

ABSTRACT
The majority of marketed seasonal influenza vaccines are prepared using viruses that are chemically inactivated and treated with a surfactant. Treating with surfactants has important consequences: it produces ‘split viruses’ by solubilizing viral membranes, stabilizes free membrane proteins and ensures a low level of reactogenicity while retaining high vaccine potency. The formulation stability and potency of split influenza vaccines are largely determined by the specifics of this ‘splitting’ process; namely, the consequent conformational changes of proteins and interactions of solubilized particles, which may form aggregates. Robust methods to quantitatively determine the split ratio need to be developed before optimal splitting conditions can be investigated to streamline production of superior influenza vaccines. Here, we present a quantitative method, based on both steady-state and time-resolved fluorescence spectroscopy, to calculate the split ratio of the virus after surfactant treatment. We use the lipophilic dye Nile Red (NR) as a probe to elucidate molecular interactions and track changes in molecular environments. Inactivated whole influenza viruses obtained from a sucrose gradient were incubated with NR and subsequently treated with increasing concentrations of the surfactant Triton X-100 (TX-100) to induce virus splitting. NR's emission spectra showed that the addition of TX-100 caused ˜27 nm red-shifts in the emission peak, indicative of increasingly hydrophilic environments surrounding NR. The emission spectra of NR at different surfactant concentrations were analyzed with multi-peak fitting to ascertain the number of different micro-environments surrounding NR and track its population change in these different environments. Results from both the emission spectra and fluorescence lifetime spectroscopy revealed that NR showed presence in 3 distinct molecular environments. The split ratio of the virus was then calculated from the percentages of NR in these environments using both fluorescence emission and lifetime data. This study can pave the way for the development of robust methods to rapidly quantify splitting extent during vaccine manufacturing.
KEYWORDS: fluorescence emission, fluorescence lifetime, influenza split virus vaccine, influenza virus, membrane-surfactant interactions, Nile Red, partitioning of dye in membranes, protein aggregation, split ratio quantification, surfactant, Triton X-100, vaccine formulation, vaccine manufacturing, virus splitting, virus-surfactant interactions
Introduction
Influenza virus has significant socioeconomic impacts with burgeoning costs of more than US$ 71 billion and ˜500,000 deaths globally per year.1 The increasing number of pandemic cases observed in the last few decades is particularly concerning.2 This stresses the importance of timely development of quality vaccines, which at present is mostly based on knowledge acquired as a result of trial-and-error approaches. The development of quality vaccines by rational formulation design instead will be possible only if a fundamental understanding of molecular interactions between vaccine constituents is acquired. This is critical considering that seasonal influenza vaccines must be prepared each year in a short time frame.
Most of the influenza vaccines in the market employ surfactants to split the whole intact virus and subsequently solubilize the fragments of lipids and proteins.3 In particular, Triton X-100 (TX-100) has been used successfully as a mild, non-denaturing surfactant to solubilize surface-exposed membrane glycoproteins hemagglutinin (HA) and neuraminidase (NA) and other virus proteins.4,5 During the manufacturing process, the required TX-100 concentration to split the virus is usually <1% (w/v) depending on the virus strain, but in general the final product contains <0.015% TX-100 after filtration steps.6 When the inner contents of virus particles are released by membrane disintegration, a vast number of biophysical interactions including protein aggregation may occur. The efficacy of split virus vaccines is largely determined by how immunogenic the solubilized particles are and whether 3D protein structures are conserved.7 The ability of TX-100 to solubilize viral proteins with preserved structures ensures that split virus vaccines exhibit sufficient immunogenicity while displaying lower reactogenicity when compared to that of whole virus vaccines.
The main reason manufacturers need to inevitably proceed with trial-and-error methods established over years is due to lack of in-depth understanding about splitting parameters and kinetics.2 Every so often this instigates alarming issues. For example, in 2010 Fluvax® was found to display increased reactogenicity causing adverse side effects like fever, malaise, shivering and febrile convulsions. Although the root cause of these adverse reactions is still debated, all the signs hint at ‘sub-optimal splitting’ of the virus. While this indicates a strong relationship between the level of splitting and reactogenicity is hypothesized, there has been little research on virus–surfactant interactions at the molecular level to establish this. Furthermore, there is currently no established method to rapidly quantify the extent of splitting or ‘split ratio’. This renders potential studies on the ‘optimization of splitting extent’ unfeasible for the time being because the split ratio of a formulation deemed as ideal (maximal immunogenicity and minimal reactogenicity) through immunological studies will not be known to establish an optimum. Also, given that factors such as intrinsic virus stability and glycosylation can lead to differences in splitting kinetics across various strains, a ‘separate optimum’ may need to be established for each different strain, especially new ones. The achievement of such a task would again depend on the development of a method for rapid and reliable quantification of the split ratio. In the case of different optima for different strains, time-consuming immunological studies mentioned above may be bypassed through the development of a database of optimum splitting ratios for a wide variety of strains with different glycosylation features; such a database can serve as a comparison tool to quickly determine the optimal splitting ratio for new strains that are similar in many ways to older counterparts. Overall, studies to improve the quality of split virus vaccines need to consider the impact of splitting extent, the determination of which requires a reliable and rapid method.
Previously many studies have employed dye-binding with fluorescence spectroscopy to investigate protein unfolding, lipid interactions and partitioning of dyes in membranes.8,9 Here, we adopted the same approach and studied the splitting process of inactivated, whole influenza/A/2009/Brisbane virus by TX-100, using Nile Red (NR) dye. Given NR's ability to bind to lipid membranes and protein aggregates,10 the splitting process was monitored quantitatively by studying molecular interactions and micro-environmental changes around dye molecules in the presence of TX-100, using fluorescence emission and lifetime spectroscopy. Qualitative monitoring was also performed by imaging viruses in different stages of splitting using transmission electron microscopy (TEM).
Results
Influence of NR concentration and surfactant on NR fluorescence
Nile red is not an efficient fluorophore in an aqueous environment even at very high concentrations, so its fluorescence intensity does not change significantly in the control sample (Fig. 1A - circles). In the presence of virus, however, the fluorescence intensity increases by 5-fold, reaching a peak value at 25 μM dye concentration, before dropping considerably most likely due to self-quenching (Fig. 1A - triangles). The effect of NR concentration on the emission wavelength was also visible; the emission peak wavelength was 600 nm at the lowest NR concentration (Fig. 1B - triangles), increasing until the maximum value of 615 nm around 10 μM dye concentration. Further dye concentration increase caused the emission wavelength to shift slightly toward lower values, most likely due to fluorescence energy transfer among the dye molecules within the viral membrane. The critical micelle concentration (CMC) of TX-100 in PBS is about 0.02% w/v and this is reflected by the NR emission peak wavelength change from 662 nm to ˜640 nm in the control sample (Fig. 1C - circles) by this TX-100 concentration. The emission wavelength of NR does not change considerably with increasing TX-100 concentration beyond this point. The shape of the spectrum also remains the same between surfactant concentrations of 0.03% w/v and above (not shown). The fluorescence intensity of NR increases with TX-100 concentration gradually, particularly after the CMC, suggesting the continuing formation of surfactant micelles that provide hydrophobic environments for NR to bind to (Fig. 1D - circles). Although the number of these hydrophobic environments increases with surfactant concentration, the hydrophobicity of NR's vicinity does not change considerably as evident from the unchanging emission wavelength after the CMC (Fig. 1C - circles). NR in the virus sample on the other hand displayed completely different fluorescence properties: the peak wavelength was initially at 613 nm in the presence of whole virus (no TX-100) and gradually shifted by ˜27 nm to reach 640 nm at 0.03% w/v TX-100 concentration, after which it remained unchanged (Fig. 1C - triangles). Change of the fluorescence intensity of NR in virus samples followed the same trend as in the control samples; it increased almost linearly as a function of surfactant concentration, after a seeming lag-phase up to the CMC (Fig. 1D - triangles).
Figure 1.
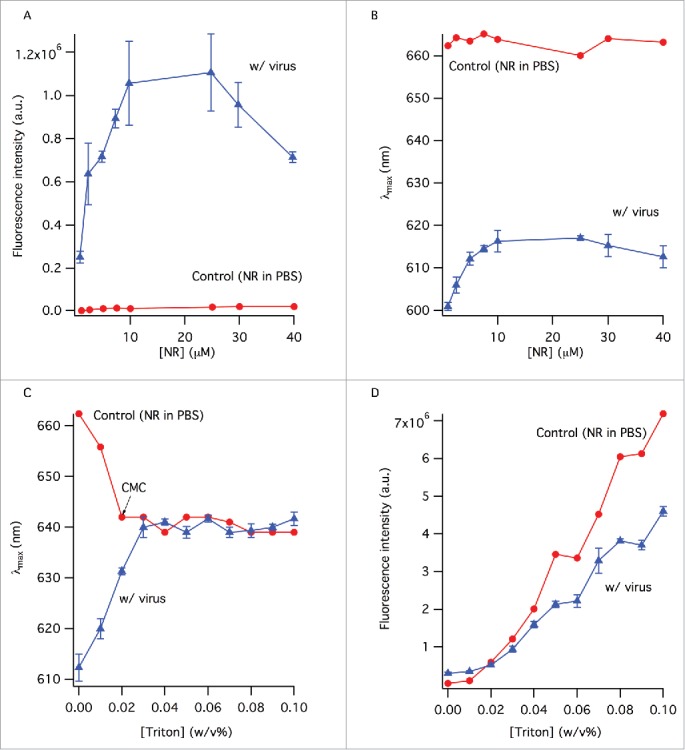
The change of the fluorescence intensity (A) and peak wavelength (B) as a function of NR concentration. Control sample contained NR in PBS. Samples with virus contained the same amount of NR as the control samples. (C) The change of peak wavelength in the absence and presence of virus with TX-100 concentration. (D) Error bars represent standard deviation (n = 3).
Virus splitting kinetics with surfactant
Splitting kinetics was also observed with NR emission over TX-100 concentrations of 0.01 – 0.1% (w/v). The change in the emission peak wavelength within our experimental timescale was insignificant for all concentrations (not shown). In comparison, the emission intensity showed exponential increase before reaching a plateau for all TX-100 concentrations except 0.01%, at which only a negligible change in the intensity occurred over 2 hours (Fig. 2A - circles). This data was analyzed by an exponential function and changes in the time component () parameters were observed. Since the splitting kinetics of virus with 0.01% TX-100 concentration was negligible for the duration of our experiment, the time component () of 0.01% TX-100 was omitted from data analysis. The rest of the surfactant concentrations demonstrated a sigmoidal time component behavior (Fig. 2B). These results, as expected, indicate that viral splitting accelerates with increasing surfactant concentration. Within our experimental time frames and virus concentration used, the critical TX-100 concentration at which major structural changes for viruses occurred was between 0.05 and 0.07% w/v (Fig. 2B).
Figure 2.
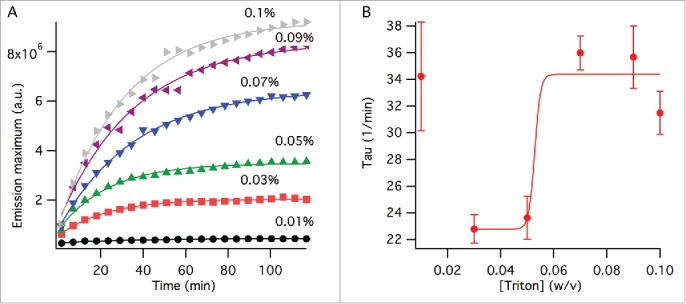
Viral splitting kinetics probed by NR fluorescence. (A) Change of the emission peak intensity with time at varying TX-100 concentrations (w/v). The lines show fits to single exponential function as outlined in the method section. (B) Time component, , obtained from the curve fits in (A). The line is a curve fit by a sigmoidal function excluding the data point at 0% TX-100 as outlined in the method section. The error bars represent standard deviation generated by the curve fits from (A).
Quantification of the split-ratio of virus after surfactant treatment using fluorescence emission spectra
Deconvolution of the total fluorescence emission into its spectral components was done by multi-peak fitting for 0–0.1% w/v TX-100 concentrations (Fig. 3). The broadness of emission spectra and appearance of shoulders in some of them indicate the presence of distinctive environments with different hydrophobicity, among which NR molecules are partitioned. Since fluorescence is additive, emission from NR residing in different molecular environments is expected to contribute to the overall spectra. We attempted 2- and 3-peak curve fits in order to identify the number of different molecular environments; only the 3-peak fit analysis was satisfactory. The presence of 3 distinct environments in the samples, even before TX-100 addition, showed that there was ‘split material’ in the samples from the very beginning. Additionally, since quantifying the amount of split material in the final formulation is more important than quantifying the ratio of whole viruses that have been split, we defined the split ratio as the total amount of split material in the final, equilibrated sample. Overall, considerable changes in the peak areas, which are proportional to the concentration of NR molecules in each environment, were observed with increasing TX-100 concentration. The three distinct peak wavelengths at 589, 630 and 670 nm are assigned to ‘peak 1’, ‘peak 2’ and ‘peak 3’, respectively, and attributed to different environments in which NR molecules reside (Fig. 3). Upon addition of 0.01% w/v TX-100, we observed minor wavelength shifts: peak 1 blue-shifted to 585 nm, peak 2 red-shifted to 635 nm and peak 3 remained unchanged (Fig. 4A). Addition of more surfactant led to a significant shift for peak 3 only, decreasing from 670 to 656 nm until 0.03% w/v TX-100 concentration (Fig. 4A). For peak 1 and peak 2, there was no significant change in wavelength with increasing TX-100 concentration over the range of concentrations we studied (Fig. 4A). Based on these distinguishable peaks, the corresponding percentages of NR in different environments were calculated from areas under the peaks (AUPs) with reference to the total peak area (Fig. 4B). The area for peak 1 gradually decreased from 43% to 5% with increasing TX-100 concentration. Peak 2′s area increased to 43% from 22%. Peak 3′s area also increased gradually, reaching 51% from 34%. Using the AUPs recovered from peaks, a split ratio was calculated from the total AUP and is shown in Figure 4C. The amount of split virus increases with increasing surfactant concentration as expected and a near-plateau is reached after 0.04% TX-100 concentration.
Figure 3.
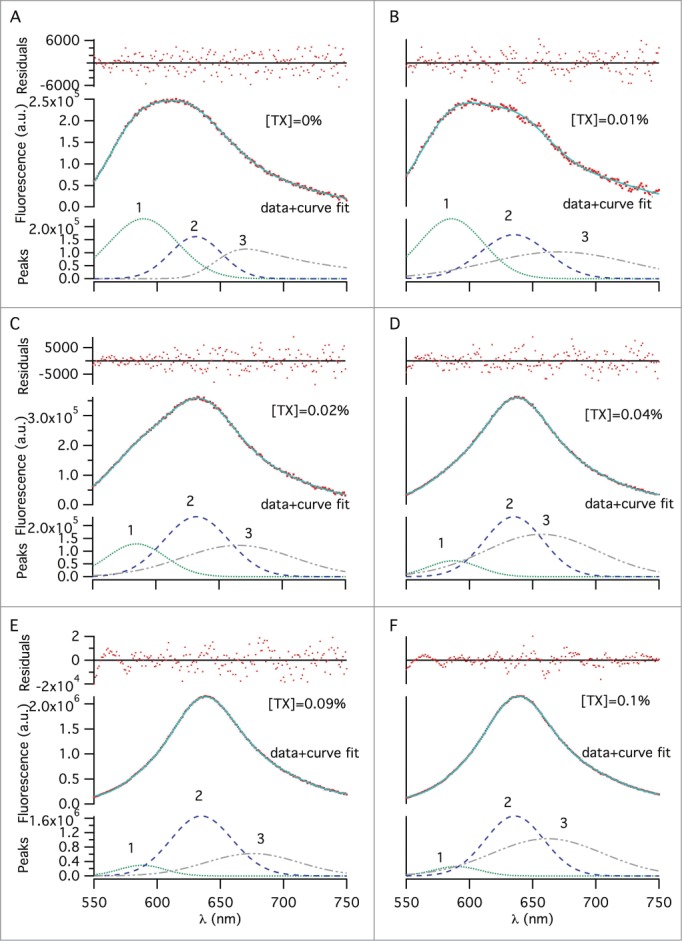
Multi–peak fitting using the exponentially–modified–Gaussian fitting function, showing dye molecules populated in 3 distinct environments, across different TX-100 concentrations (w/v): (A) 0%, (B) 0.01%, (C) 0.02%, (D) 0.04%, (E) 0.09%, and (F) 0.1%. An emission wavelength range of 550–750 nm was selected for peak fitting to exclude noise from the 2 tails. The 3 fitted peaks, representing 3 distinct dye environments, make up each spectrum. The solid lines represent the sum of the peaks 1–3. Change of the area under, and, the maximum emission wavelength of each fitted peak was determined for the different surfactant concentrations to quantify splitting extent.
Figure 4.

(A) Change of the peak wavelengths from multi-peak fitting with TX-100 concentration. (B) Change of the % areas of the peaks from (A) with TX-100 concentration. (C) Split ratio by emission, calculated as a sum of peak 2 and 3's % areas. (D) Change of the 3 fluorescence lifetimes as a function of TX-100 concentration. (E) Change of the weighted % contributions of the lifetimes from (D) to the total lifetime decay, as a function of TX-100 concentration. (F) Split ratio by lifetime, calculated as a sum of tau 1 and tau 2's % contributions. The error bars represent standard deviation (n = 3).
Quantification of the split-ratio of virus after surfactant treatment using fluorescence lifetime decays
We measured the fluorescence decay of NR that was bound to virus as a function of surfactant concentration. The fluorescence lifetime of the dye in different environments was determined by 3-exponential decay fittings (not shown). Each lifetime was assumed to be indicative of a different dye environment similar to that of the steady-state experiments.
Individual fluorescence lifetimes of NR in virus showed somewhat differing trends: between 0–0.03% w/v TX-100 concentration, both τ1 and τ2 slightly decreased from 770 ps to 500 ps and 2.5 ns to 2.0 ns, respectively, then recovered back to their original values, whereas the longest lifetime τ3 gradually decreased from 4.8 ns to 4.2 ns from 0–0.1% TX-100 concentration (Fig. 4D).
The percentage of τ1 was not really affected by the presence of surfactant and remained almost constant at 10% over the whole range of TX-100 concentrations (Fig. 4E). In comparison, the percentage of longer lifetime components revealed that τ2 and τ3 had somewhat similar starting proportions in the beginning and did not change much between 0–0.03% surfactant concentrations (Fig. 4E). At higher TX-100 concentrations, however, they displayed clear changes: τ2 gradually increased whereas τ3 progressively decreased until 0.08% w/v surfactant, after which point they were fairly constant at ˜60% and 30%, respectively. The trend of the split-ratio calculated from this method resembles that of the ratio calculated with the emission method (Figs. 4C and F). Due to considerations mentioned above in the previous section, the split ratio was defined as the amount of split material in the final, equilibrated sample.
Visual observation of whole and split virus particles
We observed the gradual splitting of whole virus particles by TX-100 using TEM (Fig. 5). Whole viruses with membrane proteins are clearly defined in the untreated sample (Fig. 5A1 and A2). The sample treated with 0.04% w/v TX-100 for 40 minutes mainly shows viruses with disrupted membranes but the viruses have not yet been completely disintegrated (Fig. 5B1 and B2). After treatment with 0.07% w/v TX-100 for 40 minutes, generally the remnants of whole viruses are seen within amorphous clusters (Fig. 5C1). Splitting is not 100% however, since agglomerates consisting of larger particles that are reminiscent of more intact viruses are also observed (Fig. 5C2).
Figure 5.

Electron micrographs of negatively stained inactivated influenza viruses; numbers 1 and 2 indicate repeats of the same samples. (A1 & A2) Whole viruses prior to the TX-100 treatment, (B1 & B2) after treatment with 0.04% TX-100 for 40 minutes, and, (C1 & C2) after treatment with 0.07% TX-100 for 40 minutes. Virus dimensions ranged from ˜100–250 nm. Scale bar is 100 nm.
Discussion
NR is a hydrophobic dye that binds preferentially to non-polar environments, with higher fluorescence intensity when bound to lipids than proteins.11 We hypothesized that split-ratio could be quantified using external NR-binding fluorescence spectroscopy if the amount of bound dye at different molecular environments could be distinguished and estimated.
NR's quantum yield is very low in aqueous environments and this is reflected in Figure 1A. The presence of viruses and other features like protein aggregates creates more hydrophobic environments for NR to bind to and leads to enhanced fluorescence. Increasing the concentration of NR beyond 25 μM leads to self-quenching (Fig. 1A – triangles). NR's emission wavelength does not change considerably with increasing NR concentration in PBS (Fig. 1B – circles) but experiences significant changes in the presence of viruses (Fig. 1B – triangles). As the NR concentration increases, the selectivity of NR molecules for hydrophobic environments is reduced, leading to an overall red-shift until quenching effects lead to slight blue-shifts.
In the absence of virus, NR's emission wavelength decreased with increasing TX-100 concentration, staying constant at ˜640 nm after the critical micelle concentration of TX-10012 (Fig. 1C - circles). The fluorescence intensity on the other hand increased substantially with increasing surfactant concentration (Fig. 1D - circles). This indicates NR molecules were burying themselves inside micelles as they were being formed, as reported previously.13
In the virus samples, NR's emission wavelength red-shifts from 613 nm to 640 nm, showing that the viruses are being split and providing NR with environments less hydrophobic than the lipid membranes of viruses 14 (Fig. 1C – triangles). The similarity of emission wavelength after the CMC between the control and virus samples suggests that particles such as protein aggregates, and, lipid-protein or surfactant-lipid assemblies formed post-splitting provide NR with a hydrophobic vicinity comparable to that of surfactant micelles. Virus splitting causes fluorescence de-quenching and leads to intensity increase with increasing surfactant concentration (Fig. 1D – triangles).
We also measured the virus splitting kinetics over a 2-hour period by observing the change in fluorescence peak intensity with time (Fig. 2A). Data analysis showed that within a 2-hour period, splitting gained significance after 0.05% w/v TX-100.15
Multi-peak fitting to the fluorescence emission spectra showed that each spectrum could be de-convoluted into at least 3 minor peaks representing different micro-environments for NR binding (Fig. 3). The addition of surfactant induced a red-shift in the overall spectra (raw data) and de-convolution of these spectra showed that this shift can be explained in terms of the changing contributions of the minor peaks (determined from the areas under the peaks) to the overall spectra. We hypothesize that peak 1 (˜590 nm) reflects populations of NR in intact phospholipid membranes, as also reported earlier in the literature.16 Peak 3 (˜660 nm) is hypothesized to be representative of NR populations in a relatively hydrophilic micro-environment, which is altered after the splitting process.11 Peak 2 (˜630 nm) is hypothesized to reflect NR micro-environments in between, namely free proteins, lipids, surfactant micelles, and, small protein aggregates. A similar wavelength maximum of 630 nm is also reported in the literature from the interaction of NR with small -casein micellar aggregates.11 Keeping these in mind, increasing surfactant concentration led to peak 1′s contribution to the overall spectra to progressively decrease and peak 2 and 3′s contributions to progressively increase (Figs. 3 and 4B), indicating whole viruses are being split and NR bound to viral lipid membranes is being released and then binding to less hydrophobic micro-environments produced as a result of splitting.
The change in the polarity of NR micro-environments suggested from emission wavelength shifts with addition of TX-100 is significant only for peak 3 (Fig. 4A). It must be stressed though that the 3 peaks are all quite broad and heavily overlap, indicating the intricacy of the molecular micro-environments. The TEM images (Fig. 5) also indicate the introduction of such complexity involving free proteins, protein aggregates of various sizes and shapes, protein-lipid complexes, surfactant micelles, and, various other particles post-splitting. Whole viruses are seen to have arrays of surface proteins, as reported in literature.17 The structural integrity of the viruses clearly degrades going from (A) to (C) and many products are formed as a result of splitting. These splitting products provide a variety of options for NR binding and give rise to complex emission spectra.
Fluorescence lifetime was employed to further investigate the populations of NR in the 3 micro-environments, represented by τ1, τ2 and τ3 (Fig. 4D). We attribute the shortest lifetime τ1 to NR populations in a relatively hydrophilic micro-environment (corresponding to peak 3 from multi-peak fitting), possessing a lifetime close to the literature value of 0.650 ns for NR in aqueous buffer.18 The slightly longer lifetime τ2 is attributed to NR populations that are bound to the various features mentioned above for peak 2, while the longest lifetime τ3 is attributed to NR molecules bound to viral membranes (peak 1 from multi-peak fitting).
The contribution of to the overall fluorescence lifetime in samples with increasing surfactant concentration remained at ˜10%, showing that only a small amount of NR molecules were present in a relatively hydrophilic environment. Because the progressive solubilization of virus membranes increases the population of protein aggregates and similar hydrophobic environments for NR binding, τ2% and τ3% were found to be inversely coupled: τ3's contribution gradually decreased from ˜50% to ˜30% showing that whole viruses are being split, while τ2's contribution increased accordingly (Fig. 4E).
Since, even in the absence of surfactant, multi-peak analysis of NR emission spectra exhibited 3 distinct peaks (Fig. 3A) and fluorescence lifetime analysis yielded 3 distinct lifetime components (Fig. 4D), it is reasonable to assume that there were at least 3 types of molecular environments with varying hydrophobicity all along. Due to this, calculation of the split ratio as the amount of all split material in the sample (peak 2% + peak 3% in emission analysis, and, τ1% + τ2% in lifetime analysis) rather than the ratio of whole viruses that have been split after surfactant treatment was more meaningful. Calculation of the split ratio showed that the 2 methods produced a similar trend (Fig. 4C and F).
In split virus vaccines, fine control of the virus splitting process is paramount not only for the formulation stability of finished vaccines but also for their potency, since split fragments are less immunogenic than whole viruses.19 A vaccine should have maximal immunogenicity and minimal reactogenicity, both of which are directly related to the splitting process in split virus vaccines. The elucidation of these relationships first requires the reliable quantification of the splitting extent or split ratio. Only after such quantification can studies on optimal splitting for maximal immunogenicity and minimal reactogenicity be feasible. Additionally, only with such quantification can a database of optimal splitting ratios for various strains with various features be developed and used to potentially bypass time-consuming and costly potency determination tests like SRID. This study has shown 2 fluorescence methods for the rapid and reliable quantification of the split ratio for potential application during split virus vaccine manufacturing.
Materials and methods
Influenza virus samples were obtained from the zonal fractions of a sucrose-gradient centrifuge and provided by Sanofi-Pasteur (Pennsylvania, USA). TX-100 (≥ 99.8%), NR, methanol and 0.1 M PBS tablets were purchased from Sigma-Aldrich (Castle Hill, AUS). A 2 mM NR stock solution was made in methanol and kept frozen as aliquots at −20°C. Whole virus samples were incubated with 1 or 10 μM NR incubated for >2 hr, followed by >24 hr incubation with TX-100 to ensure the splitting was complete and equilibrium was reached.
Steady-state fluorescence emission measurements
Steady-state fluorescence spectroscopy was undertaken on a Horiba Jobin Yvon Fluorolog FL-322 (New Jersey, USA) using a 50 μL quartz micro-cuvette from Hellma (New York, USA). All fluorescence measurements were conducted with excitation wavelength of 490 nm and the range of emission from 500–800 nm with 0.2 second integration time. Both excitation and emission slits were kept at 2 nm. All fluorescence spectra were measured 3 times, averaged and corrected for the instrument response and dark counts.
Kinetics of surfactant induced virus-splitting with fluorescence spectroscopy
Time-dependent viral splitting was observed using the same fluorometer and experimental conditions described above. The first measurement was taken 1.5 min after TX-100 addition. Subsequent emission spectra were measured at 5.5 min intervals with a total experimental time of 117 min. An exponential function was used to fit the emission maximum of each TX-100 concentration with time:
where y0, A and τ are baseline at t∞, pre-exponential factor and the rate of splitting, respectively. The τ value was obtained from kinetics experiments and its dependence on the surfactant concentration was analyzed using a sigmoidal function:
where A, B, C and D correspond to the base, maximum, concentration at the half, and, rate of the curve, respectively.
Multi-peak fitting to fluorescence emission peaks
The de-convolution of emission spectra to determine partitioning of NR in different molecular environments was realized with a multi-peak fitting method in IgorPro 6 software (Oregon, USA) as previously reported.20 Briefly, different functions were used for multi-peak fitting with the number of peaks for fitting being gradually increased. The best fit with the least number of peaks and fit parameters was achieved with ‘exponential modified Gaussian functions’.21 The relative contribution of each peak to the total spectrum was calculated from the peak areas and each peak was assumed to represent a population of NR molecules in a given environment. The goodness of fits was judged by the residuals and reduced χ2.
Fluorescence lifetime measurements
Fluorescence lifetime was measured with the time-correlated single-photon counting method using a Lifespec-blue F900 from Edinburgh Instruments (Edinburgh, UK) with a picosecond diode laser as an excitation source as previously reported.22 Emission wavelengths were selected according to the emission spectra of NR for that particular TX-100 concentration. Emission was collected at 54.7° with 4096 channels over a 50-nanoseconds time window. Fluorescence decays were analyzed by an iterative re-convolution method using a Marquardt-Levenberg algorithm23 with instrument response function and sum of exponentials as:
where τ and α represent the fluorescence lifetime and pre-exponential factor, respectively. An acceptable fit was determined when the value of χ2 was close to 1 and not reducing with a more complex model, and, a random distribution of residuals around the zero line was obtained. Average fluorescence lifetime of a 3 exponential decay was produced using the following equation:
where α and τ represent individual pre-exponential factors and fluorescence lifetimes, respectively.
Transmission electron microscopy
A JEOL JEM-1400 was used to obtain TEM images of viruses. Three whole virus samples were prepared for imaging; one of them was left untreated and 2 of them were treated with final TX-100 concentrations of 0.04% and 0.07% w/v, respectively, for 40 minutes before loading onto TEM grids. We used copper TEM grids with carbon and Formvar coating (ProSciTech, Queensland, AUS). The samples treated with TX-100 were washed with PBS after loading onto grids for better negative staining. All the samples were stained with 2% w/v ammonium molybdate and allowed to dry in air before imaging.
Conclusion
We have developed 2 quantitative methods to determine the split-ratio for split virus influenza vaccines using fluorescence spectroscopy and made qualitative observations of splitting via transmission electron microscopy. Our quantification methods are rapid and reproducible and can potentially be used for validating the splitting extent during vaccine manufacturing. They can also pave the way for studies looking into the optimization of splitting in split virus vaccines and ultimately the rapid screening of vaccine potency by helping produce an ‘optimal splitting database’.
Abbreviations
- TX-100
Triton X-100
- NR
Nile red
- CMC
critical micelle concentration
- TEM
transmission electron microscopy
- HA
hemagglutinin
- NA
neuraminidase
- AUP
area under the peak
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs Agnes Hardy-Boyer, Alain Francon and Herve Pinton for valuable discussions; Sanofi-Pasteur for financial support; and Dr Nial Wheate for the critical reading of the manuscript. The University of Sydney is acknowledged for funding this research partially and we also thank the ACMM staff members for excellent technical help.
Funding
The research was supported by Sanofi-Pasteur and The University of Sydney startup fund.
References
- [1].Fact sheet: Influenza (Seasonal) [Internet] . Switzerland: World Health Organization Headquarters; 2014 March [cited 2015 July]. [about 3 screens]. Available from: http://www.who.int/mediacentre/factsheets/fs211/en/ [Google Scholar]
- [2].Overview of vaccine regulation and safety monitoring and investigation into adverse events following 2010 seasonal influenza. Alert information: Seasonal flu vaccine: Overview of vaccine regulation and safety monitoring and investigation into adverse events following 2010 seasonal influenza vaccination in young children [Internet]. Australia: Department of Health, Therapeutic Goods Administration; 2010 Oct 8 [cited 2015 July]. [about 17 screens]. Available from: https://www.tga.gov.au/alert/seasonal-flu-vaccine-overview-vaccine-regulation-and-safety-monitoring-and-investigation-adverse-events-following-2010-seasonal-influenza-vaccination-young-children [Google Scholar]
- [3].Belanger JM, Raviv Y, Viard M, Baxa U, Blumenthal R. Orthogonal inactivation of influenza and the creation of detergent resistant viral aggregates: towards a novel vaccine strategy. Virol J 2012; 9:1–12; PMID:22214262; http://dx.doi.org/ 10.1186/1743-422X-9-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Al-Tawfiq JA, Zumla A, Gautret P, Gray GC, Hui DS, Al-Rabeeah AA, Memish ZA. Surveillance for emerging respiratory viruses. Lancet Infect Dis 2014; 14:992–1000; PMID:25189347; http://dx.doi.org/ 10.1016/S1473-3099(14)70840-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Helenius A, Soderlund H. Stepwise dissociation of the Semliki Forest Virus membrane with Triton X-100. Biochemica et Biophysica Acta 1973; 307:287–300; http://dx.doi.org/ 10.1016/0005-2736(73)90096-5 [DOI] [PubMed] [Google Scholar]
- [6].O'Hagan D. Changing TH1/TH2 balance in split influenza vaccines with adjuvants In: Patents U, ed. United States: Novartis Vaccines and Diagnostics SRL, 2009. [Google Scholar]
- [7].Geeraedts F, Bungener L, Pool J, Ter Veer W, Wilschut J, Huckriede A. Whole inactivated virus influenza vaccine is superior to subunit vaccine in inducing immune responses and secretion of proinflammatory cytokines by DCs. Influenza Other Respir Viruses 2008; 2:41–51; PMID:19453471; http://dx.doi.org/ 10.1111/j.1750-2659.2008.00038.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patois E, Capelle MA, Gurny R, Arvinte T. Stability of seasonal influenza vaccines investigated by spectroscopy and microscopy methods. Vaccine 2011; 29:7404–13; PMID:21803109; http://dx.doi.org/ 10.1016/j.vaccine.2011.07.067 [DOI] [PubMed] [Google Scholar]
- [9].Buranda T, Wu Y, Perez D, Chigaev A, Sklar LA. Real-time partitioning of octadecyl rhodamine b into bead-supported lipid bilayer membranes revealing quantitative differences in saturable binding sites in DOPC and 1:1:1 DOPC/SM/Cholesterol membranes. J Phys Chem B 2010; 114:1336–49; PMID:20043651; http://dx.doi.org/ 10.1021/jp906648q [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Greenspan P, Mayer EP, Fowler SD. Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol 1985; 100:965–73; PMID:3972906; http://dx.doi.org/ 10.1083/jcb.100.3.965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sackett DL, Wolff J. Nile Red as a polarity-sensitive fluorescent probe of hydrophobic protein surfaces. Anal Biochem 1987; 167:228–34; PMID:3442318; http://dx.doi.org/ 10.1016/0003-2697(87)90157-6 [DOI] [PubMed] [Google Scholar]
- [12].Product Information: Triton X-100 laboratory grade [Internet]. Australia: Sigma-Aldrich; cited 2015 July. [about 6 screens] . Available from: http://www.sigmaaldrich.com/catalog/product/sial/x100?lang=en®ion=AU [Google Scholar]
- [13].Anand U, Jash C, Mukherjee S. Spectroscopic determination of Critical Micelle Concentration in aqueous and non-aqueous media using a non-invasive method. J Colloid Interf Sci 2011; 364:400–6; PMID:21924731; http://dx.doi.org/ 10.1016/j.jcis.2011.08.047 [DOI] [PubMed] [Google Scholar]
- [14].Mukherjee S, Raghuraman H, Chattopadhyay A. Membrane localization and dynamics of Nile Red: Effect of cholesterol. Biochemica et Biophysica Acta 2007; 1768:59–66; http://dx.doi.org/ 10.1016/j.bbamem.2006.07.010 [DOI] [PubMed] [Google Scholar]
- [15].Lichtenberg D, Ahyayauch H, Goni FM. The mechanism of detergent solubilization of lipid bilayers. Biophys J 2013; 105:289–99; PMID:23870250; http://dx.doi.org/ 10.1016/j.bpj.2013.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Greenspan P, Fowler SD. Spectrofluorometric studies of the lipid probe, nile red. J Lipid Res 1985; 26:781–9; PMID:4031658 [PubMed] [Google Scholar]
- [17].Fontana J, Cardone G, Heymann JB, Winkler DC, Steven AC. Structural changes in influenza virus at low pH characterized by Cryo-Electron Tomography. J Virol 2012; 86:2919–29; PMID:22258245; http://dx.doi.org/ 10.1128/JVI.06698-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Datta A, Mandal D, Pal SK, Bhattacharyya K. Intramolecular charge transfer processes in confined systems. Nile red in reverse micelles†. J Phys Chem B 1997; 101:10221–5; http://dx.doi.org/ 10.1021/jp971576m [DOI] [Google Scholar]
- [19].Webster RG, Glezen WP, Hannoun C, Laver WG. Potentiation of the immune response to influenza virus subunit vaccines. J Immunol 1977; 119:2073–7; PMID:915292 [PubMed] [Google Scholar]
- [20].Kayser V, Chennamsetty N, Voynov V, Helk B, Forrer K, Trout BL. Evaluation of a non-Arrhenius model for therapeutic monoclonal antibody aggregation. J Pharmaceut Sci 2011; 100:2526–42; PMID:21268027; http://dx.doi.org/ 10.1002/jps.22493 [DOI] [PubMed] [Google Scholar]
- [21].Nikitas P, Pappa-Louisi A, Papageorgiou A. On the equations describing chromatographic peaks and the problem of the deconvolution of overlapped peaks. J Chromatograp A 2001; 912:13–29; PMID:11307976; http://dx.doi.org/ 10.1016/S0021-9673(01)00524-6 [DOI] [PubMed] [Google Scholar]
- [22].Kayser V, Chennamsetty N, Voynov V, Helk B, Trout BL. Tryptophan-Tryptophan energy transfer and classification of tryptophan residues in proteins: Therapeutic monoclonal antibody as a model. J Fluorescence 2010; 21:275–88; PMID:20886272; http://dx.doi.org/ 10.1007/s10895-010-0715-0 [DOI] [PubMed] [Google Scholar]
- [23].Bevington PR. Data reduction and error analysis for the physical sciences New York: McGraw-Hill, 1969. [Google Scholar]