

Abstract
Significance: Reactive oxygen species (ROS) reactive nitrogen species (RNS) and redox processes are of key importance in obesity- and diabetes-related kidney disease; however, there remains significant controversy in the field. Recent Advances: New data from imaging and in vivo models of obesity and diabetic kidney disease have shed new insights into this field. In the setting of obesity- and diabetes-related kidney injury, there is a growing recognition that the major moieties of ROS and RNS are hydrogen peroxide and peroxynitrite with the enzymatic sources being NADPH oxidases and nitric oxide synthase, respectively. However, the role of mitochondrial superoxide as a driver of renal complications remains unclear. Critical Issues: Several key issues that are often not discussed are the specific ROS and RNS molecules, the source of generation, the location of production, and downstream targets. Future Directions: Further understanding of the role of ROS/RNS/redox and their relationship with key signaling and metabolic pathways such as AMP-activated protein kinase (AMPK) and hypoxia-inducible factor 1-α (HIF1α) will be critical to a new understanding of kidney complications of caloric challenges and new therapeutic approaches. Antioxid. Redox Signal. 25, 208–216.
Introduction
The use of aerobic oxygen took on paramount importance as eukaryotic and multicellular organisms developed a benefit from using oxygen for more efficient adenosine triphosphate (ATP) generation and providing evolutionary advantages over other life forms. The necessity for ATP generation took on greater significance as specific cell types became responsible for specialized tasks that were required to support multicellular organisms. As fish developed the ability to extract oxygen from the water for efficient utilization of carbon sources for ATP generation, they were able to form more complex structures with specific organs. With life forms adapting to life on land, oxygen extraction was switched from gills and water to lungs and air. Indeed, the rise in oxygen concentration was beneficial for organisms with efficient means of using oxygen for survival and evolutionary advantage, whereas anaerobic organisms would not be benefited. The Great Oxidation Event is a term used to describe the first mass extinction of anaerobic organisms and the rise in eukaryotes and multicellular organisms on earth (7, 38) about 2.2 billion years ago in concordance with a rise in atmospheric oxygen (Fig. 1).
FIG. 1.
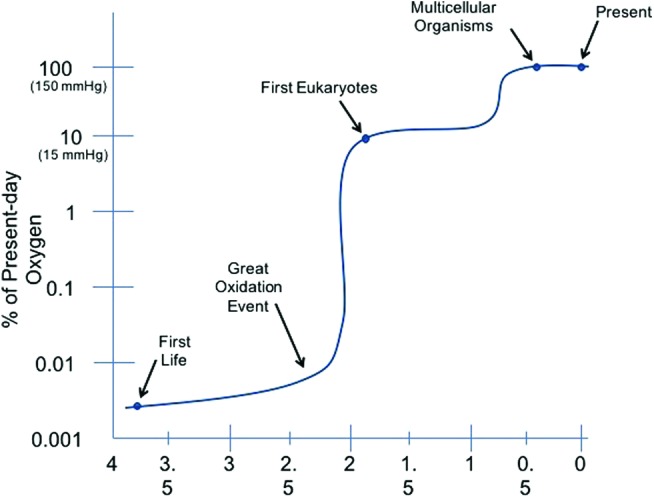
Oxygen in the atmosphere and evolutionary complexity of organisms. Ancient photosynthetic prokaryotes (cyanobacteria) released oxygen first into the ocean, then into the atmosphere. The building of oxygen allowed the evolution of bigger eukaryotes and then multicellular organisms. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Oxygen was not viewed as a toxic molecule until the recognition that oxygen radicals were developed and used as a host defense and killing mechanism by bacteria and eukaryotic cells to defend and attack potential invaders. Over the past six decades, the concept that reactive oxygen species (ROS) are toxic molecules and should be suppressed and scavenged has taken on greater popularity in the lay media and in the scientific community. The Free Radical Theory of Aging suggests that oxidants can be harmful and was initially met with scientific skepticism (21). Indeed there has been a recent major revision in the view that free radical oxidant molecules are a major cause of aging (46, 47, 49). However, the scientific community has largely accepted the theory that mitochondrial superoxide is a major unifying theory for diabetes- and obesity-related complications (41, 42). Specifically in obesity-related and diabetes-associated kidney disease, the concept that oxidant production is a major unifying theory for the development and progression of kidney disease has been widely accepted. The acceptance of this theory is somewhat surprising as several negative studies have been reported that do not support the theory, and in some cases even suggest that reducing ROS production in a nonspecific manner may be actually harmful (9, 19, 68).
In the present review, the role of oxidants and redox molecules as a cause of diabetes- and obesity-related kidney injury is examined and suggestions for more direct investigations are put forth to delineate the possible role of specific ROS molecules in the development and progression of nutrient-induced kidney disease. The term “nutrient-induced kidney disease” includes the features of both obesity-related and diabetes-associated chronic kidney disease.
Redox Balance, ROS, and Kidney Disease
The most commonly accepted definition of redox reactions includes all chemical reactions in which atoms have their oxidation state changed; in general, redox reactions involve the transfer of electrons between chemical species. The chemical species from which the electron is stripped from is “oxidized”, whereas the chemical species to which the electron is added is “reduced”. For the purpose of this review, the series of reactions linked to oxidant production is examined with their connection to kidney diseases linked to diabetes and obesity.
ROS and reactive nitrogen species (RNS) can be considered as unstable oxygen- or nitrogen-containing molecules that have a reactive electron or are an intermediate that can generate reactive molecules. The O2 or H2O molecules are considered to be relatively stable forms of oxygen. Normally, superoxide, hydrogen peroxide, hydroxyl radicals, and peroxynitrite are “oxidizing agents” that can oxidize a variety of key molecules and alter their function. Accordingly, oxidation of proteins, nucleic acids, lipoproteins, and phospholipids may be harmful in a specific context. The major sources of ROS include mitochondria (which are thought to provide about 80% of endogenously produced superoxide at the basal level), NADPH oxidases (NOXs), and nitric oxide synthases (NOSs) (Fig. 2). The precise amount and type of oxidant molecules produced by these sources under various metabolic conditions remain controversial as the methods to specifically measure each reactive oxidant moiety are not very quantitative and few have been applied in real-time in vivo states. Thus, much of the field relies on indirect measures of the footprint of oxidant production.
FIG. 2.
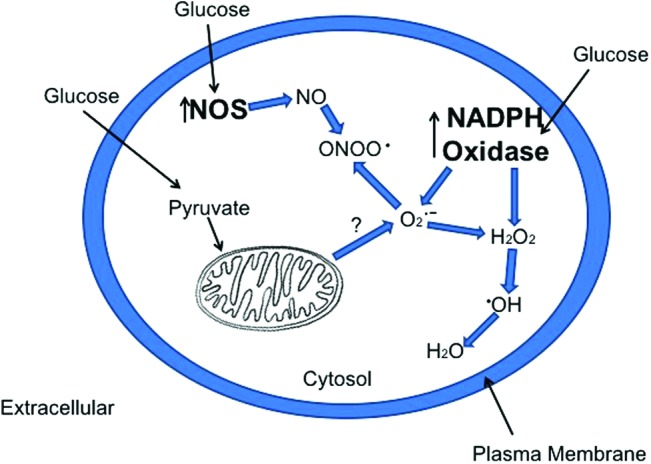
Major sources of superoxide, hydrogen peroxide, and peroxynitrite in response to elevated glucose. The major sources of ROS production in response to excess intracellular glucose are possibly NOSs, NOXs, and mitochondria. The production of peroxynitrite from NOS and hydrogen peroxide from NOX are consistently observed, whereas measurement of superoxide is difficult to measure; therefore, the mitochondrial source is unclear. H2O2, hydrogen peroxide; NO, nitric oxide; NOS, nitric oxide synthase; NOXs, NADPH oxidases; O2−, superoxide anion; ONOO•, peroxynitrite; ROS, reactive oxygen species. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In almost all published studies that examine measures of ROS in diabetic tissue, there is a uniform overproduction of ROS molecules in the tissues of diabetic animals and patients (23, 26, 45). We too have found similar results, thus there is little doubt that overall ROS production is increased in diabetic tissues. Similar data are found in models of obesity as well (10, 44). However, several major questions have emerged in recent years regarding which species of reactive oxygen are increased, and what is the source of increased ROS in diabetic tissues and with obesity. These questions are largely unresolved. To address these questions we begin with an understanding of the major source of superoxide generation in relation to mitochondrial oxidative phosphorylation.
Oxygenation, Glucose, and Fatty Acid Oxidation in Kidney and Heart
Glucose metabolism begins with glycolysis and leads to formation of pyruvate from glucose in enzymatic steps not requiring oxygen. Glycolysis is a relatively ancient pathway and is exclusively used in anaerobic organisms (Fig. 3). The glycolytic steps leading to lactate and ATP production are relatively fast and may be more prominent in states of high-glucose entry even under normoxic conditions (59). Facultative anaerobic organisms are organisms that can make ATP in the presence of oxygen via aerobic respiration and revert to fermentation or anaerobic respiration in the absence of oxygen (Fig. 3). Mammalian cells are generally considered facultative anaerobes and can switch to anaerobic glycolysis or aerobic glycolysis depending on the availability of oxygen and particular challenges. The transference of electrons is a critical step involved in aerobic oxidation of glucose within the mitochondria and specifically within the electron transport chain (ETC).
FIG. 3.

Glucose metabolism in presence of oxygen and with low oxygen. Glucose metabolism is primarily via glycolysis leading to pyruvate formation and further metabolism in the tricarboxylic acid (TCA) cycle within the mitochondria. Alternative paths include metabolism via the pentose phosphate shunt pathway and aldose reductase pathway. With hypoxic conditions there is enhanced pyruvate to lactate glycolysis and reduced pyruvate uptake into the mitochondria. With an elevation in intracellular glucose and a hypoxic or pseudohypoxic reduction in Ox-Phos activity, there could be a backup of TCA cycle metabolites leading to enhanced citrate conversion to lipid synthesis, αKG leading to amino acid synthesis, and malate leading to nucleotide synthesis. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Fatty acid oxidation also requires oxygen and takes place in the mitochondrial and peroxisomal compartments. The role of fatty acid oxidation has been well studied in the heart as the heart generally prefers beta-oxidation of fatty acid for ATP generation under baseline euglycemic and well-perfused states. The heart uses the majority of oxygen consumption for fatty acid oxidation as opposed to glucose oxidation (33, 39). Although there is a greater generation of ATP per gram from fatty acids as opposed to glucose, the relative efficiency of oxygen utilization for ATP generation actually favors glucose oxidation. Cardiac metabolism is primarily aerobic and ATP is generated via oxidative phosphorylation of fatty acids and glucose. During transient and mild ischemia as well as diminishing oxygen supply, there is a proportionate reduction in oxidative phosphorylation, although fatty acid oxidation remains the primary source of ATP generation. There is an increase in the ATP production from glycolysis in an effort to maintain the energy needs for continued contractility, but cellular integrity may be compromised partly because of increased lactate and hydrogen ion generation from glycolysis (34). The balance between energy needs and control of cytoplasmic and mitochondrial metabolites has been termed “metabolic homeostasis.” In heart tissue, the regulation of metabolic homeostasis involves a complex interplay of cytoplasmic and mitochondrial networks that regulate the rate of ATP production over a wide range of workloads. Although there is remarkable flexibility under well-perfused conditions, there is a tremendous challenge under conditions of ischemia. The additional challenge of diabetes and obesity presents an additional set of variables that are yet to be resolved.
In the kidney, the relative roles of glucose and fatty acid oxidative phosphorylation and glycolysis are even more complex as there are multiple cell types with different propensities for glucose and fatty acid metabolism under the baseline euglycemic, well-perfused state. Using in vitro studies or based on transcriptomic profiles of the major enzymes involved (hexokinase, phosphofructokinase, and pyruvate kinase), glycolysis appears to be largely present in the inner medulla and papilla. Stimulation of the pentose phosphate shunt is initiated by glucose 6-phosphate dehydrogenase (G6PD). As this enzyme is present throughout the nephron and at high levels in glomeruli and proximal tubule, it is possible that there is high activity of this pathway in these segments. In one study, using C1–C6 labeled glucose consumption, the pentose phosphate shunt accounted for 10% of glucose utilization in tissue slices and various nephron segments (48). Cortical glycolysis leading to production of lactate appears negligible in the rat kidney under fasting conditions, whereas there is evidence of ongoing glycolytic lactate production in the medullary segments (48). There also appears to be a relationship between glycolysis and sodium reabsorption in the thick ascending loop of Henle.
Glucose oxidation is also preferentially greater in medullary segments than proximal segments. Based on flux studies with 3H2O and 13CO2 generation, the ratio of glucose oxidation was found to be 3:1 in medullary tubules versus cortical tubules (48). The absolute amount has been estimated to be about 25 μmol/h/g dry weight in cortical tubules and between 100 and 180 μmol/h/g in medullary tubules. Although proximal tubules have a low ability to undergo glycolysis and glucose oxidation at basal states, the relative contributions of glycolysis and glucose oxidation are unclear under conditions of high glucose or diabetes. In contrast to glucose oxidation, the proximal tubule preferentially undergoes fatty acid oxidation much more so than distal or medullary segments (48). The proximal tubule is considered to be a major source of mitochondrial beta-oxidation of free nonesterified fatty acids and is the major source of ATP production. Oxidation of fatty acids has been identified to account for two-thirds of the overall oxygen consumption of the kidney. A recent study demonstrated that reduced fatty acid oxidation is a feature of advanced kidney disease and is linked to profibrotic pathways (28); however, the metabolic pathways that characterize the initial and late stages of disease with different nephron segments have not been well described. The regulation of these metabolic pathways in disease states will be important to identify whether oxidant stress is generated via mitochondrial or nonmitochondrial sources.
ROS Production in Diabetes- and Obesity-Related Kidney Disease
Excess mitochondrial superoxide production as a unifying cause of diabetic complications was espoused by Brownlee (42) and has been widely accepted. The theory is that cells susceptible to noninsulin-dependent glucose uptake will metabolize excess glucose via glycolysis and that pyruvate uptake into mitochondria will be enhanced. The increased activity of the tricarboxylic acid cycle will lead to increased production of electron donors, and via enhanced ETC activity there will be an excess of superoxide production. Although it is quite clear that there is likely enhanced glucose flux and increased glycolytic and possibly enhanced tricarboxylic acid (TCA) activity in cells and tissues exposed to high glucose (8, 61), the question of increased ETC activity and superoxide production emanating from the ETC remains in doubt. The initial studies by Brownlee and colleagues used aortic endothelial cells in culture to demonstrate increased ROS production by 2′,7′-dichlorofluorescein (which primarily measures H2O2 and not superoxide), and by using inhibitors of the ETC, they found that downstream pathways of protein kinase C (PKC), advanced glycation end-products, and sorbitol were reduced (42). However, similar studies were not performed in vivo (Fig. 4).
FIG. 4.
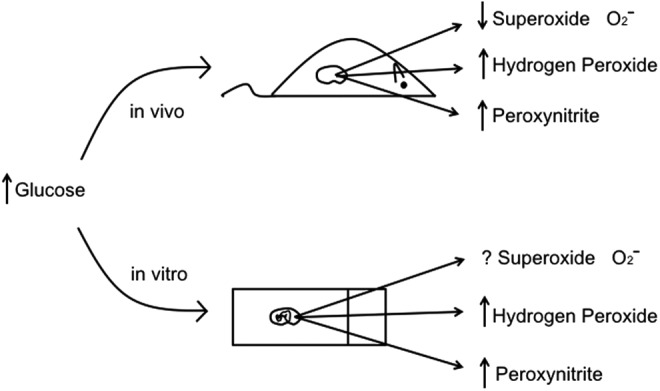
Comparison of findings on major ROS molecules in the in vivo state and in vitro cell culture in response to elevated glucose. In the in vivo state and in vitro state, there is an increase in H2O2 and peroxynitrite. However, in the in vivo state, there is a reduction in superoxide with type 1 diabetic models, whereas the regulation of superoxide by high glucose in vitro is unclear.
A major concern with in vitro studies is the change in oxygen tension from the in vivo, which will alter mitochondrial oxidative metabolism in a dramatic manner. These studies raise doubt whether data generated from cell culture can be translated to the in vivo state (38). Recent studies by our group attempted to address the question of superoxide production in the type 1 diabetic kidney using a dihydroethidium (DHE) probe as a measure of in vivo superoxide production (14). DHE is a relatively specific fluorescent indicator of superoxide when given in vivo and imaged by real-time analysis or by confocal microscopy. The specific oxidized form of DHE was found to be ethidium as measured by fluorescence lifetime decay (20). Using both real-time measurements and confocal analysis, there was a surprising reduction of DHE fluorescence in glomeruli and cortical tubules, indicating reduced superoxide production in the diabetic kidney versus the control kidney (14). A similar reduction of superoxide was also found using electron paramagnetic resonance of freshly harvested kidney cortical tissue as compared with the control kidney. In addition, H2O2 generation as measured with Amplex Red was also found to be reduced from mitochondria of the diabetic kidney in response to various substrates. Thus a combination of approaches all found that superoxide generation from the kidney and ROS production from the mitochondria of the diabetic kidney were actually reduced. Nevertheless, overall production of hydrogen peroxide, nitrotyrosine, and 8-hydroxy-2′-deoxyguanosine were all increased in the kidney or urine from diabetic mice (Fig. 4). The overall conclusion was that mitochondrial oxidative activity was reduced in the diabetic kidney and may account for the reduction in superoxide production.
The basis for reduced superoxide production is unclear. It is likely that reduced mitochondrial electron transport activity and reduced glucose oxidation may contribute to this effect. Complexes I, III, and IV activity was reduced in the diabetic kidney and associated with an overall reduction of mitochondrial content and biogenesis. Similar reductions of complexes I and III activity have been found in muscle tissue of patients with diabetes (29). It has been argued that the reduction in complex I activity is due to an initial elevation of superoxide initially generated from increased electron complex activity. However, the data for this cyclical argument are not conclusive, especially from in vivo studies. An enhancement of DHE-detected superoxide production was associated with improved complex I and III activity and reduced renal inflammation and fibrosis when AMP-activated protein kinase (AMPK) was activated in diabetic mice (14). Furthermore, studies with DHE demonstrated an enhanced DHE/superoxide response in mitochondrial SOD2-deficient mouse kidneys without any enhanced diabetic kidney disease in SOD2-deficient mice (14). These studies do not support the contention that a primary stimulation of mitochondrial superoxide is necessary for subsequent reduction of mitochondrial ETC activity (52). Similar data and conclusions were provided in studies with diabetic neuropathy by Fernyhough's group (5, 6, 16).
Several other groups have found increased ROS in mesangial and endothelial cells; however, the source has been linked to NOXs and NOSs. In endothelial cells, the major source of O2− was found to be membrane-bound oxidases, which use NADH and NADPH as substrates (27). The NOX proteins contain NADPH and FAD-binding domains in their C-terminal region. NADPH is the electron donor to molecular O2 to form superoxide anion. In contrast to other NOX isoforms, NOX4 has been found to primarily produce H2O2 (90% of ROS) and relatively less superoxide (10% of ROS) (43). NOX4 in particular has been found to be consistently upregulated in diabetic kidney disease (3, 15, 24, 54, 60, 63, 64), and the cytosolic SOD (SOD1) (17, 18) appears to be the major regulator of oxidant-mediated stress in the diabetic kidney. These studies provide convincing evidence that NOXs in the cytosolic compartment are likely the major source of harmful ROS production in diabetic kidneys. Similarly, NOX4 has been consistently found to be stimulated with obesity-related kidney disease by several groups and H2O2 production is stimulated as a major source of ROS in obesity-related kidney disease (10, 11, 54). The role of NOX4 in adipocyte differentiation appears to be well established (22) and NOX4 deficiency may be protective against high-fat-induced adipocyte expansion (32). However, the kidney appears to require NOX4 for the inflammation associated with obesity-related kidney disease and is largely dependent on AMPK activation.
Recently, the role of NOX4 as a key factor in kidney disease associated with podocyte dysfunction was demonstrated with the development of a podocyte-specific inducible NOX4 transgenic mouse (64). With induction of NOX4 in podocytes, there was a rapid development of glomerular volume expansion, glomerular basement membrane thickening, mesangial matrix expansion, and podocyte dropout, all features consistent with the glomerular features of diabetes- and obesity-related glomerulopathy. Data consistent with these findings were reported by an independent study in podocyte-specific NOX4 knockout mice (25). Induction of diabetes in these mice led to attenuation of albuminuria, mesangial matrix expansion, and glomerular basement membrane thickening. The downstream effects of NOX4-mediated ROS effects to cause diabetic kidney and obesity-related kidney disease are beginning to be revealed. NOX4-mediated stimulation of PKC-α may contribute to many of the NOX4-dependent effects in diabetic kidney disease (56). In addition, a novel pathway where NOX4 inhibits the enzyme fumarate hydratase and leads to accumulation of fumarate has also been linked to stimulation of transforming growth factor-β (TGF-β), matrix genes, and hypoxia-inducible factor 1-α (HIF1α) (64).
There is also convincing data that NOSs are stimulated in diabetic kidney disease and that endothelial nitric oxide synthase (eNOS) overproduction of NO may combine to form peroxynitrite in association with superoxide (40, 50). The source of the superoxide may be NOS itself or superoxide generated from NOX or cytosolic xanthine oxidase, peroxisomal oxidases, or endoplasmic reticular oxidases. When NOS is uncoupled with its cofactor tetrahydrobiopterin or its substrate, arginine, there can be a massive increase in superoxide formation (36). Overproduction of NOS isoforms as a contributor to diabetic neuropathy has also been recently demonstrated (1) (Fig. 2).
Redox Balance, AMPK, Pseudohypoxia, and HIF
Oxidant production and its effects on inflammation and fibrosis are linked to reversible redox. Redox can also be described to characterize the reaction by which oxidants affect critical proteins, such as a kinase or phosphatase, and antioxidants, such as glutathione (GSH), to reduce the protein back to its original state. The redox state and oxidants also appear to be potent regulators of the master energy sensor, AMPK.
Several groups have demonstrated an important role of AMPK in several renal diseases, including obesity and diabetic kidney disease. AMPK activation has been found to be beneficial in reducing the inflammation and fibrosis noted with a high-fat diet (10, 11), type 1 diabetes (14, 15), as well as in the subtotal nephrectomy model. AMPK activation may promote reduced inflammation via inhibiting NFκB activation and NOX activation (15, 51, 54, 55). In addition, AMPK activation appears to have a potent ability to reduce NOX4 production and production of H2O2 (10, 14). With respect to fibrosis, AMPK activation reduced TGF-β-mediated fibrosis and has been recently found to inhibit high glucose and TGF-β-induced nuclear translocation of Smad4 (65). The combined regulation of AMPK/Sirtuins/PGC1α and mTOR axis has been consistently found to play central roles in obesity- and diabetes-related kidney disease and is likely to be fundamental to many chronic kidney disorders. The interaction of ROS/RNS and antioxidants to explain the regulation of the combined axis is a major challenge to further understanding of obesity and diabetic kidney disease development and pathogenesis.
AMPK has been found to be reduced in the kidney from obese and diabetic animals, although the basis for reduced AMPK is unclear. It has been well established that even small changes in AMP are sensed by AMPK and in any state wherein AMP is increased, as in starvation and ischemia, are associated with marked stimulation of AMPK. However, the regulation of AMPK during nutrient stress or caloric overfeeding is less clear. There is likely to be short-term and chronic effects on the ATP/AMP ratio, which is a potent regulator of AMPK. Liver kinase B1 (LKB1) is a potent regulator of AMPK and has been found to be reduced in the diabetic kidney and may be a major basis for reduced AMPK activity in the diabetic kidney (31). Several groups have found that ROS, including H2O2 and superoxide, may stimulate AMPK activity and contribute to the stimulation of AMPK in ischemic and hypoxic states marked by overproduction of ROS (4). Similarly, there is convincing data that RNS, such as NO and peroxynitrite, are also potent stimulators of AMPK (4). However, there is concomitant reduction of AMPK in the diabetic state along with an overproduction of H2O2 and peroxynitrite. Whether there is suppression of AMPK by an excess of ATP, reduced LKB1 activity, or reduced mitochondrial superoxide production remains unclear.
Along with regulation of AMPK, there is convincing data that hypoxia and HIF1α are regulated in the diabetic kidney. Hypoxia is of course a potent stimulus for HIF1α, whereas in the diabetic state there is stimulation of HIF1α in vivo but no apparent reduction in overall oxygen content (Fig. 5). The stimulation of HIF1α is consistently observed in the in vivo state but surprisingly not replicated by high glucose in cell culture models of diabetic kidney disease. Stimulation of HIF1α would lead to stimulation of “anaerobic” glycolysis and lactate production via enhanced lactate dehydrogenase as well as suppression of mitochondrial oxidative phosphorylation and TCA activity via activating pyruvate dehydrogenase kinase (PDK). Enhanced activity of PDK will phosphorylate the pyruvate dehydrogenase E1a subunit, leading to impaired pyruvate conversion to acetyl CoA (30). The concept of pseudohypoxia to explain an increase in the NADH/NAD ratio with diabetes has been postulated by Williamson et al. (61). However, the overall increase in NADH/NAD ratio in diabetic target tissues has not been consistently reported (12), possibly because of difficulties in the accurate measurements of NADH and NAD and the lack of robust in vivo approaches. Often, there is coexistent stimulation of AMPK in hypoxic states as there would be a reduction in oxygen availability for oxidative phosphorylation and a consequent reduction in ATP and accumulation of AMP. However, as noted, AMPK is typically reduced in the diabetic state at the same time that HIF1α is stimulated (Fig. 6). A possible contribution to the stimulation of HIF1α may be due to accumulation of the metabolites fumarate and succinate that can compete with alpha-ketoglutarate to inhibit prolyl hydroxylase and thus reduce HIF1α ubiquitination and degradation (37, 62). As both fumarate (64) and succinate (57) have been found to accumulate in diabetic kidney, the basis of the pseudohypoxia stimulation of HIF1α may well be due to metabolite regulation (Fig. 6). A targeted metabolomics analysis in both experimental models (2, 64) and the human condition of diabetes- (35, 53, 58) and obesity-related kidney disease (13, 66, 67) will be very useful to unravel in vivo regulation and translation of critical and relevant pathways of renal disease progression.
FIG. 5.
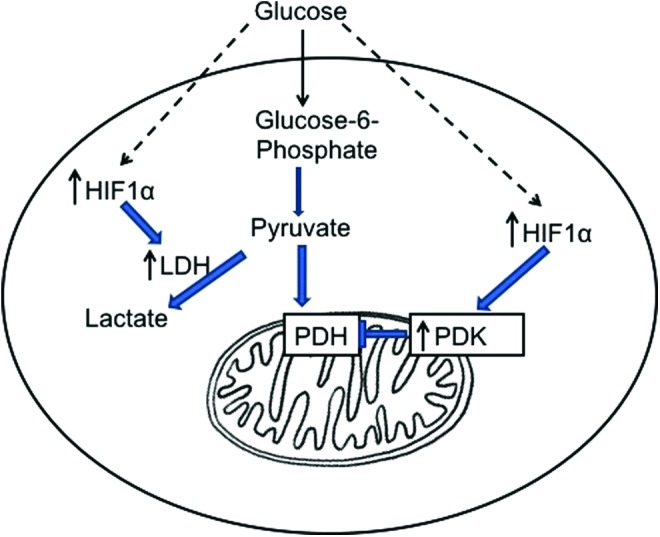
HIF1α regulation and glycolysis. HIF1α is activated with high glucose and stimulates LDH to stimulate glycolysis and lactate production and inhibits PDH via PDK to inhibit pyruvate entry to mitochondria. HIF1α, hypoxia-inducible factor 1-α; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase complex; PDK, pyruvate dehydrogenase kinase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 6.

Feed forward loop to maintain reduced AMPK and increase HIF1α in renal cells with increased intracellular glucose. The reduced activity of AMPK will enhance NOX4 leading to increased H2O2. The increased H2O2 reduces fumarate hydratase protein levels, leading to accumulation of fumarate. The increased fumarate can stimulate HIF1α accumulation, which drives the glycolytic process but suppresses oxidative phosphorylation and reduces mitochondrial superoxide. AMPK, AMP-activated protein kinase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Concluding Remarks
The role of ROS, RNS, and redox balance in obesity and diabetic kidney disease has been intensively investigated in the past decade and several consensus views are emerging. There appears to be a consistent finding of an elevation in many ROS/RNS molecules, such as H2O2 and peroxynitrite, with their sources being likely NOX4 and uncoupled NOS (eNOS). The regulation of NOX4 and eNOS uncoupling is, in part, related to reduced AMPK activity, and stimulation of AMPK activity has been consistently linked to improved outcomes. The role of superoxide has been more controversial and its link to mitochondrial function will need to be revised as additional advances in visualizing mitochondrial function in vivo are being developed. Redox relationships are central to obesity- and diabetes-related kidney disease and further analysis of specific regulators of redox with metabolic pathways will be central to the explanation of nutrient or caloric-stress responses in the kidney.
Abbreviations Used
- AMPK
AMP-activated protein kinase
- ATP
adenosine triphosphate
- eNOS
endothelial nitric oxide synthase
- ETC
electron transport chain
- HIF1α
hypoxia-inducible factor 1-α
- LKB1
liver kinase B1
- NOS
nitric oxide synthase
- NOX
NADPH oxidase
- PDK
pyruvate dehydrogenase kinase
- PKC
protein kinase C
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- TCA
tricarboxylic acid
- TGF
transforming growth factor
Acknowledgments
Funding for this project was provided by NIDDK DP3-DK094352 to K.S. The author thanks Ms. Pardis Gholami for help with figure preparation.
References
- 1.Ahlawat A, Rana A, Goyal N, and Sharma S. Potential role of nitric oxide synthase isoforms in pathophysiology of neuropathic pain. Inflammopharmacology 22: 269–278, 2014 [DOI] [PubMed] [Google Scholar]
- 2.Bain JR, Stevens RD, Wenner BR, Ilkayeva O, Muoio DM, and Newgard CB. Metabolomics applied to diabetes research: moving from information to knowledge. Diabetes 58: 2429–2443, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Block K, Gorin Y, and Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A 106: 14385–14390, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cardaci S, Filomeni G, and Ciriolo MR. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J Cell Sci 125: 2115–2125, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Chowdhury SK, Dobrowsky RT, and Fernyhough P. Nutrient excess and altered mitochondrial proteome and function contribute to neurodegeneration in diabetes. Mitochondrion 11: 845–854, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chowdhury SK, Smith DR, and Fernyhough P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol Dis 51: 56–65, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Crowe SA, Dossing LN, Beukes NJ, Bau M, Kruger SJ, Frei R, and Canfield DE. Atmospheric oxygenation three billion years ago. Nature 501: 535–538, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Dabkowski ER, Williamson CL, Bukowski VC, Chapman RS, Leonard SS, Peer CJ, Callery PS, and Hollander JM. Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am J Physiol Heart Circ Physiol 296: H359–H369, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Heerspink HJ, McMurray JJ, Meyer CJ, Parving HH, Remuzzi G, Toto RD, Vaziri ND, Wanner C, Wittes J, Wrolstad D, and Chertow GM. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 369: 2492–2503, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Decleves AE, Mathew AV, Cunard R, and Sharma K. AMPK mediates the initiation of kidney disease induced by a high-fat diet. J Am Soc Nephrol 22: 1846–1855, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Decleves AE, Zolkipli Z, Satriano J, Wang L, Nakayama T, Rogac M, Le TP, Nortier JL, Farquhar MG, Naviaux RK, and Sharma K. Regulation of lipid accumulation by AMK-activated kinase in high fat diet-induced kidney injury. Kidney Int 85: 611–623, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diederen RM, Starnes CA, Berkowitz BA, and Winkler BS. Reexamining the hyperglycemic pseudohypoxia hypothesis of diabetic oculopathy. Invest Ophthalmol Vis Sci 47: 2726–2731, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding W. and Mak RH. Early markers of obesity-related renal injury in childhood. Pediatr Nephrol 30: 1–4, 2015 [DOI] [PubMed] [Google Scholar]
- 14.Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE, Andreyev A, Quach T, Ly S, Shekhtman G, Nguyen W, Chepetan A, Le TP, Wang L, Xu M, Paik KP, Fogo A, Viollet B, Murphy A, Brosius F, Naviaux RK, and Sharma K. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest 123: 4888–4899, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eid AA, Ford BM, Block K, Kasinath BS, Gorin Y, Ghosh-Choudhury G, Barnes JL, and Abboud HE. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J Biol Chem 285: 37503–37512, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernyhough P. Mitochondrial dysfunction in diabetic neuropathy: a series of unfortunate metabolic events. Curr Diab Rep 15: 89, 2015 [DOI] [PubMed] [Google Scholar]
- 17.Fujita H, Fujishima H, Chida S, Takahashi K, Qi Z, Kanetsuna Y, Breyer MD, Harris RC, Yamada Y, and Takahashi T. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J Am Soc Nephrol 20: 1303–1313, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita H, Fujishima H, Takahashi K, Sato T, Shimizu T, Morii T, Shirasawa T, Qi Z, Breyer MD, Harris RC, Yamada Y, and Takahashi T. SOD1, but not SOD3, deficiency accelerates diabetic renal injury in C57BL/6-Ins2(Akita) diabetic mice. Metabolism 61: 1714–1724, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez-Cabrera MC, Ristow M, and Vina J. Antioxidant supplements in exercise: worse than useless? Am J Physiol Endocrinol Metab 302: E476–E477; author reply E478–E479, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Hall DJ, Han SH, Chepetan A, Inui EG, Rogers M, and Dugan LL. Dynamic optical imaging of metabolic and NADPH oxidase-derived superoxide in live mouse brain using fluorescence lifetime unmixing. J Cereb Blood Flow Metab 32: 23–32, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harman D. About “Origin and evolution of the free radical theory of aging: a brief personal history, 1954–2009”. Biogerontology 10: 783, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Higuchi M, Dusting GJ, Peshavariya H, Jiang F, Hsiao ST, Chan EC, and Liu GS. Differentiation of human adipose-derived stem cells into fat involves reactive oxygen species and Forkhead box O1 mediated upregulation of antioxidant enzymes. Stem Cells Dev 22: 878–888, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu T, Ramachandrarao SP, Siva S, Valancius C, Zhu Y, Mahadev K, Toh I, Goldstein BJ, Woolkalis M, and Sharma K. Reactive oxygen species production via NADPH oxidase mediates TGF-beta-induced cytoskeletal alterations in endothelial cells. Am J Physiol Renal Physiol 289: F816–F825, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HH, and Jandeleit-Dahm KA. Genetic targeting or pharmacologic inhibition of NADPH oxidase Nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25: 1237–1254, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jha JC, Thallas-Bonke V, Banal C, Gray SP, Chow BS, Ramm G, Quaggin SE, Cooper ME, Schmidt HH, and Jandeleit-Dahm KA. Podocyte-specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia 59: 379–389, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jun M, Venkataraman V, Razavian M, Cooper B, Zoungas S, Ninomiya T, Webster AC, and Perkovic V. Antioxidants for chronic kidney disease. Cochrane Database Syst Rev 10: CD008176, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalinowski L. and Malinski T. Endothelial NADH/NADPH-dependent enzymatic sources of superoxide production: relationship to endothelial dysfunction. Acta Biochim Pol 51: 459–469, 2004 [PubMed] [Google Scholar]
- 28.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, and Susztak K. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelley DE, He J, Menshikova EV, and Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51: 2944–2950, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Kim JW, Tchernyshyov I, Semenza GL, and Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Lee MJ, Feliers D, Sataranatarajan K, Mariappan MM, Li M, Barnes JL, Choudhury GG, and Kasinath BS. Resveratrol ameliorates high glucose-induced protein synthesis in glomerular epithelial cells. Cell Signal 22: 65–70, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Mouche S, Sajic T, Veyrat-Durebex C, Supale R, Pierroz D, Ferrari S, Negro F, Hasler U, Feraille E, Moll S, Meda P, Deffert C, Montet X, Krause KH, and Szanto I. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int J Obes (Lond) 36: 1503–1513, 2012 [DOI] [PubMed] [Google Scholar]
- 33.Lopaschuk GD. Optimizing cardiac fatty acid and glucose metabolism as an approach to treating heart failure. Semin Cardiothorac Vasc Anesth 10: 228–230, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Lopaschuk GD. Targets for modulation of fatty acid oxidation in the heart. Curr Opin Investig Drugs 5: 290–294, 2004 [PubMed] [Google Scholar]
- 35.Lu J, Xie G, Jia W, and Jia W. Metabolomics in human type 2 diabetes research. Front Med 7: 4–13, 2013 [DOI] [PubMed] [Google Scholar]
- 36.Luo S, Lei H, Qin H, and Xia Y. Molecular mechanisms of endothelial NO synthase uncoupling. Curr Pharm Des 20: 3548–3553, 2014 [DOI] [PubMed] [Google Scholar]
- 37.MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, and Gottlieb E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol 27: 3282–3289, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mailloux RJ. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol 4: 381–398, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Masoud WG, Abo Al-Rob O, Yang Y, Lopaschuk GD, and Clanachan AS. Tolerance to ischaemic injury in remodelled mouse hearts: less ischaemic glycogenolysis and preserved metabolic efficiency. Cardiovasc Res 107: 499–508, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montezano AC. and Touyz RM. Reactive oxygen species and endothelial function—role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin Pharmacol Toxicol 110: 87–94, 2012 [DOI] [PubMed] [Google Scholar]
- 41.Nishikawa T, Brownlee M, and Araki E. Mitochondrial reactive oxygen species in the pathogenesis of early diabetic nephropathy. J Diabetes Investig 6: 137–139, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, and Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404: 787–790, 2000 [DOI] [PubMed] [Google Scholar]
- 43.Nisimoto Y, Diebold BA, Cosentino-Gomes D, and Lambeth JD. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 53: 5111–5120, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Percy CJ, Brown L, Power DA, Johnson DW, and Gobe GC. Obesity and hypertension have differing oxidant handling molecular pathways in age-related chronic kidney disease. Mech Ageing Dev 130: 129–138, 2009 [DOI] [PubMed] [Google Scholar]
- 45.Pollock JS. and Pollock DM. Endothelin, nitric oxide, and reactive oxygen species in diabetic kidney disease. Contrib Nephrol 172: 149–159, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med 20: 709–711, 2014 [DOI] [PubMed] [Google Scholar]
- 47.Ristow M. and Schmeisser K. Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response 12: 288–341, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ross BD, Espinal J, and Silva P. Glucose metabolism in renal tubular function. Kidney Int 29: 54–67, 1986 [DOI] [PubMed] [Google Scholar]
- 49.Sanz A. and Stefanatos RK. The mitochondrial free radical theory of aging: a critical view. Curr Aging Sci 1: 10–21, 2008 [DOI] [PubMed] [Google Scholar]
- 50.Satoh M, Fujimoto S, Haruna Y, Arakawa S, Horike H, Komai N, Sasaki T, Tsujioka K, Makino H, and Kashihara N. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am J Physiol Renal Physiol 288: F1144–F1152, 2005 [DOI] [PubMed] [Google Scholar]
- 51.Sharma K. The link between obesity and albuminuria: adiponectin and podocyte dysfunction. Kidney Int 76: 145–148, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Sharma K. Mitochondrial hormesis and diabetic complications. Diabetes 64: 663–672, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma K, Karl B, Mathew AV, Gangoiti JA, Wassel CL, Saito R, Pu M, Sharma S, You YH, Wang L, Diamond-Stanic M, Lindenmeyer MT, Forsblom C, Wu W, Ix JH, Ideker T, Kopp JB, Nigam SK, Cohen CD, Groop PH, Barshop BA, Natarajan L, Nyhan WL, and Naviaux RK. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol 24: 1901–1912, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, Falkner B, and Goldstein BJ. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 118: 1645–1656, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song P. and Zou MH. Regulation of NAD(P)H oxidases by AMPK in cardiovascular systems. Free Radic Biol Med 52: 1607–1619, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thallas-Bonke V, Jha JC, Gray SP, Barit D, Haller H, Schmidt HH, Coughlan MT, Cooper ME, Forbes JM, and Jandeleit-Dahm KA. Nox-4 deletion reduces oxidative stress and injury by PKC-alpha-associated mechanisms in diabetic nephropathy. Physiol Rep 2: pii: , 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, Hanner F, Meer E, and Peti-Peterdi J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest 118: 2526–2534, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van der Kloet FM, Tempels FW, Ismail N, van der Heijden R, Kasper PT, Rojas-Cherto M, van Doorn R, Spijksma G, Koek M, van der Greef J, Makinen VP, Forsblom C, Holthofer H, Groop PH, Reijmers TH, and Hankemeier T. Discovery of early-stage biomarkers for diabetic kidney disease using ms-based metabolomics (FinnDiane study). Metabolomics 8: 109–119, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vazquez A, Liu J, Zhou Y, and Oltvai ZN. Catabolic efficiency of aerobic glycolysis: the Warburg effect revisited. BMC Syst Biol 4: 58, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams KJ, Qiu G, Usui HK, Dunn SR, McCue P, Bottinger E, Iozzo RV, and Sharma K. Decorin deficiency enhances progressive nephropathy in diabetic mice. Am J Pathol 171: 1441–1450, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, Nyengaard JR, van den Enden M, Kilo C, and Tilton RG. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 42: 801–813, 1993 [DOI] [PubMed] [Google Scholar]
- 62.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, and Guan KL. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26: 1326–1338, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.You YH, Okada S, Ly S, Jandeleit-Dahm K, Barit D, Namikoshi T, and Sharma K. Role of Nox2 in diabetic kidney disease. Am J Physiol Renal Physiol 304: F840–F848, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.You YH, Quach T, Saito R, Pham J, and Sharma K. Metabolomics reveals a key role for fumarate in mediating the effects of NADPH oxidase 4 in diabetic kidney disease. J Am Soc Nephrol 27: 466–481, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao J, Miyamoto S, You YH, and Sharma K. AMP-activated protein kinase (AMPK) activation inhibits nuclear translocation of Smad4 in mesangial cells and diabetic kidneys. Am J Physiol Renal Physiol 308: F1167–F1177, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao YY, Cheng XL, and Lin RC. Lipidomics applications for discovering biomarkers of diseases in clinical chemistry. Int Rev Cell Mol Biol 313: 1–26, 2014 [DOI] [PubMed] [Google Scholar]
- 67.Zhao YY, Cheng XL, Lin RC, and Wei F. Lipidomics applications for disease biomarker discovery in mammal models. Biomark Med 9: 153–168, 2015 [DOI] [PubMed] [Google Scholar]
- 68.Zoja C, Corna D, Nava V, Locatelli M, Abbate M, Gaspari F, Carrara F, Sangalli F, Remuzzi G, and Benigni A. Analogs of bardoxolone methyl worsen diabetic nephropathy in rats with additional adverse effects. Am J Physiol Renal Physiol 304: F808–F819, 2013 [DOI] [PubMed] [Google Scholar]