

Abstract
Mesoporous silica nanoparticle supported-lipid bilayers, termed ‘protocells,’ represent a potentially transformative class of therapeutic and theranostic delivery vehicles. The field of targeted drug delivery poses considerable challenges that cannot be addressed with a single ‘magic bullet’. Consequently the protocell has been designed as a modular platform composed of interchangeable biocompatible components. The mesoporous silica core can have variable size and shape to direct biodistribution and controlled pore size and surface chemistry to accommodate diverse cargos. The encapsulating supported lipid bilayer can be modified with targeting and trafficking ligands as well as PEG to effect selective binding, endosomal escape of cargo, drug efflux prevention, and potent therapeutic delivery, while maintaining in vivo colloidal stability. This mini-review describes the individual components of the platform, including the mesoporous silica nanoparticle core and supported lipid bilayer, their assembly (by multiple techniques) into a protocell, and the combined, often synergistic, performance of the protocell based on in vitro and in vivo studies including assessment of biocompatibility and toxicity. We close by commenting on the many emerging variations of the protocell theme and the future directions for protocell research.
Keywords: mesoporous silica, supported lipid bilayer, protocell, nanoparticle, targeted drug delivery
1. Introduction
Targeted delivery of drugs incorporated within nanoparticles can potentially ameliorate a number of problems exhibited by conventional ‘free’ drugs, including poor solubility, limited stability, rapid clearing, and, in particular, lack of selectivity, which results in non-specific toxicity to healthy cells and precludes dose escalations needed to combat multiple drug resistance. An ideal targeted nanoparticle drug carrier, or “nanocarrier” should have the following combined features: 1) the capacity for carrying high levels of multiple diverse molecular cargos (small molecules, drugs with varying physiochemical properties, siRNAs, peptides, imaging agents); 2) the ability to circulate in the blood in vivo for extended periods without elimination by the immune or excretory systems; 3) specificity for binding only to target disease cells; 4) controlled release and intracellular trafficking of the cargo; and 5) low immunogenicity and toxicity. Additionally, as the optimal biodistribution and biological interactions of the nanocarrier can vary between different diseases (and individuals), an ideal nanocarrier should also have physical and chemical properties that can be controlled and essentially tuned for the specific application. Finally, the potential to include imaging agents as well as therapeutics presents the possibility of creating theranostics, which could allow both drug delivery and the monitoring of the course of therapy to be achieved with a single nanocarrier. In the context of creating a tunable nanocarrier that can address this wide range of requirements, protocell constructs have a distinctive combination of features that could potentially enable their development as a ‘universal’ nanocarrier that is both drug and disease agnostic.
2. Challenges in nanomedicine for nanostructured platforms
A wide variety of nanocarrier systems have been developed for the delivery of therapeutic cargo all of which have both advantages and disadvantages, which present challenges for their ultimate clinical use. Major challenges to the successful development of nanotherapeutics include: biocompatibility, ability to load and release varied therapeutic cargos, high cargo loading capacity, the ability to circulate in blood for extended periods of time, evasion of elimination by the immune or excretory systems, specific targeting of and delivery to diseased cells, and low immunogenicity. One of the most successful nanocarrier-based approaches to date is liposomal-based drug delivery, for which there are over a dozen FDA approved formulations and 5 approved for use in cancer.[1–4] The advantages to liposomal nanocarriers are their high biocompatibility, low immunogenicity, flexible formulation, and easy and scaleable synthesis.[5–7] Additionally, the specificity of liposomal formulations can be increased by addition of targeting moieties, such as antibodies, directly to the surface of the liposomes.[3, 5–9] However, it has proven difficult to identify stable lipid formulations that allow drug encapsulation but prevent leakage, making liposomes poor ‘universal’ nanocarriers.[10, 11] Polymeric based therapeutic nanocarriers have also been developed, and several formulations are currently undergoing clinical trials.[3] Similar to lipid formulations, many polymer based nanocarriers are biocompatible and easy to manufacture, however they also suffer from limited stability in in vivo systems and dose dependent toxicity.[12, 13] In addition to the issues specific to each carrier type, both liposomes and polymer based nanoparticles share the issues of invariant size and shape, uncontrollable release profiles, and highly interdependent properties, whereby changing one property, such as loading efficiency, affects numerous other properties, such as size, charge, and stability.[5–7, 9]
Many of the challenges of nanocarrier delivery can be addressed by mesoporous silica nanoparticles (MSNP). MSNP have controllable size and shape and exhibit a high internal surface area (>1000 m2/g) resulting from uniform periodic arrangements of internal nanopores (ranging in diameter from 2 to >20nm) embedded within a silica framework.[14, 15] The major advantage of using MSNP as therapeutic nanocarriers is that their pore size and pore surface chemistries can be easily modified to accommodate a variety of cargos and that their high surface areas result in high loading capacities (vide infra).[15] Additionally, MSNP are biocompatible and degrade overtime with in a biological system into non-toxic silicic acid (Si(OH)4) by-products.[16] However, MSNP use as a nanocarrier is limited by the rapid clearance of the particles by immune and excretory systems after injection.[16–18]
To address the limitations of liposomes, polymer conjugates and MSNP, while taking advantage of their strengths, we developed a flexible modular nanocarrier we term a “protocell” (Figure 1A).[15, 19–24] Protocells are formed by encapsulation of MSNP cores within supported lipid bilayer (SLB) membranes which can then be modified by conjugation with targeting/trafficking ligands and PEG.[18–20, 22, 25–32] They synergistically combine the advantages of liposomes (low inherent toxicity, immunogenicity, and long circulation times) with mesoporous silica nanoparticles (stability and enormous capacity for multiple cargos and disparate cargo combinations).[19, 20, 22, 24] In addition to combining the independent advantages of the MSNP and the liposome systems, the adhesion energy between the MSNP and SLB suppresses large scale membrane bilayer fluctuations responsible for liposome instability and leakage, while the SLB serves to retain soluble cargos within the MSNP. The earliest conceptual protocell was synthesized using micron-sized mesoporous silica particles.[33, 34] The first-generation nanosized protocell consisted of a hydrophilic, spherical MSNP core prepared by aerosol-assisted evaporation-induced silica-surfactant self-assembly[35] fused with either zwitterionic/cationic (DOPC/DOTAP) or zwitterionic/anionic (DOPC/DOPS) liposomes[24] which served to simultaneously load and seal negatively charged cargo within the MSNP and allow it to be delivered across the cell membrane. Since that time many variations of the protocell design have been reported including: lipid monolayer encapsulated hydrophobic MSNP,[18, 30] covalent attachment of lipids to enable chemically triggered release under disulfide reducing conditions,[36] polymer additives to the SLB or monolayer,[29, 30] native cell membrane encapsulated particles,[37, 38] and red blood cell mimicking lipid compositions.[39]
Figure 1.

A) Schematic illustration of the protocell construct. Disparate types of therapeutic and diagnostic agents, such as smaller nanoparticles, toxins, oligonucleotides and drugs, can be loaded within the mesoporous silica core. Targeting ligands, such as peptides or antibodies, and fusogenic peptides can be chemically conjugated to phosphatidylethanolamine (DOPE or DPPE), present in the limited amounts (usually 1–5%) in the supported lipid bilayer (SLB), by a heterobifunctional crosslinker with a PEG spacer arm. The SLB can be composed of either fluid (DOPC) or non-fluid (DPPC) zwitterionic phosphatidylcholine lipids along with cholesterol and can be further modified with PEG-2000 PE, or other agents, to enhance colloidal stability and decrease nonspecific interactions. B) Cryogenic TEM image of the protocell, the white arrows highlight the lipid bilayer on the surface of the MSNP core. Scale bar=25nm. C) Recruitment of Alexa Fluor 647-labelled peptides (white) to the surface of a HCC cell when peptides are displayed on a mesoporous silica thin film-supported lipid bilayer (green) composed of fluid DOPC (open circles) or solid DPPC (closed circles). Cells were labeled with CellTracker Red CMTPX (red) and Hoechst 33342 (blue). Inset scale bars=5μm. Adapted with permission from Ashley 2011.[5]
3. Modular Design and Combined Functions of Protocells
The modular design and synergistic characteristics of the protocell confer a unique combination of properties that can be further independently engineered or tuned for specific applications (Figure 1A): 1) the MSNP core size can be varied from 25-nm to over 250-nm and the MSNP shape can be varied from prismatic to spherical to torroidal to rod-like;[17, 26, 35, 40–43] 2) through self-assembly, the MSNP pore diameter can be varied from 2-nm to over 20-nm,[14] and, using silane coupling chemistry, the pore surface chemistry can be varied to accommodate high concentrations of disparate cargos;[14, 44–46] 3) SLB formation by spontaneous liposome fusion with the silica core seals and protects sensitive cargo (Figure 1B), while SLB destabilization under acidic conditions provides for pH triggered cargo release from the endosome;[19, 20, 22, 27–29] 4) lateral bilayer diffusivity enables recruitment of targeting ligands to cell surface receptors thereby achieving high avidity with low targeting ligand density and reducing immunogenicity and non-specific binding (Figure 1C);[19, 47] 5) the re-configurable SLB surface supports complex biomolecular interactions with the cell surface, involving, for example, targeting, immune cell evasion, and endosomal escape ligands;[18–20, 22, 27–29] 6) the silica dissolution rate and hence release of cargo can be modulated by controlling the extent of siloxane condensation during the synthesis of MSNP;[48–51] 7) both therapeutic compounds and imaging agents can be incorporated to create a theranostic nanocarrier, allowing assessment of protocell stability, biodistribution, co-localization with target cells, toxicity, and efficacy at the cellular/intracellular level as well as in the whole organism.[19, 27, 28, 52, 53]
3.1. MSNP core synthesis
Mesoporous silica nanoparticles are synthesized by colloidal or aerosol-based self-assembly employing surfactants or block co-polymers as structure directing agents. Using solution based colloidal self-assembly processes derived, for example, from the original Stöber process for preparing spherical colloidal silica particles, the synthesis of micrometer- and submicrometer-size spheres of ordered mesoporous oxide MCM-41,[54] or a dendritic process referred to as colloidal stable mesoporous silica nanoparticles (CMS),[55] it is possible to synthesize uniformly sized populations of MSNP with spherical, prismatic, torroidal, rod-like, or hollow shapes with dimensions spanning 25-nm to over 250-nm,[26, 40–43] while in many cases maintaining low polydispersity indices <0.1.[23] Using evaporation induced self-assembly,[35] it is possible to generate in a single step spherical MSNP with a predictable power law particle size distribution spanning 25-nm to over 250-nm. The highly tunable synthesis of MSNP allows for the selection of the size, size distribution, and shape most applicable based on the proposed delivery route and target biodistribution. The MSNP pore and particle surface chemistry can be readily modified via reactions with silanol groups (≡Si-OH) present both within the pore interiors and on the outer surface. Silanol groups (which are partially deprotonated to form anionic ≡Si-O−) are chemically accessible and can be reacted with alkoxy or chlorosilane derivatives to introduce organic functionality. Modification performed in single step or multi-step procedures provides an unlimited ability to ‘tune’ the charge, polarity, and hydrophobic/hydrophilic character of the pore and exterior particle surfaces as well as provide sites for further chemical conjugation or chelation with targeting and control ligands as well as imaging agents.
3.2. Cargo Content and Loading
The controlled pore size and surface chemistry allow multiple cargo types to be efficiently loaded within the MSNP, where the loading efficiency scales with the drug accessible surface area for surface chemistries with attractive drug interactions arising from electrostatic, hydrophobic, hydrogen-bonding or other generally non-covalent forces.[15] The most common cargos are small molecule drugs like doxorubicin (DOX) that can access and interact electrostatically with the negatively charged ~2-nm diameter pores characteristic of MCM-41-type MSNP.[25, 27–29] Loading of hydrophilic small molecule drugs is typically done by incubating the MSNP core with the drug of interest prior to centrifugation, re-suspension in buffer and fusion of liposomes to the surface of the MSNP.[19, 27–29] To load negatively charged cargos, such as nucleic acid cargos or proteins, the MSNP framework can be modified with aminosilanes to produce a positively charged framework.[19, 20, 22] Once the MSNP is positively charged, cargo can be loaded by incubation of the modified MSNP with the cargo of interest prior to liposome fusion. Other methods of drug loading have also been explored, including simultaneous drug loading and liposome fusion [24] and simultaneous SLB assembly and drug loading using a solvent exchange method.[25] To facilitate loading of larger cargo, such as plasmid DNA,[56] larger pore sizes can be achieved with block copolymer templating agents, micro-emulsion procedures,[22] and swelling agents.[57]
The loading of hydrophobic cargo can be achieved in several ways. Using the standard protocell formulation with a SLB, the hydrophobic cargo can be loaded in the hydrophobic domain of the SLB.[27] The use of the hydrophobic domain of the SLB limits the amount of hydrophobic cargo that can be loaded, but does allow for the loading of both a hydrophilic drug in the MSNP core as well as a hydrophobic drug within the same protocell.[27] Hydrophobic cargos can also be loaded from organic solvents like DMSO (or mixtures of DMSO and alcohol) followed by vacuum drying and re-suspension in buffer for liposome fusion.[58] Hydrophobic drug cargos have also been loaded in hybrid protocells composed of organosilane modified MSNP with a single lipid monolayer interacting with molecules directly on the surface of the MSNP.[18, 30, 59]
3.3. Liposomal components and protocell assembly
The earliest protocell lipid formulations consisted zwitterionic/cationic (DOPC/DOTAP),[24, 25] zwitterionic/anionic (DOPC/DOPS),[24] or zwitterionic lipids alone (POPC or DOPC).[25] Since these initial formulations, the complexity of protocell lipid formulations has increased and, as depicted in Figure 1, a large variety of lipid and membrane bound components can be incorporated into the SLB. Most commonly, the major component of the SLB remains a zwitterionic lipid,[19, 20, 22, 27–29, 31, 39] although the cationic lipid DOTAP is occasionally still utilized.[32] In the selection of the primary lipid component, the important design considerations are the lipid melting transition temperature, which controls the SLB fluidity/diffusivity and stability,[19] and charge, which controls interactions with cells and tissues and can affect the fusion of the SLB to the MSNP.[21, 24, 25, 60] The lipid melting transition temperature depends mainly on the length and degree of saturation of the alkane tails, ranging from 55°C for the saturated 18 carbon chain lipid DSPC to −17°C for the single unsaturated 18 carbon chain lipid DOPC. Higher transition temperature lipids increase stability and reduce leakage but can limit the diffusion of targeting ligands conjugated to the lipid head groups (vide infra) reducing multivalent interactions and binding avidity with the target cell surface.[19]
In addition to the primary lipid composition, auxiliary components can be added to control the fluidity of the SLB, increase the colloidal stability and circulation time in vivo, or add functionality to the protocell. In many formulations, cholesterol is added to control the fluidity and leakage of the SLB,[19, 20, 22, 27, 28, 39] and polyethylene glycol (PEG) modified lipids are commonly added to increase colloidal stability and circulation time in vivo.[19, 20, 22, 27, 28, 32] In addition to cholesterol and PEGylated lipids, functional lipids which provide a site for chemical conjugation are included in protocell lipid formulations to allow addition of targeting ligands.[19, 20, 22, 28, 32] The most common functional lipids utilized for addition of targeting are ethanolamine lipids,[19, 20, 22, 28, 32] although other functional lipids incorporating nickel chelating agents have been demonstrated.[31] In addition to adding targeting ligands, lipids modified with polymers such as pluronic P123 and D-α-tocopherol polyethylene glucol 1000 succinate (TPGS) have been used to block drug resistance proteins and thereby add functionality to the lipid bilayer itself.[29, 30]
The most common method of protocell assembly is liposomal fusion, in which mixtures of lipids suspended in organic solvents are dried then hydrated in aqueous buffer, followed by extrusion through a filter to produce liposomes of the desired size. Liposomes spontaneously fuse to the surface of MSNP upon mixing due to the highly lipophilic nature of silica.[60] This phenomenon has been demonstrated using cryoTEM to observe the successive steps of SLB formation on a solid silica nanoparticle.[60] After fusion, excess liposomes are removed by centrifugation and the resulting protocells are resuspended.[19, 22, 28] Protocells can also be assembled by solvent exchange, wherein MSNP are dissolved in ethanolic solution followed by addition of water, which causes transfer of the lipid bilayer directly to the MSNP.[25] A third method of assembly involves adding MSNP in saline to a dried lipid film accompanied by probe sonication, wherein an SLB forms directly or through a liposomal pathway.[27] A critical consideration is the extent of drug leakage during the assembly process and the integrity of the supported lipid bilayer that can be assessed by drug leakage after assembly.[19, 27]
3.4. Targeting chemistry
The multifunctionality of the protocell platform allows for the presentation of targeting agents, including peptide, molecule, and/or antibodies via lipid head group modification, while maintaining biocompatibility and prolonging circulation times by the incorporation of PEGylated or other modified lipids. The major concerns surrounding targeting chemistry are the choice of ligand, the chemical conjugation method, and determination of what stage in the protocell assembly process to perform the conjugation. To date small molecule ligands, such as folate and hyaluronan,[18, 28, 32, 59] and peptides [19, 20, 22] have been used for protocell targeting. Although antibodies have not yet been utilized for protocell targeting, full antibodies, as well as partial antibodies, have been utilized for liposome targeting and could be applied to protocells as well.[3, 8, 61, 62]
After selection of the targeting ligand, a conjugation strategy can then be employed to link the ligand to the protocell. Multiple conjugation strategies that covalently or non-covalently associate the targeting ligand with the functional lipid are possible and the conjugation strategy should be carefully selected to maintain functionality of the targeting ligand. The most common functional lipids utilized for addition of targeting ligands to protocells are ethanolamine lipids, which are linked to the ligand utilizing covalent heterobifunctional linkers.[19, 20, 22, 28, 32] Additionally non-covalent association of a targeting ligand with the protocell has been performed utilizing lipids with incorporated nickel chelating agents and His-tagged targeting ligands.[31] This conjugation method is convenient because it occurs in a single step with good yields, but the ligand-protocell binding interaction is weaker than other covalent strategies, risking the potential dissociation of the targeting ligand. Although only a limited number of conjugation chemistries have been utilized for targeting protocells, additional targeting strategies have been employed for liposome targeting and could be applied to protocells as well. For example, simple thiol groups can be added to both the targeting antibodies and lipids in the liposomes, these thiol groups can be used to conjugate the targeting antibody to the liposome through disulfide bonds. This method has been utilized with targeting antibodies such as anti-HER2 or anti-My9, to create targeted liposomes.[63, 64] Other potential chemistries include click chemistry [65] and avidin/biotin chemistries that have previously been employed with liposome based carriers and could readily be translated to protocells.[66, 67]
Finally the timing of lipid modification with the targeting moiety must be selected; lipids can be modified before creation of the liposome, before liposomal fusion with the MSNP, or after formation of the supported lipid bilayer. Among the earliest reported methods for the addition of targeting moieties involved modification of lipids with a folate derivative, prior to addition to the MSNP.[18, 59] This approach allows the synthesis of large quantities of targeted lipids and works well for the creation of lipid monolayer coated MSNP. However, this method is problematic for the creation of protocells as it is impossible to control the inward and outward orientation of the folate modified lipids on the liposome. To address this concern, preformed liposomes were modified prior to fusion with MSNP.[28] This method assumes the original liposome orientation to be maintained throughout MSNP fusion which may not be the case. Most commonly, targeting moieties are conjugated to completely assembled protocells, resulting in surface-only displayed targeting ligands.[19, 22] Modification of completely assembled protocells is also amenable to non-covalent conjugation chemistries employing, for example, nickel chelating lipids that bind to histidine-terminated ligands.[31] Another unique approach to targeting ligand modification involves the insertion of ligand-functionalized lipids, e.g. folate-modified lipids, after the protocell assembly.[32] Although addition of functionalized lipids after assembly is potentially simple and economical, it is difficult to predict and control the final proportion of ligand-modified lipid incorporation. Perhaps the most promising method for complete outward orientation of targeting ligand display involves a hybrid bilayer constructed by lipid monolayer deposition on an organosilane modified MSNP.[18, 59] Hybrid bilayers form via tail group interaction with the hydrophobic MSNP cores, positioning targeting ligands with the correct outward orientation. This approach was demonstrated using folate modified lipids and resulted in selective uptake in vitro. While many methods of protocell surface modification have been described, many other methods have yet to be reported. Thus, with the multitude of different lipid head group modifications and numerous unexplored functionalization techniques, protocell targeting remains an active area of research.
4. In vitro performance of protocells
Figure 2A depicts the successive stages of protocell binding (step 1), internalization (step 2), endosomal escape (step 3), and nuclear targeting of desired cargo(s) (step 4) by which targeted protocells selectively deliver encapsulated cargos to a cell of interest. Importantly, the fluid but stable SLB promotes lateral diffusivity and enables targeting peptides introduced at low concentrations (important for avoiding non-specific binding and immunogenicity) to be recruited to cell surface receptors (see Figure 1C), promoting high avidity multivalent binding and internalization by receptor mediated endocytosis (Figure 2A, step 1). Dissociation constants (Kd, where Kd is inversely related to affinity) were used to quantify surface binding of SP94-targeted protocells to Hep3B, normal hepatocytes, endothelial cells, and immune cells.[19] Protocells modified with only six SP94 peptides per particle exhibit a 10,000-fold greater affinity for Hep3B than for normal hepatocytes, and other control cells suggesting the specificity necessary for efficacious targeted delivery in vivo. Furthermore, SP94-modified protocells have a 200-fold higher affinity for Hep3B than free SP94, a 1000-fold higher affinity for Hep3B than nanoparticles bearing a non-targeting control peptide, and a 10,000 fold higher affinity for Hep3B than unmodified particles.[19] The affinity of protocells is a function of both peptide density and the fluidity of the supported lipid bilayer; therefore the dissociation constant (Kd) can be precisely controlled by changing the composition of the bilayer to include varying amounts of fluid and non-fluid lipid components (e.g. Figure 1A), which is envisioned to be important for translation to in vivo conditions.[19] To demonstrate that binding results in internalization and cytosolic delivery (Figure 2A, steps 2 and 3) of multiple cargos, Figure 2B shows hyperspectral confocal images of four categories of fluorescently labelled cargo mimics delivered by a single targeted protocell. After 15 minutes (ref. [19] data not shown) calcein (a drug mimic), ds-DNA (an siRNA mimic), red fluorescent protein (a toxin mimic), and quantum dots appear as punctate spots co-localized with fluorescently-labelled silica and lipid indicating incorporation into endosomes consistent with the receptor-mediated endocytotic process depicted in Fig. 2A steps 1 and 2. Within 12 hours (Fig. 2B), calcein, ds-DNA, red fluorescent protein, and quantum dots are delivered into the cytosol, and calcein and dsDNA (both conjugated with a nuclear localization sequence) are further delivered into the nucleus (Figure 2A, step 4). Delivery of the drug from the endosome into the cytosol is crucial for therapeutic efficacy and has emerged as a major problem in nanocarrier-based drug delivery. For the protocell, the natural acidification of the endosome initiates three pH-triggered events insuring endosomal escape (Figure 2A, step 3). First, it reduces the SLB adhesion energy allowing leakage of cargo as confirmed in vitro, second, below its pKa, the endosomolytic peptide H5WYG serves as a proton sponge resulting in endosome swelling and disruption, third, for partially aminated silica cores, lowered pH increases the silica solubility, which along with diffusion controls cargo release. Thus, for protocells, delivery profiles may be tuned/optimized through variation of pore size, charge, and solubility of the silica core along with the extent of SLB modification with the endosomolytic peptide. In vitro delivery has also been demonstrated for other cargos including: siRNA,[22] a variety of anti-cancer drugs,[19, 25, 27–29] a photodynamic therapeutic,[59] and multiple anticancer drugs within a single protocell.[19, 27]
Figure 2.

A) Schematic diagram depicting the successive steps of the multivalent binding [1], internalization [2], endosomal escape [3] and delivery of cargo to the nucleus [4] of peptide targeted protocells. B) Hyperspectral confocal imaging of targeted delivery of multicomponent cargo by protocells to Hep3B cells for 12 hours at 37°C. Alexa Fluor 532-labeled mesoporous silica cores (yellow) were loaded with calcein, dsDNA oligonucleotide (magenta), Red Fluorescent Protein (orange), and CdSe/ZnS quantum dots (teal). Cargos were sealed in the cores by fusion of Texas Red-labeled DOPC liposomes (red). The calcein and dsDNA oligonucleotide were modified with a nuclear localization signal and show accumulation in the nucleus by 12 hours, while the RFP and the quantum dots remain in the cytosol. Scale bars=20μm. C) Left axis (bars in grey and black): The percentage of multidrug resistant positive (MDR+) Hep3B hepatocellular carcinoma cells and normal hepatocytes that remained viable after exposure to free doxorubicin (DOX), targeted-protocell encapsulated DOX, liposomal DOX, targeted-protocells containing a cocktail of chemotheraputics or liposomes containing a cocktail of chemotherapeutics for 24 hours at 37°C at the LC90 value for free DOX. Right axis (bars in red): The percentage of MDR+ Hep3B cells that remain viable after exposure to free doxorubicin (DOX), protocell encapsulated DOX, liposomal DOX, targeted-protocells containing a cocktail of chemotheraputics or liposomes containing a cocktail of chemotherapeutics for 24 hours at 37°C at the LC50 value for free DOX. D) Confocal microscopy images of Hep3B hepatacellular carcinoma cells and normal hepatocytes after exposure to an excess peptide targeted protocells loaded with an anti-cyclin siRNA cocktail for 1 or 48 hours at 37°C. Cells were fix and then imaged by confocal microscopy, protocells are shown in white, cyclins in green and nuclei in blue. Scale bar=20μm . E) Induction of apoptosis by exposure to peptide targeted protocells loaded with an anti-cyclin siRNA cocktail. Cells were classified for early apoptosis (annexin V positive) or late apoptosis (annexin V and Propidium Iodide positive). A, B and C adapted with permission from Ashley 2011.[5] D and E adapted with permission from: Delivery of Small Interfering RNA by Peptide-Targeted Mesoporous Silica Nanoparticle-Supported Lipid Bilayers. Carlee E. Ashley, Eric C. Carnes, Katharine E. Epler, David P. Padilla, Genevieve K. Phillips, Robert E. Castillo, Dan C. Wilkinson, Brian S. Wilkinson, Cameron A. Burgard, Robin M. Kalinich, Jason L. Townson, Bryce Chackerian, Cheryl L. Willman, David S. Peabody, Walker Wharton, and C. Jeffrey Brinker. ACS Nano 2012 6 (3), 2174–2188. Copyright 2012 American Chemical Society.
In addition to delivery of cargo, visualization of protocells in an in vitro system has been utilized to demonstrate targeting specificity and functional cellular response to drug delivery (e.g. Figure 2D). Addition of targeting peptides was shown to provide specificity by demonstrating binding and internalization of protocells, shown in white in the merged images in Figure 2D, to HCC cells but not to normal hepatocytes.[22] These peptide targeted protocells delivered a cocktail of siRNAs to knockdown the expression of a selection of cyclin proteins. Confocal microscopy showed not only specific binding and uptake only in HCC cells but also reduction in cyclin protein expression only in the targeted cancer cells, while leaving the normal hepatocytes unaffected (Figure 2D). In vitro imaging has been used to demonstrate specific targeting of protocells utilizing small molecule ligands, such as folic acid,[18, 32, 59] soluble protein ligands, such as epidermal growth factor,[32] polysaccharides, such as hyaluronan [28] and complex proteins, such as cell surface receptors, Ephrin-B2 and Ephrin-B3.[31]
In vitro systems have also been used to evaluate the therapeutic efficacy of small molecule chemo therapuetic cargo delivery. Delivery of doxorubicin (DOX) or a cocktail of DOX, 5-fluorouracil and cisplatin by peptide targeted protocells demonstrated killing of multidrug resistant HCC cells while sparing normal hepatocytes (Figure 2C).[19] When the targeted-protocells were compared to liposomes containing the same drugs, the liposomes resulted in reduced cell killing of the HCC cells and increased toxicity to normal hepatocytes compared to targeted-protocell delivery (Figure 2C), presumably due to the leakiness of liposomal formulations.[19] The delivery of a variety of drugs by non-targeted protocells has been demonstrated in vitro including: colchicine,[25] gemcitabine,[27] paclitaxel,[27] docetaxel,[28] irinotecan,[29] and DOX,[30] both as single drugs[25, 28–30] and as drug cocktails.[27] In vitro delivery of 8-hydroxyquinoline,[28] DOX,[19] 5-fluorouracil[19], protoporphyrin IX,[59] and cisplatin[19] by targeted protocells both individually[19, 28, 59] and as cocktail[19] has also been demonstrated. In addition to the therapeutic delivery of drugs, in vitro efficacy and cytotxicity of siRNA cargo has also been examined (Figure 2E).[22] Peptide targeted protocells were able to deliver a cocktail of siRNAs to reduce expression of cyclin proteins. The reduction in cyclin proteins resulted in apoptosis of HCC cells but did not cause apoptosis of the normal hepatocytes.[22] In vitro systems have also been used to assess the efficacy of potential therapeutic additions to the lipid bilayer such as Pluronic 123 and D-α-tocopherol polyethylene glucol 1000 succinate (TPGS).[29, 30] Pluronic123 and TPGS, when released from the lipid bilayer on the surface of the protocell, blocks drug pumps present in tumor cells that result in multidrug resistance and results in increased killing of cancer cells in vitro.[29, 30] In addition to delivery of therapeutic cargo, in vitro testing has been utilized to test novel therapeutic technologies which utilize protocells such as specific cargo release by red-light photoactivation,[32] and even the expansion of protocell technology beyond cancer treatment to antiviral therapy.[31]
Finally, in vitro assessment provides a rapid method to evaluate the biocompatibility of various lipid compositions and protocell components prior to progressing to evaluation of toxicity, biocompatibility or therapeutic delivery in an in vivo system. For example, hemotoxicity testing demonstrated that a combination of phosphatidyl choline, phosphatidyl serine and cholesterol, designed to mimic red blood cell membranes, was more biocompatible than phosphatidyl choline alone for the SLB formulation.[39] In vitro testing for biocompatibility, such as hemocompatibility, serves two major functions in the evaluation of a nanocarrier such as the protocell. First, in vitro testing, including hemocompatibility, is required prior to FDA approval. Secondly, in vitro testing can be used to select only those formulations likely to be biocompatible for testing in in vivo systems.
5. In vivo use and testing of protocells
5.1 In vivo Biocompatibility and Toxicity
A critical issue for any nanocarrier is in vivo toxicity and biocompatibility. Toxicity from protocells can arise from either the MSNP core or from the supported lipid bilayer. Although the formulations may vary and individual formulations will need to be tested for biocompatibility and toxicity, liposomes that are biocompatible and non-toxic are FDA-approved for delivery of chemotherapeutic drugs.[8] The toxicity of silicon dioxide has been studied for more than a century and amorphous silica is Generally Recognized as Safe (GRAS) by the FDA. Recently, the toxicity of silica nanoparticles has been extensively investigated, because the high surface to volume ratio of nanoparticles could lead to enhanced cellular interactions and different pathways of toxicity compared with coarse grained silica.[68] Based on the high surface to volume ratio of silica NPs, it might be anticipated that they would show higher toxicity compared with their bulk counterparts. However, the bulk of evidence supports lack of toxicity and the biocompatibility of silica nanoparticles prepared by low temperature colloidal synthesis. Recently amorphous silica nanoparticle ‘C-dots’ (Cornell Dots) were FDA approved for diagnostic applications in a stage I human clinical trial.[69] The FDA approval for a clinical trial of silica nanoparticles should accelerate the acceptance of amorphous, colloidally derived silica particles in medical applications.
In the case of MSNP, the intrinsic porosity of the MSNP surface reduces the extent of hydrogen bonding or electrostatic interactions with cell membranes, a potential cause of silica nanoparticle toxicity.[50] Although the porosity of MSNP should decrease their toxicity, studies of MSNP toxicity have shown variable and occasionally high toxicity. One potential reason for the variability in toxicity studies is the surfactant used to template the pores is toxic and variable amounts of this surfactant can remain within the pores of the MSNP depending on the processing.[49] However, a study which used Fourier transform infrared spectroscopy (FTIR) to confirm that the template surfactant had been removed prior to toxicity testing of the MSNP found survival of all mice treated with up to 1000mg/kg by IV injection and followed for 14 days.[48] The survival of all the animals treated with a very high dose of MSNP that did not retain surfactant shows the lack of intrinsic toxicity of the silica framework of the MSNP.
In addition to toxicity, biocompatibility must also be taken into account. In this area, the porous structure of the MSNP further enhances their biocompatibility as the high surface area and low extent of condensation of the MSNP siloxane framework promotes a high rate of dissolution into soluble silicic acid species, which are nontoxic.[49] The breakdown of the MSNP overtime into nontoxic species supports the potential of repeat and long term use of protocells to deliver drugs as the MSNP core can be cleared from the biological system overtime in a nontoxic way. Examination of animals treated with both PEG coated and unmodified MSNP showed excretion of the silica in both feces and urine without any signs of significant organ damage.[17] Although assessment of toxicity and biocompatibility will be important for each individual protocell formulation that is developed, biocompatibility of the individual components of the protocell should greatly reduce the potential toxicity and enhance the biocompatibility of the complete protocell. Potential toxicity is further mitigated by the high drug loading capacity of MSNP and protocells, which greatly reduces needed dosages and therefore the potential for toxicity. Finally, the ability to add cell specific targeting will further mollify potential toxicity as the protocells are directed specifically to the cells or tissues of interest and will have reduced nonspecific interactions within the body.
5.2 In vivo application of protocell technology
The most common area of research for therapeutic protocells is cancer due to the highly toxic and non-specific nature of most cancer therapeutics. Increased specific delivery of encapsulated cancer therapeutics would address the lack of selectivity, which results in non-specific toxicity to healthy cells and prevents the dose escalation necessary to eradicate diseased cells and overcome drug resistance. The tunable nature of protocells makes them highly adaptable nanocarriers which can be easily altered to fit the needed biodistribution and drug release profile of the specific cancer. Therapeutic protocells have been used to take advantage of the enhanced permeability and retention (EPR) effect which leads to accumulation of nanosized materials in tumors. The EPR effect is due to the rapid growth of tumor vessels which are often abnormal in form and architecture. Due to the abnormal architecture, nanosized carriers are released into the tumor tissue and trapped due to impaired lymphatic drainage often present in tumors.
Although many nanocarriers can take advantage of the EPR effect, the addition of a lipid bilayer to the surface of a drug loaded MSNP greatly increased the EPR effect compared to uncoated MSNP.[29] In addition, a comparison of uncoated MSNP to protocells both loaded with irinotecan showed a significant increase in survival and a reduction in tumor growth in mice treated with the protocells.[29] Protocells created to treat breast cancer have been further modified to address a common breast cancer resistance pathway. The tumor cells were modified to express the breast cancer resistance protein (BCRP), which confers resistance by pumping chemotherapy agents out the cells. To counteract this pump, the protocell lipid bilayer was modified with Pluronic 123, which can block the action of the BCRP. The combined delivery of Pluronic 123 with the chemotherapy agent resulted in the greatest reduction in tumor growth as well as an increase in survival [29]. The ability to both load drugs as well as modify the components in the surface of the protocell to address the BCRP demonstrates the customizability of the protocell platform. Protocell therapeutic delivery has been applied to pancreatic cancer, a notoriously difficult to treat disease, with dual drug loaded protocells.[27] Gemcitabine and paclitaxel were loaded into protocells to treat mice with both subcutaneous and orthotopic pancreatic cancer xenografts. The combined therapy protocells were more effective than single agent gemcitabine protocells or the combined free drug treatment at tumor growth retardation in the subcutaneous tumors and in prevention of metastasis in the orthotopic model.[27] The dual therapeutic protocells utilize the high surface area MSNP core to carry the hydrophilic drug gemcitabine and the hydrophobic space within the lipid bilayer to encapsulate the hydrophobic drug paclitaxel, which can be used in low quantities in combination therapy.
In addition to studies focusing on the EPR effect, targeting moieties have been used to enhance the delivery of protocells (Figure 3). Protocells containing 8-hydroxyquinoline (8-HQ) were optimized to target breast cancer xenografts by the addition of hyaluronan to the surface.[28] Hyaluronan targets CD44, a surface marker highly expressed in breast cancer stem cells. 8-HQ alone is not toxic, but has a synergistic effect when present with a secondary chemotherapy agent and may help overcome chemoresistance. In addition to the targeted protocells loaded with 8-HQ, a non-targeted set of protocells containing docetaxel and relying on the EPR effect were also used as combination therapy. While the docetaxel loaded protocells and the targeted protocells loaded with 8-HQ had limited effect on tumor growth alone, when combined, the effect was stronger than the combined free drugs. Additionally, free docetaxel was very toxic, resulting in significant weight loss, and this toxicity was avoided by inclusion of the drug within the protocell.[28] Targeted delivery, utilizing folate, of photodynamic therapeutic protoporphyrin IX loaded protocells to mice bearing subcutaneous melanoma tumors has also been demonstrated. Delivery of the free protoporphyrin IX alone or in the absence of phototherapy resulted in no response. However, delivery of the protoporphyrin in the protocell in combination with light irradiation resulted in significant reduction in tumor burden.[59]
Figure 3.

Targeted and non-targeted protocells for treatment of breast cancer xenografts. A) Schematic of protocells (MSS) both targeted with hyaluronan (HA-MSS) and loaded with 8-hydroxyquinoline (8-HQ) and non-targeted protocells loaded with docetaxel (DTX). B) Antitumor activity of free drugs in comparison to drug loaded protocells alone or in combination with each other and free drug as shown by tumor size after 43 days. Treatments were given 4 times between day 15 and day 25. C) Assessment of toxicity by the change in body weight ratio. At day 43, the body weight change ratio was compared to saline injection by the Student’s t test. **P< 0.01, *** P<0.001 and n.s. represents not significant (P> 0.05). Adapted with permission from Wang 2013. [15]
6. Future directions
The modular design of the protocell platform has led to rapid advancement in this field; however there are still many areas in which improvements can be made. Many areas of improvement can be achieved by utilizing the recent developments to each modular part of the protocell but which have not yet been combined and applied to protocell technology. For example modifications can be made to the MSNP core, the external lipid layer, the targeting chemistry or to the cargo to create unique protocells for specific applications. Although the protocell has been utilized for therapeutic delivery in cancer in in vivo systems,[27–29] technological improvements could be utilized to increase the therapeutic effectiveness. Increases in effectiveness could be achieved by utilizing advances in MSNP synthesis or lipid conjugations developed independently of protocell development. Recent improvements in MSNP synthesis have allowed for the creation of monosized or narrow size range protocells, which is important for optimizing delivery of cancer therapeutics utilizing the EPR effect. Additionally the flexible lipid formulations available for protocell creation allow a variety of surface chemistries for the addition of targeted ligands. With the emergence of new chemical reactions more commonly known as click chemistry combined with the ability to custom manufacture peptide, single chain variable fragment antibodies (scFv), antibodies and other targeting ligands, opportunities for utilizing targeting of protocells have greatly increased.[65] In particular the potential to use copper-catalyzed or copper free click chemistry for the insertion of targeted ligands has the potential to reach quantitative yields and avoid immunogenicity due to non-reactive lipids, while still allowing the addition of a wide variety of targeting ligands (Figure 4). While neither of these technological advances has yet been applied to protocells in an in vivo system, they have the potential to greatly increase the utility of the protocell.
Figure 4.
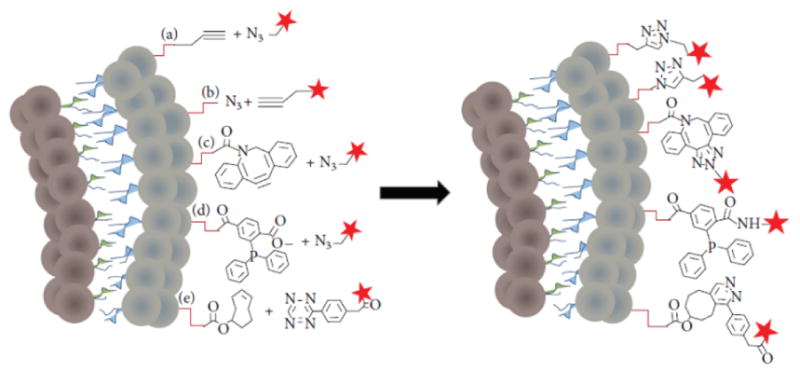
New chemistry approaches for coupling ligands to the lipid bilayer of the protocell. Schematic shows a cross section of a lipid bilayer containing functional groups that form the basis for: copper (I)-catalyzed Huisgen 1,3-dipolar cycloaddition ligation (a and b), copper-free click chemistry ligation (c), Staudinger ligation (d), and tetrazine/trans-cyclooctene inverse electron demand Diels-Alder cycloaddition (e). Coupling ligands for each reaction are represented by a red star. Adapted from Marqués-Gallego 2014.[42]
A second area for advancement is the development of protocells for the delivery of a wider range of therapeutic cargo. Although the delivery of many cargos by protocells has been demonstrated in in vitro systems, these have not been translated to in vivo systems yet and the cargo that has been delivered in vivo by protocells to date is limited. In addition to testing those cargos demonstrated in in vitro systems in an in vivo model, there is a great need to develop new protocells for the delivery of more varied cargo. For example, development of protocells designed to deliver hydrophobic drugs would greatly increase the utility of the protocell technology. To date, only limited research has been done on utilizing modified protocells for the delivery of hydrophobic drugs.[18, 30, 36]
A third area for potential advancement of protocell technology is the modification of protocells to limit the potential for premature drug leakage which can lead to toxicity and limit therapeutic effectiveness. The simplest method to achieve this is the addition of hydrophobic chains to the surface of the MSNP prior to the addition of a lipid layer which prevents premature leakage of hydrophilic drugs from the MSNP core.[18, 30] However even greater control of drug release can be developed utilizing some of the recent technologies that would allow the protocells to react to the tumor microenvironment. For example, lipids can be covalently attached to the MSNP surface through disulfide bonds to provide controlled release of cargo only in the presence of disulfide reducing conditions likely to be present in tumors.[36] Other potential agents such as complex nanomachines or molecular valves can be incorporated to increase the specificity of drug release from the MSNP core.[15] These technologies have only been tested in in vitro systems to date, but have the potential to greatly reduce the toxicity of therapeutic chemotherapy delivered by the protocell.
In addition to their potential as nanocarriers for therapeutics, the modular nature makes protocells ideal imaging and theranostic agents. To date, protocells with incorporated fluorophores have been utilized in in vivo biodistribution in a tumor model.[27] The inclusion of a fluorophore allows real-time assessment of the biodistribution of the protocells in both normal and tumor tissue, which can provide invaluable information for the development of protocells as therapeutic carriers. However, the ability to readily create MSNP cores that incorporate a metallic or magnetic core,[15, 70–72] fluorophores,[27–29] PET imaging agents,[73] or even other nanoparticles such as quantum dots[19] would allow the development of protocells as imaging agents for a variety of imaging technologies. One of the greatest potentials of the large porosity and easily modifiable nature of the MSNP core is the as yet untested potential to add both a clinical imaging agent, such as PET or MRI contrast agent, in addition to a therapeutic cargo to create a truly theranostic nanocarrier that could deliver cargo and monitor the course of therapy simultaneously. Finally, the field of protocells is advancing into other medical areas in addition to cancer therapeutics such as antiviral therapy [31] and will likely advance further into other as yet untested areas of medical research.
7. Conclusion
The modular design of protocell constructs promises a new drug and disease agnostic platform for customized delivery and controlled release of multiple types of cargos and cargo combinations. Packaging drugs within MSNP core protected by the supported lipid bilayer may enable the re-purposing of drugs that have to date failed clinical trials due to poor solubility, high toxicity, and/or susceptibility to degradation. The supported bilayer can retain and protect fragile and/or highly soluble cargos and enable triggered release of the cargo upon acidification within the tumor or tumor microenvironment. The ability to add imaging agents to the core of the protocells gives rise to the potential for more specific imaging agents and even the development of theranostics which can provide both imaging and therapy simultaneously. The modularity of the protocell size, shape, pore architecture and surface chemistry further suggest applications in personalized medicine requiring individualized cargo combinations, targeting, and release profiles. However the modularity and versatility of protocell technology means that there are many factors which must be accounted for in assessing biocompatibility, toxicity, drug release and utility in vivo before protocell technology can be applied in patient populations.
Acknowledgments
This work was supported by NIH National Cancer Institute (NCI) Alliance Grant UO1 CA151792-01, the Lymphoma and Leukemia Society (LLS) Specialized Center of Research (SCOR) Award 7010-14, and Oncothyreon, Inc. P.N.D. was funded by a fellowship from the New Mexico Cancer Nanoscience and Microsystems Training Center (CNTC) and by the George D. Montoya Research Scholarship, Edmund J. & Thelma W. Evans Charitable Trust Scholarship, and the Charlotte and William Kraft Graduate Fellowship. C.J.B acknowledges additional support from U.S. Department of Energy (DOE), Office of Basic Energy Sciences (BES), Division of Materials Sciences and Engineering for supporting research on mesoporous silica materials. CJB also acknowledges support from Sandia National Laboratories’ Laboratory Directed Research and Development (LDRD) program, the Air Force Office of Scientific Research grant FA 9550-1-14-066, National Science Foundation, 21 Environment Protection Agency, and University of California’s Center for Environmental Implications of Nanotechnology. CJB, CEA and ECC acknowledge support from the Defense Threat Reduction Agency grant DTRA JSTO-CBD NATV.
Contributor Information
Kimberly S. Butler, Center for Micro-Engineered Materials, The University of New Mexico, Albuquerque, NM 87131 USA.
Paul N. Durfee, Department of Chemical and Biological Engineering, The University of New Mexico, Albuquerque, NM 87131 USA.
Christophe Theron, Center for Micro-Engineered Materials, The University of New Mexico, Albuquerque, NM 87131 USA.
Carlee E. Ashley, Bioenergy and Defense Technology Department, Sandia National Laboratories, Livermore, CA 94551 USA
Eric C. Carnes, Nanobiology Department, Sandia National Laboratories, Livermore, California 94551
Prof. C. Jeffrey Brinker, Department of Chemical and Biological Engineering, The University of New Mexico, Albuquerque, NM 87131 USA. Center for Micro-Engineered Materials, The University of New Mexico, Albuquerque, NM 87131 USA. Advanced Materials Laboratory, Sandia National Laboratories, Albuquerque, New Mexico 87185.
References
- 1.Pérez-Herrero E, Fernández-Medarde A. Eur J Pharm Biopharm. 2015;93:52–79. doi: 10.1016/j.ejpb.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 2.Akbarzadeh A, Rezaei-Sadabady R, Davaran S, Joo SW, Zarghami N, Hanifehpour Y, Samiei M, Kouhi M, Nejati-Koshki K. Nanoscale Res Lett. 2013;8:102. doi: 10.1186/1556-276X-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Egusquiaguirre SP, Igartua M, Hernandez RM, Pedraz JL. Clin Transl Oncol. 2012;14:83–93. doi: 10.1007/s12094-012-0766-6. [DOI] [PubMed] [Google Scholar]
- 4.Iwamoto T. Biol Pharm Bulletin. 2013;36:715–718. doi: 10.1248/bpb.b12-01102. [DOI] [PubMed] [Google Scholar]
- 5.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 6.Torchilin VP. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 7.Farokhzad OC, Langer R. ACS Nano. 2009;3:16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 8.Deshpande PP, Biswas S, Torchilin VP. Nanomedicine (Lond) 2013;8 doi: 10.2217/nnm.13.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nano. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 10.Drummond DC, Noble CO, Guo Z, Hayes ME, Connolly-Ingram C, Gabriel BS, Hann B, Liu B, Park JW, Hong K, Benz CC, Marks JD, Kirpotin DB. J Control Release. 2010;141:13–21. doi: 10.1016/j.jconrel.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Noble CO, Guo Z, Hayes ME, Marks JD, Park JW, Benz CC, Kirpotin DB, Drummond DC. Cancer Chemo Pharm. 2009;64:741–51. doi: 10.1007/s00280-008-0923-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Draz MS, Fang BA, Zhang P, Hu Z, Gu S, Weng KC, Gray JW, Chen FF. Theranostics. 2014;4:872–892. doi: 10.7150/thno.9404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williford JM, Wu J, Ren Y, Archang MM, Leong KW, Mao HQ. Annu Rev Biomed Eng. 2014;16:347–370. doi: 10.1146/annurev-bioeng-071813-105119. [DOI] [PubMed] [Google Scholar]
- 14.Xiong L, Du X, Shi B, Bi J, Kleitz F, Qiao SZ. J Mater Chem B. 2015;3:1712–1721. doi: 10.1039/c4tb01601g. [DOI] [PubMed] [Google Scholar]
- 15.Tarn D, Ashley CE, Xue M, Carnes EC, Zink JI, Brinker CJ. Acc Chem Res. 2013;46:792–801. doi: 10.1021/ar3000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Q, Zhang Z, Gao F, Li Y, Shi J. Small. 2011;7:271–280. doi: 10.1002/smll.201001459. [DOI] [PubMed] [Google Scholar]
- 17.Huang X, Li L, Liu T, Hao N, Liu H, Chen D, Tang F. ACS Nano. 2011;5:5390–5399. doi: 10.1021/nn200365a. [DOI] [PubMed] [Google Scholar]
- 18.Wang L-S, Wu L-C, Lu S-Y, Chang L-L, Teng IT, Yang C-M, Ho J-aA. ACS Nano. 2010;4:4371–4379. doi: 10.1021/nn901376h. [DOI] [PubMed] [Google Scholar]
- 19.Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, Carroll NJ, Jiang X, Dunphy DR, Willman CL, Petsev DN, Evans DG, Parikh AN, Chackerian B, Wharton W, Peabody DS, Brinker CJ. Nat Mater. 2011;10:389–397. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Epler K, Padilla D, Phillips G, Crowder P, Castillo R, Wilkinson D, Wilkinson B, Burgard C, Kalinich R, Townson J, Chackerian B, Willman C, Peabody D, Wharton W, Brinker CJ, Ashley C, Carnes E. Adv Healthc Mater. 2012;1:348–353. doi: 10.1002/adhm.201200022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J, Jiang X, Ashley C, Brinker CJ. J Am Chem Soc. 2009;131:7567–7569. doi: 10.1021/ja902039y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashley CE, Carnes EC, Epler KE, Padilla DP, Phillips GK, Castillo RE, Wilkinson DC, Wilkinson BS, Burgard CA, Kalinich RM, Townson JL, Chackerian B, Willman CL, Peabody DS, Wharton W, Brinker CJ. ACS Nano. 2012;6:2174–2188. doi: 10.1021/nn204102q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin YS, Haynes CL. J Am Chem Soc. 2010;132:4834–4842. doi: 10.1021/ja910846q. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Stace-Naughton A, Jiang X, Brinker CJ. J Am Chem Soc. 2009;131:1354–1355. doi: 10.1021/ja808018y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cauda V, Engelke H, Sauer A, Arcizet D, Bräuchle C, Rädler J, Bein T. Nano Lett. 2010;10:2484–2492. doi: 10.1021/nl100991w. [DOI] [PubMed] [Google Scholar]
- 26.Du L, Liao SJ, Khatib HA, Stoddart JF, Zink JI. J Am Chem Soc. 2009;131:15136–15142. doi: 10.1021/ja904982j. [DOI] [PubMed] [Google Scholar]
- 27.Meng H, Wang M, Liu H, Liu X, Situ A, Wu B, Ji Z, Chang CH, Nel AE. ACS Nano. 2015;9:3540–57. doi: 10.1021/acsnano.5b00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Huang J, Wang X, Yu Y, Zhang H, Chen Y, Liu J, Sun Z, Zou H, Sun D, Zhou G, Zhang G, Lu Y, Zhong Y. Biomaterials. 2013;34:7662–73. doi: 10.1016/j.biomaterials.2013.06.042. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X, Li F, Guo S, Chen X, Wang X, Li J, Gan Y. Biomaterials. 2014;35:3650–65. doi: 10.1016/j.biomaterials.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 30.Han N, Zhao Q, Wan L, Wang Y, Gao Y, Wang P, Wang Z, Zhang J, Jiang T, Wang S. ACS Appl Mater Interfaces. 2015;7:3342–3351. doi: 10.1021/am5082793. [DOI] [PubMed] [Google Scholar]
- 31.Porotto M, Yi F, Moscona A, LaVan DA. PLoS ONE. 2011;6:e16874. doi: 10.1371/journal.pone.0016874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackowiak SA, Schmidt A, Weiss V, Argyo C, von Schirnding C, Bein T, Bräuchle C. Nano Lett. 2013;13:2576–2583. doi: 10.1021/nl400681f. [DOI] [PubMed] [Google Scholar]
- 33.Buranda T, Huang J, Ramarao GV, Ista LK, Larson RS, Ward TL, Sklar LA, Lopez GP. Langmuir. 2003;19:1654–1663. [Google Scholar]
- 34.Bayerl TM, Bloom M. Biophys J. 1990;58:357–62. doi: 10.1016/S0006-3495(90)82382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu Y, Fan H, Stump A, Ward TL, Rieker T, Brinker CJ. Nature. 1999;398:223–226. [Google Scholar]
- 36.Roggers RA, Lin VS-Y, Trewyn BG. Mol Pharm. 2012;9:2770–7. doi: 10.1021/mp200613y. [DOI] [PubMed] [Google Scholar]
- 37.Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Proc Natl Acad Sci U S A. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu C-MJ, Fang RH, Wang K-C, Luk BT, Thamhiwatana S, Dehaini D, Nguyen P, Angantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu SJ, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L. Nature. 2015 doi: 10.1038/nature15373. epub ahead of print. [DOI] [Google Scholar]
- 39.Roggers RA, Joglekar M, Valenstein JS, Trewyn BG. ACS Appl Mater Interfaces. 2014;6:1675–81. doi: 10.1021/am4045713. [DOI] [PubMed] [Google Scholar]
- 40.Chen Y, Chen H, Zhang S, Chen F, Zhang L, Zhang J, Zhu M, Wu H, Guo L, Feng J, Shi J. Adv Funct Mater. 2011;21:270–278. [Google Scholar]
- 41.Han L, Zhou Y, He T, Song G, Wu F, Jiang F, Hu J. J Mater Sci. 2013;48:5718–5726. [Google Scholar]
- 42.Meng H, Yang S, Li Z, Xia T, Chen J, Ji Z, Zhang H, Wang X, Lin S, Huang C, Zhou ZH, Zink JI, Nel AE. ACS Nano. 2011:4434–4447. doi: 10.1021/nn103344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trewyn BG, Nieweg JA, Zhao Y, Lin VSY. Chem Eng J. 2008;137:23–29. [Google Scholar]
- 44.Du X, Qiao SZ. Small. 2015;11:392–413. doi: 10.1002/smll.201401201. [DOI] [PubMed] [Google Scholar]
- 45.Nandiyanto ABD, Kim SG, Iskandar F, Okuyama K. Micropor Mesopor Mat. 2009;120:447–453. [Google Scholar]
- 46.Huh S, Wiench JW, Yoo J-C, Pruski M, Lin VSY. Chem Mater. 2003;15:4247–4256. [Google Scholar]
- 47.Stachowiak JC, Hayden CC, Sasaki DY. Proc Natl Acad Sci U S A. 2010;107:7781–7786. doi: 10.1073/pnas.0913306107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He L, Lai H, Chen T. Biomaterials. 2015;51:30–42. doi: 10.1016/j.biomaterials.2015.01.063. [DOI] [PubMed] [Google Scholar]
- 49.He Q, Zhang Z, Gao Y, Shi J, Li Y. Small. 2009;5:2722–2729. doi: 10.1002/smll.200900923. [DOI] [PubMed] [Google Scholar]
- 50.Slowing II, Wu CW, Vivero-Escoto JL, Lin VS. Small. 2009;5:57–62. doi: 10.1002/smll.200800926. [DOI] [PubMed] [Google Scholar]
- 51.Brinker CJ, Scherer GW. Sol-gel science: the physics and chemistry of sol-gel processing. Gulf Professional Publishing; 1990. [Google Scholar]
- 52.Li C, Yang D, Ma P, Chen Y, Wu Y, Hou Z, Dai Y, Zhao J, Sui C, Lin J. Small. 2013;9:4150–4159. doi: 10.1002/smll.201301093. [DOI] [PubMed] [Google Scholar]
- 53.Yanes RE, Tarn D, Hwang AA, Ferris DP, Sherman S, Thomas CR, Lu J, Pyle AD, Zink JI, Tamanoi F. Small. 2013;9:697–704. doi: 10.1002/smll.201201811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grün M, Lauer I, Unger KK. Adv Mater. 1997;9:254–257. [Google Scholar]
- 55.Möller K, Kobler J, Bein T. Adv Funct Mater. 2007;17:605–612. [Google Scholar]
- 56.Du X, Shi B, Tang Y, Dia S, Qiao SZ. Biomaterials. 2014;35:5580–5590. doi: 10.1016/j.biomaterials.2014.03.051. [DOI] [PubMed] [Google Scholar]
- 57.Shen D, Yang J, Li X, LZ, Zhang R, Li W, Chen L, Wang R, Zhang F, Zhao D. Nano Lett. 2014;14:923–932. doi: 10.1021/nl404316v. [DOI] [PubMed] [Google Scholar]
- 58.Ferris DP, Lu J, Gothard C, Yanes RE, Thomas CR, Olsen J, Stoddart JF, Tamanoi F, Zink JI. Small. 2011;7:1816–1826. doi: 10.1002/smll.201002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teng IT, Chang YJ, Wang LS, Lu HY, Wu LC, Yang CM, Chiu CC, Yang CH, Hsu SL, Ho JA. Biomaterials. 2013;34:7462–70. doi: 10.1016/j.biomaterials.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 60.Mornet S, Lambert O, Duguet E, Brisson A. Nano Lett. 2005;5:281–285. doi: 10.1021/nl048153y. [DOI] [PubMed] [Google Scholar]
- 61.Reynolds JG, Geretti E, Hendriks BS, Lee H, Leonard SC, Klinz SG, Noble CO, Lücker PB, Zandstra PW, Drummond DC, Olivier KJ, Jr, Nielsen UB, Niyikiza C, Agresta SV, Wickham TJ. Toxicol Appl Pharmacol. 2012;262:1–10. doi: 10.1016/j.taap.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 62.Sapra P, Allen TM. Prog Lipid Res. 2003;42:439–62. doi: 10.1016/s0163-7827(03)00032-8. [DOI] [PubMed] [Google Scholar]
- 63.Shaik MS, Kanikkannan N, Singh M. J Control Release. 2001;76:285–295. doi: 10.1016/s0168-3659(01)00450-3. [DOI] [PubMed] [Google Scholar]
- 64.Park JW, Hong K, Kirpotin DB, Colbern G, Shalaby R, Baselga J, Shao Y, Nielsen UB, Marks JD, Moore D, Papahadjopoulos D, Benz CC. Clinical Cancer Res. 2002;8:1172–1181. [PubMed] [Google Scholar]
- 65.Marqués-Gallego P, de Kroon AIPM. BioMed Res Int. 2014;2014:12. doi: 10.1155/2014/129458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Torchilin VP, Goldmacher VS, Smirnov VN. Biochem Biophys Res Comm. 1978;85:983–990. doi: 10.1016/0006-291x(78)90640-x. [DOI] [PubMed] [Google Scholar]
- 67.Papadia K, Markoutsa E, Antimisiaris SG. J Biomed Nanotechnol. 2014;10:871–876. doi: 10.1166/jbn.2014.1792. [DOI] [PubMed] [Google Scholar]
- 68.Meng H, Xue M, Xia T, Ji Z, Tarn DY, Zink JI, Nel AE. ACS Nano. 2011;5:4131–4144. doi: 10.1021/nn200809t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y, Humm J, Gönen M, Kalaigian H, Schöder H, Strauss HW, Larson SM, Wiesner U, Bradbury MS. Sci Transl Med. 2014;6:260ra149. doi: 10.1126/scitranslmed.3009524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen Y, Wang X, Liu T, Zhang DS, Wang Y, HG, Di W. I J Nanomed. 2015;10:2579–2594. doi: 10.2147/IJN.S78774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim J, Kim HS, Lee N, Kim T, Kim H, Yu T, Song IC, Moon WK, Hyeon T. Angew Chem Int Ed. 2008;47:8438–8443. doi: 10.1002/anie.200802469. [DOI] [PubMed] [Google Scholar]
- 72.Wu S-H, Lin Y-S, Hung Y, Chou Y-H, Hsu Y-H, Chang C, Mou C-Y. ChemBioChem. 2008;9:53–57. doi: 10.1002/cbic.200700509. [DOI] [PubMed] [Google Scholar]
- 73.Kumar R, Roy I, Ohulchanskky TY, Vathy LA, Bergey EJ, Sajjad M. ACS Nano. 2010;4:699–708. doi: 10.1021/nn901146y. [DOI] [PMC free article] [PubMed] [Google Scholar]