKY was the best substitution model, and the molecular clock model chosen the Relaxed Clock: Uncorrelated Log-normal. Using the Bayesian Markov Chain Monte Carlo framework, 80 million steps were run, sampling every 8000 and removing 10 % as burn-in. Convergence was assessed using Tracer (v1.6), and effective sample size (ESS) values above 200 were accepted.
Results
Phylogenetic analysis of CV-A16
A total of 708 sequences (including six complete VP1 genome sequences determined by our laboratory) with complete VP1 region of CV-A16 were included in the phylogenetic analysis. Samples collected from 14 discrete locations in Southeast Asia (China, Japan, Malaysia, Russia, Australia, Cameroon, South Africa, France, Saudi Arabia, South Korea, Sweden, Taiwan, Thailand, and Viet Nam) between 1951 and 2013. Phylogenetic analyses were performed with Bayesian Markov chain Monte Carlo (MCMC) method. All CV-A16 strains identified could be classified into five major genogroups, denoted by GI–GV (Fig. 1). Further, the genogroup GIV could be divided into sub-genogroups GIV-1, GIV-2, and GIV-3. The genogroup GI, which no longer played a dominating role in contemporary epidemics, was represented by a prototype isolate (G-10), isolated in South Africa approximately 60 years earlier. Following its initial isolation, the genogroup GI was detected in China in 2008 and 2010 (EU812514 and JQ315094). Phylogenetic analysis revealed that genogroup GII was first reported from Japan in 1981 (AB465366). Among the 19 GII grouptypes, 15 were isolated from Japan during 1981–1998, and the other four from Malaysia during 1998–2000, with no further no genogoups reported thereafter. Interestingly, CV-A16 strains isolated in 1995 in Yamagata, Japan, were uniquely grouped into genogroup GIII, except for Y95-2260 (AB634302). After 1995, genogroups GIV and GV showed widespread co-circulation in many countries; 16 GIV-1 sub-grouptypes were composed of strains in Yamagata in Japan (1997), Taiwan (1998 and 2005), and in Russia (2004). Following 2005, this genigroup emerged in Wuhan, China, until 2011. Whereas 29 strains from Malaysia in 2005–2007, Russia in 2009–2010, France in 2010, and Japan (Yamagata) in 2011 were composed of GIV-2. From 1998 to 2013, the GIV-3 was distributed across eight countries, including Australia, Saudi Arabia, Japan, Malaysia, Russia, Cameroon, Taiwan, and China. The genogroup GV (353/708) originated from Malaysia in 1997, where the epidemic lasted 10 years (Table 1). Around 2007, the widely prevalent genogroups (GIV-3 cluster 1 and GV cluster 1) that caused major HFMD epidemics after 1995 appeared all to have been replaced by the new cluster (cluster 2), which has since dominated and circulated endemically in the Asia–Pacific region, generating major CV-A16-associated HFMD outbreaks (Fig. 1).
Fig. 1.

The maximum clade credibility (MCC) tree was estimated by Bayesian analysis of 708 CV-A16 complete VP1 sequences between 1951 and 2013. The phylogeny include 6 sequences from our laboratory as well as 702 sequences from GenBank with known collection dates and isolate country. Branches arecolor-coded according to the different genogroups. All sequences were classified into five genogroups, denoted GI–GV, Further, genogroup GIV could be divided into sub-genogroups GIV-1, GIV-2, and GIV-3. For the year and isolate country for each sequence, see Additional file 4: Figure S2 in the supplemental material
Table 1.
Global distribution of genogroups of CV-A16
| Isolated time and location | MCC | NJ |
|---|---|---|
| 1951 South Africa | GI | A |
| 2008 China: Fuyang | ||
| 2010 China: Ningbo | ||
| 1981–1998 Japan | GII | B2 |
| 1998–2000 Malaysia | ||
| 1995 Japan: Yamagata | GIII | B1a |
| 1995, 1997 Japan: Yamagata | GIV-1 | B1a |
| 1998, 2005 Taiwan | ||
| 2004 Russia | ||
| 2011China: Wuhan | ||
| 2005–2007 Malaysia | GIV-2 | B1c |
| 2009–2010 Russia | ||
| 2010 France | ||
| 2011 Japan: Yamagata | ||
| 2000–2001 Australia | GIV-3 cluster1 | B1b |
| 2000 Malaysia | ||
| 2001, 2003 Arabia; | ||
| 2000, 2002–2003 Japan: Toyama | ||
| 2007–2008 China | ||
| 2007 Malaysia | GIV-3 cluster2 | B1b |
| 2009 Cameroon | ||
| 2007–2010 Russia | ||
| 2011 Japan: Yamagata | ||
| 2007–2013 China | ||
| 1997–2003, 2005–2007 Malaysia | GV cluster1 | B1a |
| 1998 Sweden | ||
| 2000–2004, 2006, 2008 Japan: Yamagata | ||
| 1999 Australia | ||
| 2000–2002 Thailand | ||
| 2008 South Korea | ||
| 2002 Japan Toyama | ||
| 2010 France | ||
| 2005 Australia | GV cluster2 | B1a |
| 2006 Taiwan | ||
| 2005, 2007 Malaysia | ||
| 2005 Thailand | ||
| 2008–2010 Japan: Yamagata | ||
| 2007 Japan: Toyama | ||
| 2005 Vietnam | ||
| 2010 Thailand | ||
| 2007–2012 China |
Note: The time and spatial distribution of all genogroups were shown in this table. The comparison the genogroups is constructed by maximum likelihood
Origin and distribution of CV-A16 from Japan, Malaysia, and China
We, furthermore, studied country-specific epidemic strains to evaluate the trend of CV-A16 epidemic genogroups associated with serious HFMD outbreaks in Japan, Malaysia, and China. From the data sets shown in Fig. 2, we collected 255 CV-A16 strains from Japan and identified that the CV-A16 causing HFMD originated in Toyama in 1981. Subsequently, across 12 years, all CV-A16 epidemic strains belonged to genogroup GII in Japan; thereafter, genogroup GIII became the new epidemic strain until 1995. Shortly thereafter, genogroups GIV and GV replaced GIII and became the predominant genogroups since 1997. The epidemic strains GIV and GV circulated in 1997–2002 (85/255) and 2000–2010 (139/255), respectively. Interestingly, the Japanese epidemic strains (e.g., 1779-Yamagata-2011) in 2011 showed a close relationship to strains isolated during 2010–2012 from China (Figs. 2 and 5), as they all belonged to the GV genogroup, suggesting that the China epidemic strain invaded Japan in 2011.
Fig. 2.
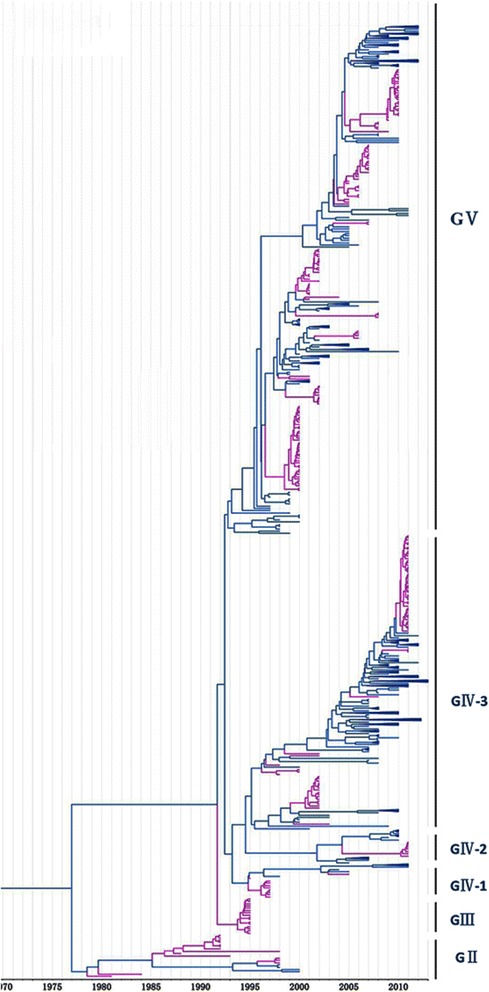
Phylogenetic analysis of CV-A16 from Japan. The scale is in units of evolutionary time in years. Phylogenetic tree of 255 Japan strains from 1981 to 2010. The branches of sequences from Japan are highlighted in purple
Fig. 5.
Origin and distribution of CV-A16 from Japan, Malaysia, and China. Different background colors distinguish countries. The blue color depicts Japan (n = 255), and green and pink colors display Malaysia (n = 82) and China (n = 315), respectively
In our study, 81 CV-A16 epidemic strains from Malaysia were collected, of which genogroup GV was the most dominant, accounting for 83 % (68/82) (Fig. 3). Four strains belonging to genogroup GII were detected in Malaysia only in 1998–2000. The GIV genogroup (9/82) was a minor epidemic strain discovered in Malaysia in 2006–2007 and closely resembles the strains (Additional file 3: Figure S1, Fig. 5) detected in France, Japan, and Russia around 2010.
Fig. 3.
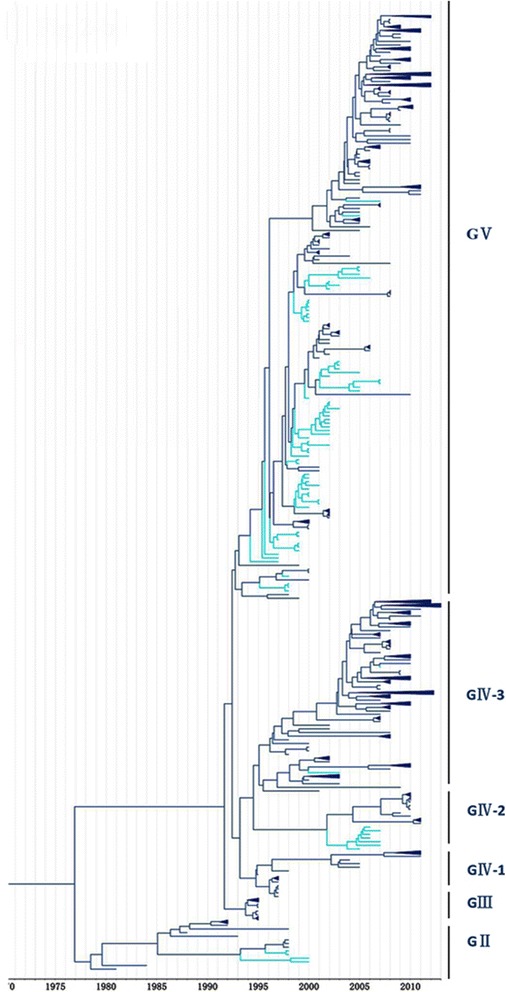
Phylogenetic analysis of CV-A16 from Malaysia. The scale is in units of evolutionary time in years. Phylogenetic tree of 81 Malaysia strains from 1998 to 2007. The branches of sequences from China are highlighted in blue
It was shown that the CV-A16 that co-circulated in China since 2007 were clustered strains of genogroups GIV-3 and GV, except for four strains of genogroup GIV (Wuhan0232/HuB/CHN/2011, Wuhan0289/HuB/CHN/2011, Wuhan0158/HuB/CHN/2011, and Wuhan0136/HuB/CHN/2011) and two strains of genogroup GI (FY18 and CA16v-2010221). Among the 315 strains isolated in China, 195 and 118 strains could be assigned to genogroups GIV and GV, respectively, by phylogenetic analysis (Fig. 4). The first strain of sub-genogroup GIV-3 (shzh00-1) documented in mainland China was detected in 2000 in Guangzhou province [25]. The GIV-3 epidemic of 1998–2000 in China originated from a Japanese genogroup (Fig. 5). Genogroup GV is another dominant genogroup, which has been co-circulating with GIV-3 since 2006 that closely resembles the Japanese strains. Figure 1 shows that the CV-A16 serotypes circulating recently in China have the same ancestor, suggesting that the epidemic strains are native and have not invaded the region since 2007. The CV-A16 epidemic strain isolated in the Jilin province, China, can be classified in the GIV-3 sub-genogroup.
Fig. 4.
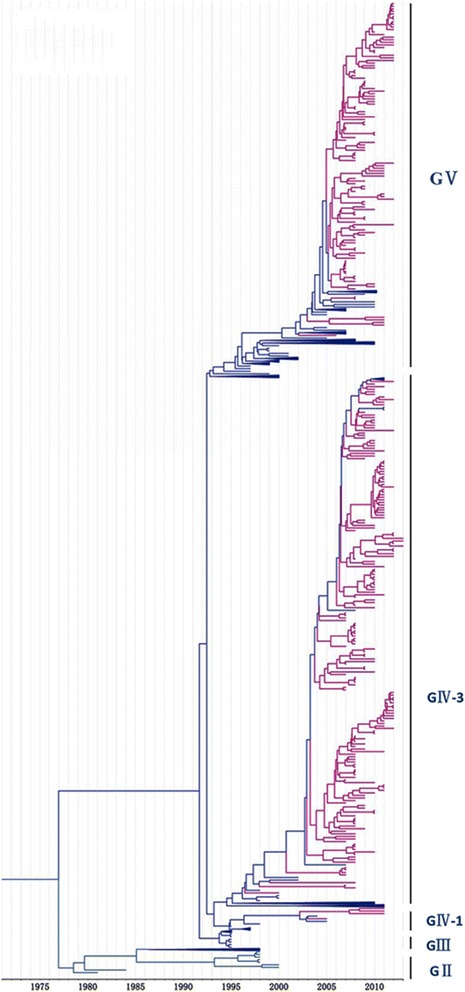
Phylogenetic analysis of CV-A16 from China. The scale is in units of evolutionary time in years. Phylogenetic tree of 81 China strains from 2000 to 2013. The branches of sequences from China are highlighted in red
Genetic diversity analysis with Bayesian skyline plot
To reconstruct the evolutionary epidemiology of CV-A16, we used Bayesian skyline plot analysis by measuring the dynamics of CV-A16 VP1 genetic diversity over time (Fig. 6). It can be observed from the evolutionary epidemiology that CV-A16 VP1 genetic diversity continuously increased since the mid-1990s, indicating evolution from genogroup GII to GIII. This trend coincided with the major HFMD outbreak, mostly caused by the CV-A16 pathogen, in Japan in 1995. A sharp but transient increase in relative genetic diversity was observed for the CV-A16 VP1 gene between 1996 and 1997. Since 1997, several large epidemics of HFMD have been reported in the Asia–Pacific region, especially in Southeast Asia. Outbreaks with multiple cases have occurred in Taiwan, Malaysia, and Singapore [11, 25, 26]. These revealed the evolution of CV-A16 genogroup GIII to genogroups GIV and GV around 1996–1997. From 1999 to 2001, the genetic diversity of CV-A16 VP1 underwent a smooth and steady rise, reflecting the transient but sporadic occurrence of CV-A16 outbreaks in various parts of the world. In late 2000, a recurrence of an outbreak of HFMD occurred in Malaysia, with eight deaths in peninsular Malaysia [25]. From 2006 to 2007, another sharp increase emerged for the genetic diversity of CV-A16 VP1, representing the emergence of a new branch of the genogroups GIV and GV. After 2007, the cluster 1 of genogroups GIV-3 and GV were replaced by cluster 2, and transmission of these led to a new HFMD outbreak in China and Malaysia [20, 27]. CV-A16 is reported to have accounted for 90 % (399/417) of all enteroviruses causing HFDM in Malaysia in 2007 [27]. Since 2007, the Bayesian skyline plot of CV-A16 showed that its genetic diversity decreased and was maintained corresponding to the MCC (Fig. 1) no new sub-genogroup emerged. Due to the stability of CV-A16 after 2010 and accumulated population immunity, CVA6 has become the main pathogen of HFMD disease substituting for CV-A16 in recent years [13, 28, 29].
Fig. 6.
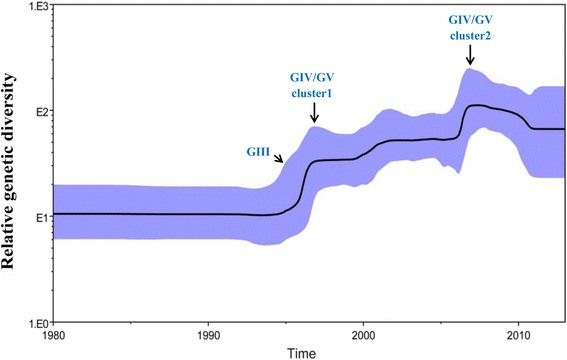
The genetic diversity dynamics of the CV-A16 VP1 gene estimated by a Bayesian skyline plot through time. A dashed line indicates the mean, whereas shaded areas show the upper and lower 95 % HPD values. The horizontal axis is in the unit year, and the vertical axis is the Net (the product of the effective population size and the generation length in radiocarbon years). The plot for CV-A16 VP1 shows a continuous increase since the mid-1990s, with sharp increases in genetic diversity in 1997 and 2007. Very low genetic diversity existed in the early 1990s and in 2010
Codon substitution and evolution rates of CV-A16 VP1 gene
To understand the evolution of CV-A16, we estimated codon substitution and evolution rates using the Bayesian MCMC method. All three codon positions of the CV-A16 VP1 gene had different relative substitution rates (Table 2). The mean values of the first, second, and third codon positions were 0.244, 0.018, and 2.67, respectively. Among these codon positions, the relative substitution rate of the third codons was highest. Simultaneously, our analysis showed that the CV-A16 VP1 gene evolutionary rate was estimated to be 6.656E-3 substitutions per site per year (3.978E-4, 2.456E-3; 95 % HPD), inferred by the models of HKY, approximated to estimates of EV-A71 and much less frequently than the polio virus (1.036 E-2) that was estimated also by the MCMC method previously [18, 30].
Table 2.
Estimates of the relative substitution rates for the core gene of all three codon positions
| Summary statistic | CP1.mu | CP2.mu | CP3.mu |
|---|---|---|---|
| Mean | 0.268 | 0.108 | 2.624 |
| 95 % HPD lower | 0.234 | 0.091 | 2.592 |
| 95 % HPD upper | 0.295 | 0.124 | 2.656 |
| Effective sample size (ESS) | 7502 | 8586 | 7621 |
Discussion
This study establishes the phylogenetic relationship, genomic diversity, and the evolutionary rate of CV-A16 for the first time using the Bayesian Markov chain method, providing new sights into the relationship of evolutionary history of virus population and disease periodicity. We reconstructed the epidemic history of CV-A16 and found that the CV-A16 virus, prevalent between 1980 and 2013, is a pathogen that originated around mid-twentieth century.
Bayesian derivation and the maximum likelihood method have similar characteristics in that both have excellent statistical characteristics. However, one difference is that the Bayesian method can use posterior probability, which is derived from the Markov chain to optimize criterion [31]. A very accurate posterior probability can be obtained using the Bayesian MCMC method due to the rigorous control criterion of every link. Against this background, we established the phylogenies of the CV-A16 gene based on Bayesian derivation combined with the Markov chain model method. Compared with the phylogenetic analysis reported in the previous study [8] using the Neighbor-joiningmethod, the CV-A16 causing the HFMD outbreak in Yamagata, Japan, in 1995 can be independently classified under genogroup GIII using the Bayesian method (Fig. 1, Table 1). There were swift but sporadic occurrences of HFMD in Japan in various years such as 1984, 1988, and 1991 [32]. However, the nucleotide sequence of CV-A16 were relatively stable in this period, reflecting that the epidemic disease that occurred every several years was determined by the cumulative proportion of unvaccinated children and not by the viral antigen’s evolution [32]. Four years late, in 1995, an HFMD outbreak was reported in Japan, which was different from the past episodes; CV-A16 became the main pathogen that replaced EV71 [32, 33]. This suggested that a new CV-A16 genogroup emerged different from former CV-A16 epidemic strains. From the results of our Bayesian skyline plot (Fig. 6), we can also survey the great change in the genetic diversity of CV-A16 in 1995. Therefore, the MCC we reconstructed on the basis of spatiotemporal divergence and genetic diversity is consistent with the trends of epidemic disease. It is very interesting that genogroup GI disappeared for almost 60 years and then it is detected again in 2010 both in our and previous reports [12]. We do not know the reasons for such large-scale changes for genogroup GI, but they may be associated with the G-10’s weakly pathogenicity which didn’t cause enough attention and lead to a lack of continuity monitor data.
From the Bayesian skyline plots (Fig. 6), we can see that every sharp change of genetic diversity resulted in a large-scale HFMD outbreak. To some extent, the increase of genetic diversity corresponding to this characteristic since mid-1990 was a marker for the emergence of a new CV-A16 genogroup. The data set also indicated agreement between genetic diversity dynamics and emergent genogroups, which reflect the earlier HFMD outbreaks. Since 2007, the genetic diversity of CV-A16 stabilized and slightly decreased, and no novel genogroup emergence was reported [12, 13, 33]. However, there were some HFMD outbreaks caused by CV-A16 in various provinces of China that may be attributable to the cumulative proportion of unvaccinated children and increased detection intensity. Since 2010, genetic diversity dynamics tended to be gentler, CVA6 replaced CVA16 become the second pathogene in Shenzhen and Guangdong, China [34, 35].
Conclusions
CV-A16 has long been the main pathogen of HFMD, seriously threatening human health [8, 11, 12, 33]. The reports of some deaths caused by CV-A16 infection [2, 36, 37], suggesting that more attention should be paid to the detection and prevention of CV-A16. From the data we obtained, we predicted the dynamic phylogenetic trends, which indicate outbreak trends of CV-A16, and provide theoretical foundations for clinical prevention and treatment of HFMD which caused by a CV-A16. The relatively stable nucleotide sequence will provide a great opportunity to develop a vaccine for this disease. So the development and administration of its vaccine should be accelerated.
Acknowledgements
This work was supported in part by grants from National Natural Science Foundation of China (#81271897, #81611130074), foundation of Jilin Province Science and Technology Department (#20140414048GH), the Norman Bethune Program of Jilin University (#2012219) and Research funds from the Jilin Key Laboratory of Biomedical Materials and Jilin University - Xinjiang Medical University joint research project. We would like to extend our special thanks to the two anonymous reviewers for their helpful comment on our manuscript.
Funding
National Natural Science Foundation of China (#81271897, #81611130074), Foundation of Jilin Province Science and Technology Department (#20140414048GH), The Norman Bethune Program of Jilin University (#2012219) and Research funds from the Jilin Key Laboratory of Biomedical Materials and Jilin University - Xinjiang Medical University joint research project.
Authors’ contributions
FL conceived the study. GZ designed the experiments, analyzed the data and wrote the manuscript.GW guided the experiment process. XZ and CW contributed in data collection. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
This work was approved by the Ethics Committee of Jilin University, to ensure the consent of all research objects.
Additional files
Table S1. Primers, probes and PCR amplification condition in this study. (DOCX 17 kb)
Table S2. List of the 708 complete VP1 sequences of CVA16 strains available from GenBank which were selected to generate the CVA16 MCC. (XLSX 38 kb)
160 typical CV-A16 VP1 used to reconstruct the population history of CV-A16.(PDF 38 kb)
The exact time and location of 708 sequences between 1951 and 2013.(PDF 36 kb)
Contributor Information
Guoqing Wang, Email: qing@jlu.edu.cn.
Fan Li, Email: lifan@jlu.edu.cn.
References
- 1.Yi L, Lu J, Kung HF, He ML. The virology and developments toward control of human enterovirus 71. Crit Rev Microbiol. 2011;37:313–27. doi: 10.3109/1040841X.2011.580723. [DOI] [PubMed] [Google Scholar]
- 2.Shekhar K, Lye MS, Norlijah O, Ong F, Looi LM, Khuzaiah R, Marzuki I, Hussein I, Wong SL, Mohan J, et al. Deaths in children during an outbreak of hand, foot and mouth disease in Peninsular Malaysia--clinical and pathological characteristics. Med J Malaysia. 2005;60:297–304. [PubMed] [Google Scholar]
- 3.Zhu F, Xu W, Xia J, Liang Z, Liu Y, Zhang X, Tan X, Wang L, Mao Q, Wu J, et al. Efficacy, safety, and immunogenicity of an enterovirus 71 vaccine in China. N Engl J Med. 2014;370:818–28. doi: 10.1056/NEJMoa1304923. [DOI] [PubMed] [Google Scholar]
- 4.Ding NZ, Wang XM, Sun SW, Song Q, Li SN, He CQ. Appearance of mosaic enterovirus 71 in the 2008 outbreak of China. Virus Res. 2009;145:157–61. doi: 10.1016/j.virusres.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Tao ZX, Li Y, Wang HY, Song LZ, Liu GF, Liu Y, Lin XJ, Feng L, Yang H, Fan QY, Xu AQ. Genotype distribution of enterovirus A species isolated in Shandong Province, China. Bing Du Xue Bao. 2009;25:410–4. [PubMed] [Google Scholar]
- 6.Zhu JP, Xu ZG, Chen H, Zhang X, Fan DY, Wang J. Primary detection of pathogen from children with hand, foot, and mouth disease in Beijing, 2007. Bing Du Xue Bao. 2009;25:23–8. [PubMed] [Google Scholar]
- 7.AbuBakar S, Chee HY, Al-Kobaisi MF, Xiaoshan J, Chua KB, Lam SK. Identification of enterovirus 71 isolates from an outbreak of hand, foot and mouth disease (HFMD) with fatal cases of encephalomyelitis in Malaysia. Virus Res. 1999;61:1–9. doi: 10.1016/S0168-1702(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 8.Chen X, Tan X, Li J, Jin Y, Gong L, Hong M, Shi Y, Zhu S, Zhang B, Zhang S, et al. Molecular epidemiology of coxsackievirus A16: intratype and prevalent intertype recombination identified. PLoS One. 2013;8:e82861. doi: 10.1371/journal.pone.0082861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson CR, Doane FW, Rhodes AJ. Report of an outbreak of febrile illness with pharyngeal lesions and exanthem: Toronto, summer 1957; isolation of group A Coxsackie virus. Can Med Assoc J. 1958;79:615–21. [PMC free article] [PubMed] [Google Scholar]
- 10.Alsop J, Flewett TH, Foster JR. “Hand-foot-and-mouth disease” in Birmingham in 1959. Br Med J. 1960;2:1708–11. doi: 10.1136/bmj.2.5214.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perera D, Yusof MA, Podin Y, Ooi MH, Thao NT, Wong KK, Zaki A, Chua KB, Malik YA, Tu PV, et al. Molecular phylogeny of modern coxsackievirus A16. Arch Virol. 2007;152:1201–8. doi: 10.1007/s00705-006-0934-5. [DOI] [PubMed] [Google Scholar]
- 12.Song JH, Park K, Shim A, Kwon BE, Ahn JH, Choi YJ, Kim JK, Yeo SG, Yoon K, Ko HJ, et al. Complete sequence analysis and antiviral screening of medicinal plants for human coxsackievirus a16 isolated in Korea. Osong Public Health Res Perspect. 2015;6:52–8. [DOI] [PMC free article] [PubMed]
- 13.Lu J, Zeng H, Zheng H, Yi L, Guo X, Liu L, Sun L, Tan X, Li H, Ke C, Lin J, et al. Hand, foot and mouth disease in Guangdong, China, in 2013: new trends in the continuing epidemic. Clin Microbiol Infect. 2014;20:O442–5. [DOI] [PubMed]
- 14.Varon A, Wheeler WC. Local search for the generalized tree alignment problem. BMC Bioinformatics. 2013;14:66. doi: 10.1186/1471-2105-14-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei C, Shi J, Liu B, Shi Y, Zheng J, Xu G, Ma J, Wang G, Li F. Molecular characterization of the measles virus genotypes in JiLin Province, China. PLoS One. 2012;7:e46011. doi: 10.1371/journal.pone.0046011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature. 2009;459:1122–5. doi: 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- 18.Tee KK, Lam TT, Chan YF, Bible JM, Kamarulzaman A, Tong CY, akebe Y, Pybus OG. Evolutionary genetics of human enterovirus 71: origin, population dynamics, natural selection, and seasonal periodicity of the VP1 gene. J Virol. 2010;84:3339–50. [DOI] [PMC free article] [PubMed]
- 19.Xu M, Su L, Cao L, Zhong H, Dong N, Dong Z, Xu J. Genotypes of the Enterovirus Causing Hand Foot and Mouth Disease in Shanghai, China, 2012–2013. PLoS One. 2015;10:e0138514. doi: 10.1371/journal.pone.0138514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu J, Luo Z, Wang J, Xu Z, Chen H, Fan D, Gao N, Ping G, Zhou Z, Zhang Y, An J. Phylogenetic analysis of Enterovirus 71 circulating in Beijing, China from 2007 to 2009. PLoS One. 2013;8:e56318. doi: 10.1371/journal.pone.0056318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, He Y, Yang H, Zhu J, Xu X, Dong J, Zhu Y, Jin Q. Genetic characteristics of human enterovirus 71 and coxsackievirus A16 circulating from 1999 to 2004 in Shenzhen, People’s Republic of China. J Clin Microbiol. 2005;43:3835–9. doi: 10.1128/JCM.43.8.3835-3839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin DP. Recombination detection and analysis using RDP3. Methods Mol Biol. 2009;537:185–205. doi: 10.1007/978-1-59745-251-9_9. [DOI] [PubMed] [Google Scholar]
- 23.Negrisolo E, Minelli A, Valle G. The mitochondrial genome of the house centipede scutigera and the monophyly versus paraphyly of myriapods. Mol Biol Evol. 2004;21:770–80. doi: 10.1093/molbev/msh078. [DOI] [PubMed] [Google Scholar]
- 24.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 25.Chan YF, Wee KL, Chiam CW, Khor CS, Chan SY, Amalina WM, Sam IC. Comparative genetic analysis of VP4, VP1 and 3D gene regions of enterovirus 71 and coxsackievirus A16 circulating in Malaysia between 1997–2008. Trop Biomed. 2012;29:451–66. [PubMed] [Google Scholar]
- 26.Huang SW, Hsu YW, Smith DJ, Kiang D, Tsai HP, Lin KH, Wang SM, Liu CC, Su IJ, Wang JR. Reemergence of enterovirus 71 in 2008 in taiwan: dynamics of genetic and antigenic evolution from 1998 to 2008. J Clin Microbiol. 2009;47:3653–62. doi: 10.1128/JCM.00630-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chua KB, Kasri AR. Hand foot and mouth disease due to enterovirus 71 in Malaysia. Virol Sin. 2011;26:221–8. doi: 10.1007/s12250-011-3195-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung WH, Shih SR, Chang CF, Lin TY, Huang YC, Chang SC, Liu MT, Ko YS, Deng MC, Liau YL, et al. Clinicopathologic analysis of coxsackievirus a6 new variant induced widespread mucocutaneous bullous reactions mimicking severe cutaneous adverse reactions. J Infect Dis. 2013;208:1968–78. doi: 10.1093/infdis/jit383. [DOI] [PubMed] [Google Scholar]
- 29.Tan X, Li L, Zhang B, Jorba J, Su X, Ji T, Yang D, Lv L, Li J, Xu W. Molecular epidemiology of coxsackievirus A6 associated with outbreaks of hand, foot, and mouth disease in Tianjin, China, in 2013. Arch Virol. 2015;160:1097–104. doi: 10.1007/s00705-015-2340-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jorba J, Campagnoli R, De L, Kew O. Calibration of multiple poliovirus molecular clocks covering an extended evolutionary range. J Virol. 2008;82:4429–40. doi: 10.1128/JVI.02354-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwai M, Masaki A, Hasegawa S, Obara M, Horimoto E, Nakamura K, Tanaka Y, Endo K, Tanaka K, Ueda J, et al. Genetic changes of coxsackievirus A16 and enterovirus 71 isolated from hand, foot, and mouth disease patients in Toyama, Japan between 1981 and 2007. Jpn J Infect Dis. 2009;62:254–9. [PubMed] [Google Scholar]
- 33.Mizuta K, Abiko C, Aoki Y, Ikeda T, Matsuzaki Y, Hongo S, Itagaki T, Katsushima N, Ohmi A, Nishimura H, Ahiko T. Molecular epidemiology of Coxsackievirus A16 strains isolated from children in Yamagata, Japan between 1988 and 2011. Microbiol Immunol. 2013;57:400–5. doi: 10.1111/1348-0421.12041. [DOI] [PubMed] [Google Scholar]
- 34.Zeng H, Lu J, Zheng H, Yi L, Guo X, Liu L, Rutherford S, Sun L, Tan X, Li H, et al. The Epidemiological Study of Coxsackievirus A6 revealing Hand, Foot and Mouth Disease Epidemic patterns in Guangdong, China. Sci Rep. 2015;5:10550. doi: 10.1038/srep10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He YQ, Chen L, Xu WB, Yang H, Wang HZ, Zong WP, Xian HX, Chen HL, Yao XJ, Hu ZL, et al. Emergence, circulation, and spatiotemporal phylogenetic analysis of coxsackievirus a6- and coxsackievirus a10-associated hand, foot, and mouth disease infections from 2008 to 2012 in Shenzhen, China. J Clin Microbiol. 2013;51:3560–6. doi: 10.1128/JCM.01231-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang CY, Li Lu F, Wu MH, Lee CY, Huang LM. Fatal coxsackievirus A16 infection. Pediatr Infect Dis J. 2004;23:275–6. doi: 10.1097/01.inf.0000115950.63906.78. [DOI] [PubMed] [Google Scholar]
- 37.Legay F, Leveque N, Gacouin A, Tattevin P, Bouet J, Thomas R, Chomelt JJ. Fatal coxsackievirus A-16 pneumonitis in adult. Emerg Infect Dis. 2007;13:1084–6. doi: 10.3201/eid1307.070295. [DOI] [PMC free article] [PubMed] [Google Scholar]