

abstract
The year 2015 sees the fiftieth anniversary of the publication of a research paper that underpins much of our understanding of fungal prion biology, namely “ψ, a cytoplasmic suppressor of super-suppressor in yeast” by Brian Cox. Here we show how our understanding of the molecular nature of the [PSI+] determinant evolved from an ‘occult’ determinant to a transmissible amyloid form of a translation termination factor. We also consider the impact studies on [PSI] have had – and continue to have - on prion research. To demonstrate this, leading investigators in the yeast prion field who have made extensive use of the [PSI+] trait in their research, provide their own commentaries on the discovery and significance of [PSI].
Keywords: yeast, prion, Brian Cox, non-Mendelian inheritance, [PSI+] prion, nonsense suppression
In the Beginning…
Fifty years ago this year Brian Cox, then at the University of Liverpool, published an article in the journal Heredity on ψ (psi), an unusual genetic trait he had observed in the yeast Saccharomyces cerevisiae1 (Fig. 1). The ‘mutant’ he identified abolished nonsense suppression of the ade2–1 ochre (UAA) mutation. Remarkably, in mutant by wild-type back-crosses, the underlying mutation was not inherited since the suppression of the ade2–1 mutation was recovered in the diploids and in all of their meiotic progeny. Such a non-Mendelian pattern of inheritance was diagnostic of a cytoplasmically-inherited trait. Further crosses with strains obtained from Don Hawthorne showed that the suppression was what was then sometimes called “super-suppression,” and suppressed a suite of ochre mutations in addition to ade2–1. The genetics also showed that suppression depended on two factors, one being a Mendelian gene segregating 2:2 in tetrads (SUQ5 now SUP16, coding for a mutant serine-inserting tRNA) and the other inherited in a non-Mendelian fashion: both suppressors were dominant. The non-Mendelian determinant was given a Greek letter, ψ, as was the convention at the time for cytoplasmic traits in yeast. Naming the determinant ψ was not a reference to any paranormal nature, but rather was inspired by a catch phrase used to end the, then highly popular, BBC radio comedy show ‘The Goons’, namely “It's all in the mind, you know.”
Figure 1.

Brian Cox and the first Ψ/[PSI] paper published in 1965.
Cox assigned ψ+ as the dominant enhanced suppression state and ψ− the “normal” state allowing efficient polypeptide chain termination. Efficient chain termination in ψ− cells causes the translation of the ade2–1 mRNA to terminate at the mutant UAA codon, and causes a precursor (P-ribosylaminoimidazole or AIR) of the adenine biosynthesis pathway to accumulate. This precursor oxidises to form a red pigment and the resultant red colony phenotype, as well as adenine requirement, have been used as a marker for most genetic studies of ψ ever since. Colonies of well-suppressed strains are white and adenine independent.
ψ was not the first non-Mendelian trait to be described in yeast, for example the petite mutant described 16 years earlier and designated ρ (rho)2,3 is caused by a defect in or deficiency of mitochondrial DNA. A few years later came [URE3], which modifies the way yeast takes up ureidosuccinic acid, a precursor in the pathway synthesising uracil.4,5 A further example is the killer phenotype that is determined by a dsRNA containing virus-like particle (VLP) that infest many strains of yeast.6,7 Neither the ψ+ nor [URE3] determinants could be linked with mitochondrial DNA,8 or with cytoplasmically transferred nucleic acids such as dsRNA or the 2μm circle dsDNA.9
All of these discoveries were serendipitous, and quite independent of previous publications: the question of priority is quite meaningless. The genetic behavior of these factors was, and still is, variously described as “cytoplasmic,” “extrachromosomal” or “non-Mendelian.” The 2μm circle has not been associated with any easily recognized phenotype and such plasmids are sometimes described as“cryptic,” implying you can see them, but have no idea what they do except replicate themselves. For some 30 years we tried to promote the epithet ‘occult’ for ψ and [URE3] for something you know is there but cannot see - but it never caught on.
For nearly 30 years the ψ determinant remained a mystery, baffling numerous graduate students and postdocs in the Cox laboratory, which by now had moved to the Botany School, University of Oxford. During this time, all speculation about the nature of ψ was done in the overwhelmingly dominant paradigm of inheritance, as introduced by Watson and Crick. Disease, likewise, was expected always to be traceable to changes in native DNA or invasion by parasites introducing foreign DNA or RNA. Then in 1994 Reed Wickner proposed that the non-Mendelian [URE3] determinant in yeast was a heritable conformational variant of the cellular protein Ure2 i.e. the [URE3] determinant was a prion.10 Furthermore, Wickner proposed that ψ most likely also had a prion-like determinant and concluded, from the then limited available data, that Sup35 was the strongest candidate. As is now well documented, his conjecture on ψ was correct; ψ+ is the prion form of the translation termination factor Sup35 (eRF3) encoded by the SUP35 gene. In keeping with the accepted modern nomenclature, the ψ+ trait is now referred to as [PSI+] with cells lacking the determinant being designated [psi−].
The ‘protein only’ prion concept, adding a novel paradigm, was first proposed by Prusiner in 1982 as an explanation for the transmi-ssion of scrapie in sheep and goats11 and then eventually for the human analogs kuru, Creutzfeldt-Jakob disease (CJD), fatal familial insomnia and Gerstmann-Straussler-Scheinker syndrome; all pathologies associated with the same PrP protein.11,12 By the time of Wickner's publication in 1994 there was considerable evidence for the prion hypothesis for transmissible spongiform encephalopathies. The prion protein had been identified, the genes determining it in man, in mice, in hamsters and in sheep had been sequenced, and the existence of 2 conformations of the PrP protein in prion and normal forms had been demonstrated, as had some critical details of the molecular structure. It is undeniable that Prusiner introduced a new paradigm into the understanding of disease and heredity. Equally, one cannot deny that the acceptance of the ‘protein only’ prion concept was both slow and grudging, even after Charles Weissmann's laboratory in Zurich reported that mice without the PrP gene were totally resistant to scrapie, and that the species barrier to transmission between mice and hamsters disappeared if the recipient was transgenic with the PrP gene of the donor.13
That the discovery that two phenotypically distinct heritable traits in a completely different and only remotely-related organism could be explained by the prion hypothesis, was one of the factors that promoted the acceptance of the ‘protein only’ phenomenon. Another factor of course, was the emergence in Britain in 1995 of “mad cow disease” in man. This variant of CJD (vCJD) was apparently caught from cows and the possible involvement of the bovine equivalent of PrP caught the attention not only of the public, but of the medical establishment. It is unforgivable that even in 1988 we published a review of ψ 14 that discussed in detail various models we thought might account for the ψ phenomenon without even considering prions, in spite of the growing body of evidence for them from the Prusiner laboratory.
In this short article we illustrate how our understanding of the molecular nature of the [PSI+] determinant evolved and the impact studies on [PSI] have had – and continue to have - on prion research. To illustrate this further, several leading investigators in the yeast prion field who have made extensive use of the [PSI+] trait in their research, provide their own commentaries on the discovery and significance of [PSI] (see Boxes 1–5).
Early Thoughts and Speculations on the Nature of [PSI+]
During the 30 y following the 1965 paper, a considerable body of genetic data on [PSI+] were gathered largely by the Cox (Oxford), McLaughlin (University of California at Irvine), Inge-Vechtomov (St. Petersburg) and Sherman (Rochester NY) laboratories. For example, [PSI+] was shown to suppress all 3 classes of nonsense mutation, in some cases in the absence of a known suppressor tRNA.15 As we now know, and consistent with these early observations and inferences, the efficiency of translation termination is significantly impaired in a [PSI+] strain. On the biochemical side, Tuite and McLaughlin established an in vitro translation termination assay, and using yeast cell-free extracts and globin mRNAs, found that the extracts from [PSI+] yeast promoted read-through of nonsense codons. This read-through was poisoned by adding extracts from [psi−] cells16,17 leading to the conclusion that [psi−] cells contained an unidentified“fidelity factor” that enhanced chain termination. It was not until much later, with the publication of papers by Doel et al.18 and Ter-Avanesyan et al.19 that Sup35 (eRF3) was implicated as that fidelity factor, and by extension as the prion-forming protein,20 as Wickner had suggested.10 We wish to acknowledge that even in 1994 we were unable to discuss the role of Sup35 and [PSI] in terms of the prion hypothesis. And so it was that, leading up to 1994, much information about the genetics of [PSI+] and how it affected the biochemistry of translation termination in yeast had accumulated, from at most 3 or 4 laboratories around the world, without any understanding of who the players were or how they worked.
In 1994, with the emergence of Reed Wickner's seminal paper,10 and followed by publications two years later from the Lindquist21 and Ter-Avanesyan22 laboratories demonstrating the alternative states of the Sup35 (eRF3) protein in [PSI+] and [psi−] strains, the curtain was drawn and all was revealed. It was one of science's rare moments and it constituted a paradigm shift if only in the parochial field of yeast genetics. In the preceding moment, all explanations of a set of phenomena were based on one perception of how things worked, and then a new window opened, and with it a new set of information became relevant, while the existing data all acquired different weights, without necessarily losing their truth.
The effect of this paradigm shift is illustrated in our review published six years before Wickner's paper.14,23 In this review we summarized the known properties of [PSI] and discussed various models for how it might work, without presuming to identify the actual molecular factor. The purported models were limited to pre-existing and well-understood examples of irregular genetics based on DNA organization or regulation. Those we favored were more or less elaborate episomal theories or positive feed-back regulation systems, similar to that proposed by Novick for example, to explain an environmentally-triggered self-sustaining inhe-rited ‘ON’ state of the lac operon in E. coli23 or to the inheritance of methylated sites in DNA, like those determining restriction-modification systems in bacteria.24,25 The most elegant and economical model came from Jeff Strathern at one of the annual Cold Spring Harbor courses on Yeast Genetics (Fig. 2). The gene is hypothetical of course and the model does not work for Sup35, which at the time was not one of the suspects, but it is very clever.
Figure 2.
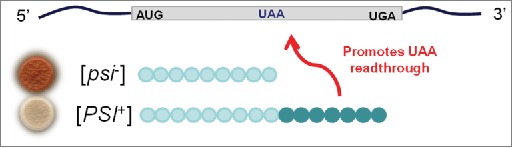
The ‘Strathern model’ for [PSI] inheritance. Fortuitous or induced read-through of the UAA codon generates a protein (hypothetical) that sustains itself by promoting reading through its own internal stop codon and also, incidentally, any other codon mutations to UAA in other genes. The suppression ('[PSI+]') phenotype so generated would be dominant and inherited in non-Mendelian fashion since it does not depend on any change in DNA sequence, only in its activity.
The Cox et al.14 review summarized various models in these categories and assessed them in the light of the documented facts about [PSI], listing those “pro' and those ‘con’ for each model, without coming to any conclusions. The subsequent realization of the prion nature of the [PSI] determinant have now made it possible to re-categorise our observations and knowledge about [PSI] as follows:
- Observations directly supporting the theory:
- Identification of a mutation in Sup35 which abolished inheritance of [PSI+], namely PNM218;
- [psi−] could be converted to [PSI+] by over-expression of SUP3526;
- Sup35 (eRF3) had a function required of the“fidelity factor” (i.e., recognition of stop codons and chain termination) as demonstrated by in vitro assays of stop codon readthrough in [psi−] lysates16;
- The sequence of Sup35 in various guises (GST1, SUP2, SUF12, SAL3) with an N-terminal region containing a polyQN tract and a series of oligopeptide repeats necessary to maintain [PSI+].27–29
- Observations newly explained by the theory and previously recognized as significant but that did not clarify much:
- Observations not enlightened by the theory, but which were thought to deserve further investigation, or were otherwise deemed irrelevant:
- [PSI+]-mediated lethal interactions; [PSI+] sal3, [PSI+] sal4 (coding for eRF1) and sal3-sal4 double mutants33 and Sup35 overexpression.
- 100% curing of [PSI+] by growth in millimolar concentrations of guanidine hydrochloride.30,35
- Curing of [PSI+] by recessive mutations in the HSP104 gene encoding a molecular chaperone.36
- Dominant PNM mutations.18,37
- Observations which made no sense whatsoever:
- Transformation of [psi−] to [PSI+] by super-coiled DNA co-purifying with 3-micron (rDNA) circles.38
- Mutagenesis of [PSI+] to [psi−] by DNA mutagens with, in the case of UV, single-hit kinetics and subject to repair by all the known yeast DNA-repair mechanisms: photoreactivation (specifically targeting pyrimidine dimers), excision-repair (now known as bulky-damage repair) and recombination/mutagenic repair.14,32,39
Confirming [PSI+] is a Transmissible Amyloid
The original amyloid-prion identity of [PSI+] was reinforced by subsequent prion-theory-driven studies. For example, the ability of recombinant Sup35 to form amyloid in vitro which can then be further applied in vitro to seed solutions of monomer to form more amyloid.40,41 The amyloid products of this can then convert in vivo [psi−] protoplasts of yeast cells to [PSI+] protoplasts to generate stable [PSI+] clones of cells.42,43 This latter discovery was particularly gratifying since it was a predicted property of prions that for many years had resisted attempts to confirm it using natural and recombinant forms of the mammalian PrPSc protein. In addition, just as [PSI+] cultures arising spontaneously comprise a set of phenotypically different “variants” (the amyloid equivalent of allelomorphs in DNA- and RNA-based heredity), so in vitro-generated Sup35 amyloid promoted the formation of such variants. Reg-ardless of in vivo or in vitro generation, the [PSI+] variant of the source strain was promoted when [psi−] protoplasts were transfected.
An essential role for Hsp104 for the propagation of [PSI+] in vivo was initially demonstrated through genetic inactivation of the HSP104 gene36 and subsequently by chemical inhibition of the ATPase activity of this molecular chaperone with very low concentrations of guanidine hydrochloride.35,44-46 Likewise roles for the Hsp70 (Ssa1/2) and Hsp40 (Sis1) chaperones were subsequently uncovered.47 Hsp104 is a requirement common to nearly all yeast prions, to the extent that it is taken to be a defining feature in establishing their prion nature. Hsp104-dependency has been used as an easily verifiable first criterion for putative prions identified by prion-associated sequences, such as poly-QN domains48 or by their ability to mediate the de novo formation of other prions i.e. to act as [PIN+] factors.49,50 These properties have been used to help identify several other prions in S. cerevisiae although it is very likely that many others exist.48,51
It is worth pointing out that, in addition to the effect of Reed Wickner's 1994 paper on the direction of [PSI+] prion studies, it “changed the water on the beans” for prion biology in a more general way. As we note above, we think it is fair to say that acceptance of Stanley Prusiner's prion hypothesis had until then been grudging, in spite of papers from his laboratory and others illuminating the molecular basis of spongiform encephalopathies.12,52 With the demonstration of the [URE3] and [PSI+] prions in yeast, many more people involved in fields ranging from biomedical sciences to biophysics and particularly neurodegenerative diseases took up research in the field of amyloids and prions. The outbreak of mad cow disease in Britain in the early 1990s alerted the general public, not to mention the health industry, to the dangers of this novel form of infection and heredity. It is common for many new paradigms to languish in obscurity or odium for a long time before coming back to light: take gene linkage, gene conversion, epigenetics or Lamarkism for example? This article is therefore as much a celebration of the 21st birthday of Wickner's paper promoting acceptance of the ‘protein only’ theory, as it is of [PSI+]'s 50th.
What is the Point of [PSI+]?
Our understanding of fungal prions leads us to predict a role for chaperones in the maintenance and propagation of amyloids, however no such intervention or requirement has yet been found. Indeed, by contrast to fungal prions, almost all mammalian amyloids and prions are pathogenic and therefore the existence of a mammalian enzyme system to maintain them would perhaps be counter-selected. This highlights the question of whether yeast and fungal prions have selective advantages. Heather True and Susan Lindquist have demonstrated that [PSI+] is advantageous in certain stress environments (the presence of heavy metals, for example); equally, there are conditions in which being [psi−] provides a selective advantage e.g. growth at 12–15°C.31,32 It may also be significant that several of yeast's prion-forming proteins are transcription factors, allowing the prion plus-to-minus switch (or vice versa) to be adaptive. There may be an evolutionary balance-point involved, since there are circumstances in which the prion form of Sup35 appears to be toxic.33,53,54 There seem to be no obviously pathogenic prions in yeast, however this may be an illusion since pathogenic prions could result in cell death and therefore remain inconspicuous, unlike the disease-related prions and amyloids of humans and animals.
What Can We—Have We—Learned from Studying [PSI+]?
Naturally, the question arises as to whether fungal prions have much to offer in the way of illuminating the nature, or more importantly the control, of other prions in general or mammalian prions in particular. The cellular environments of unicellular microorganisms and com-plex multicellular mammalian cells are radically different. Yeast and bacteria are rapidly proliferating, relatively undifferenti-ated organisms, whereas mammalian cells are mostly reproductively arrested, highly differentiated from one another, and presented with different and more varied opportunities and challenges for prion propagation than are yeast cells. Fundamentally, yeast amyloids have to be prions to survive cell division, whereas mammalian amyloids may have other options.
As we have pointed out above, nearly all prions and amyloids within mammalian cells are pathogenic, and we have argued in contrast that most have been recruited to useful functions within fungal hosts. The implication of this difference must be profound, and the differing relationships between these proteins and their host cells, in terms of maintenance or switching between one state and the other, at first sight would seem to position yeast as a poor model for better understanding mammalian prion behavior and properties. However, the underlying agents have the common molecular structure of aggregates for their pathogenic and/or sub-functional forms, as opposed to monomer or oligomers for the active “normal” form and it is the pathogenic form which is self-perpetuating. The aggregates, in most cases, are assemblies of amyloid fibers formed of parallel or anti-parallel β-sheets and protein sequences rich in hydrophilic amino acids such as Gln and Asn that drive nucleation and subsequent growth of the amyloid fiber.55–57 Other sequence features of amyloidogenic proteins such as the oligopeptide repeats found in Sup35, are largely dispensable for nucleation and fiber growth, although they contribute to the mechanism of chaperone-dependent propagation.58–60 It is plausible therefore that continued studies of yeast prions will result in treatments or procedures applicable to preventing the formation or propagation of prions and amyloids in general. To our benefit, yeast prions reside in an organism which has proven to be very tractable to experimentation, quick and easy to handle, with much of the technology simple, cheap and not, it seems, ethically disturbing. Genes from other organisms, including animals, are routinely expressed in yeast and mammalian prions and amyloid can be propagated as such.61,62
One example we have recently explored for the use of yeast as a model organism which seems likely to be relevant for most amyloid- and prion-forming proteins stems from the work of Ricardo Marchante.63 His work involved the isolation of all possible alleles of PNM2–1 (i.e., Sup35-G58D; 18 screening for those alleles showing the ‘PNM’ phenotype i.e. an inability to propagate [PSI+]. The responses of the G58X mutations in the formation or maintenance of [PSI+] were documented and correlated with NMR-determined conformations of an oligopeptide of the region. The outcome was the discovery that the amyloid-forming tendency of mutants correlated with high entropy (disorder) of the polypeptide studied.
Genetic crosses with a selection of the Sup35 G58X mutants can be used to identify the interactions of these mutants with each other or with wild-type Sup35 protein via their effects on the diploid phenotypes and patterns of inheritance in meiosis. A variety of such interactions and patterns of inheritance has already been observed, some of which have not previously been seen for Sup35. For example, as originally reported in 1965, when either [PSI+] or [psi−] strains are crossed with a [PSI+] strain, the resulting diploid is [PSI+],1 in accordance with the rules of prion biology. However, we have recently found that 2 of the sup35 alleles so constructed, G58W and G58A, can exist like wild type Sup35 as either [PSI+] or [psi−] but when either of these mutants is crossed with wild type [PSI+], a [psi−] diploid results (Fig. 3). In these cases, a phenotypically [psi−] cell arises from the fusion of 2 [PSI+] cells! The meiotic progeny of such crosses are variously and irregularly either [PSI+] or [psi−], so once one of the 2 alleles is removed by haploidisation, either can regenerate and support a prion form, although Sup35 G58A usually manifests as a weak [PSI+] variant (B.S.Cox, unpublished data).
Figure 3.
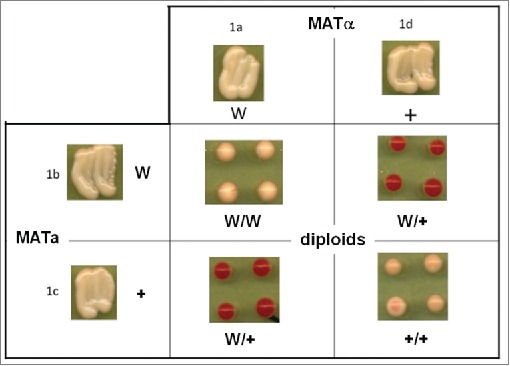
The significance of prion heterozygosity: the unusual interactions between some Sup35 G58X mutants. In this example the suppressible mutation is ade1–14 (UGA) and there is no tRNA suppressor in any of the strains. [PSI+] cells form white colonies while [psi−] cells form red colonies. Shown are colonies arising from single zygote cells picked from mixes of the haploid MATa and MATα strains as indicated. The four parent haploids differed at the Gly58 residue of the protein; + is G58 (wild type) and ‘W’ is a G58W mutant. All four parent haploids taken from a single tetrad are [PSI+]. Unlike normal [PSI+] x [PSI+] crosses involving wild-type Sup35 parents, the diploid progeny heterozygous for the two sup35 alleles are clearly [psi−]. The epigenetic segregation makes its own interpretation of the underlying Mendelian ratios.
The situation described above is reminiscent of the common M129V polymorphism in the PrP gene of human populations. Although either allele can support the PrPSc prion when in a homozygous state, when in a heterozygous state individuals are resistant to infection by many (but not all) PrPSc variants.64,65 Many similar interactions are seen in the PrP gene of sheep, affecting scrapie. In yeast, we see a variety of interactions between heteroallelic proteins, ranging from a total failure for one to interact with the other (frigid), through failure to reproduce (sterile), mutual modification, but separate variant forms (dating but living apart) to co-integration (happily married).
The unexpected interactions we observe between different forms of Sup35 are interesting for many reasons. Firstly, because they are single-residue alterations, they may be instructive about structural molecular reasons for amyloid formation; for example they may make it possible to classify the effects of heteroallelic situations; what interactions may promote amyloid formation and what enhance it; what interactions affect amyloid fragmentation and thus proliferation; what interactions may affect inheritance from cell to cell or what, if any, promote the enzymatic destruction of amyloid. Further study of their behavior may also prove to be a route toward designing a common strategy of therapy for the widely divergent amyloid-forming proteins in man and animals. Finally, it is worth remarking that the G58 residue is not the only residue within Sup35 which has yielded ‘PNM’ mutations. Angela de Pace and Jonathan Weissman described dozens of PNM mutants located in the N-terminal polyQN region of Sup35.66
To conclude; our recent experience with the [PSI+] prion has drawn our attention to the significance of heterozygosity. Man, in spite of the bottlenecks in his genetic history, is highly heterozygous and many of his domesticated animals are too, if rather less so, and it is apparent that heterozygosity is an important factor in the resistance to, and diagnosis and prediction of, diseases.67–69 With the exception of Alzheimer's, Parkinson's and cancer, amyloidoses in man are rare and sporadic and although often highly morbid, not the subject of much pharmacological or medical attention. However, if their common molecular aetiology offers the possibility of a common approach to their control, anatomical diversity may not be a hindrance to their treatment.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Box 1:
Reed Wickner
The secret handshake: After learning about Brian Cox's discovery of the non-chromosomal genetic element [PSI] when I took the Cold Spring Harbor Yeast Course in 1972, I was elated to meet him at various Yeast Genetics meetings in the '70s and '80s. I was working on the double-stranded RNA viruses determining the ‘killer character’ of yeast, another non-chromosomal system, so I was always interested in [PSI]. Brian is always the gentleman, but with a great sense of humor, so I always think of him smiling.
Brian's discovery of the [PSI+] non-chromosomal genetic element started the yeast prion story, and his careful characterization of [PSI+], and particularly its genetic properties, made our later work possible. The fact that curing of [PSI+] was reversible 70 and that mutation of the SUP35 gene could lead to loss of [PSI+] 18 were critical facts leading to our explanation of [PSI+] as a prion of Sup35 10. At the end of Brian's chapter in“The Early Days of Yeast Genetics” 71, he wrote, “The club of yeast geneticists is still in existence but has suffered a degree of dispersion and lost its exclusivity. However, a number of clubs within the club have formed informally, like eddies in a tide pool, and one of the most esoteric is the little coterie interested in cytoplasmic or extrachromosomal inheritance, and the more obscure the particle, the greater the bond. We do not have a handshake, but sometimes an eyebrow is raised. One of the pleasures that endures at the huge modern conventions on yeast genetics is meeting each other, especially those who did and still do genetics with their phenotypes, to exchange details of the latest rococo experiments. Here is to the killer people, especially Reed Wickner and Alan Bevan, to the mitochondrial people and their doyen, Piotr, to Jim Haber for giving us 20S and congratulations to them all on their graduation into molecular respectability. Ψ and URE3, I am sure Francois Lacroute and Michel Aigle would agree, te salute cumque gratias.” I was pleased at the time to be included in Brian's club along with Francois Lacroute. And it is true that those interested in non-chromosomal inheritance had common interests and approaches that made me look forward to developments in [PSI+], [URE3], 2 micron DNA, the killer factor and the various RNA viruses that were our main interest before prions. The orphan non-Mendelian genetic elements were particularly exciting and mysterious. Brian always had interesting new data that stimulated our thinking.
Brian has continued to work on [PSI+], the prion, and has made several important recent contributions, such as his characterization of prion ‘seeds’, defining them by a direct and simple assay method based on his discovery (with Mick Tuite) of the curing of [PSI+] by millimolar concentrations of guanidine.
Box 2:
Susan Liebman
I first learned of the ψ+ mystery in 1971, when I entered Fred Sherman's laboratory as a graduate student at the University of Rochester. I was to determine the efficiency of“super-suppressor” read-through and the amino acids inserted at nonsense codons, in ψ+ and ψ− cells. At that time posters and talks on ψ+ were largely ignored in favor of work on promoters. Luckily for me Brian Cox became a frequent visitor to Rochester serving as a de facto second advisor. Discussions of what ψ+ might be consumed by us and a small core of scientists whenever we met. Due to Brian's influence, my 1973 PhD thesis states, “… according to this hypothesis yeasts having identical genotypes may exist in the ψ+ or ψ− state depending upon whether or not they have been exposed to a particular substance which induces a series of chain events resulting in the continued production of that substance.” One focus in my laboratory was super-suppressors and their genetic modifiers. By 1989 this led me to seek funding for a structure function analysis of Sup35, whose role as a translational release factor and ψ+ was still unknown. After three such proposals failed I instead studied rRNA's role in translational fidelity. This enabled me to recruit Yury Chernoff who brought a side project that eventually showed deletion or overexpression of the Hsp104 chaperone cured cells of ψ+. This initially esoteric topic became of broad interest when Reed Wickner, combining his data on [URE3] with that of scientists in Russia that had continued to study Sup35, convincingly proposed that ψ+ and [URE3] were, respectively, the prion forms of the SUP35 and URE2 gene products. Thereafter, together with Irina Derkatch we showed the existence of different heritable variants of [PSI+] and that another prion, [PIN+], affected the appearance and maintenance of [PSI+]. Biochemical and transfection data have now proven that [PSI+] is a self-perpetuating amyloid form of Sup35. Self-propagation of [PSI+] requires chaperones to break prion fibers continually providing ends where soluble Sup35 molecules join and convert to prion. Other yeast proteins also exist in prion forms. These lessons are now being used to characterize human proteins with prion-like properties. Since many of these proteins are associated with neurodegenerative disease, the legacy of the 1965 paper continues.
Box 3:
Susan Lindquist
My favourite part of the Brian Cox story, an inextricable piece of what I believe will prove a profoundly important scientific legacy, is the object lesson it provides on the very nature of scientific inquiry. I remember hearing a lecture by Susan Liebman (then University of Illinois) on the non-chromosomal inheritance of an omnipotent nonsense suppressor, when I was a postdoc at the University of Chicago. I always loved to think about compelling puzzles that needed tackling. I walked out of Sue's lecture realizing I had no way to tackle that one, but having a sense of real excitement from the sheer craziness of the story. A heritable change in translation termination – one of the key elements of the central dogma – didn't obey the rules of DNA-based inheritance. Wow! But as I was walking back to the lab with 2 fellow postdocs, they surprised me by complaining about how ridiculous it was to study something so odd and uninteresting. So I briefly delved into Brian's papers (which laid the entire foundation!), and that work, a brilliant investigation of an odd chance observation, was creatively and rigorously laid. I asked a friend who was attending a yeast meeting where Brian was speaking to check it out. My friend was not happy about the waste of time. I forgot about it.
Fast forward many years later. My lab was deep in the throes of deciphering the problems of protein misfolding. We had reported that Hsp104 saves cells from extreme stresses. No problem. But when we discovered that it did so by using its 12 ATPase domains to disaggregate the aggregates of other proteins, the reaction was much the same as Brian had gotten: can't be right; this is a stupid idea. While we were struggling to get the story published, I got a call from Yury Chernoff, who was working with Sue Liebman. He'd found that Hsp104 over-expression cures cells of this mysterious heritable suppressor element. Did I have any idea what Hsp104 might be doing? Time to reread Brian's papers!
Prions break previous dogmas, expand our paradigms for inheritance, and provide entirely new understandings of self-perpetuating states in biology. As interesting and as vitally important as their roles in disease may be, I think it will be their roles in normal biology that will have the biggest impact. It's been 50 y since Brian's first work on this system and, believe me, we have only seen the tip of the iceberg. In another 50 y (or maybe just 10?), Brian's astonishing story of tackling the inexplicable with beautifully reasoned and rigorously controlled experiments will be in all of the textbooks. Meanwhile, it provides a number of object lessons. I ask today's anonymous manuscript reviewers with their demands for mechanistic understandings prior to publication, I ask students and postdocs who want only to publish in the top journals, and I ask granting agencies who want to fund only work that is important for human disease: where would be we if Brian hadn't published those papers?
Box 4:
Michael Ter-Avanesyan
To be honest, Brian Cox's 1965 publication describing [PSI+], a novel non-chromosomally-inherited determinant in Saccharomyces cerevisiae, passed by me unnoticed. Indeed, though at that time I had already decided to link my life with genetics, I did not think that yeast would be the focus of my future work. Even later, when I had already become a yeast geneticist, I did not plan to study [PSI+]. Of course, studying translation termination in Sergey Inge-Vechtomov's laboratory, I knew that such a determinant existed, but this phenomenon seemed too elusive and formally-genetic, while at that time I was attracted by things which had molecular interpretations. This is why [PSI+] and I peacefully co-existed without touching each other until the late 1980s. However, about twenty years after its discovery, [PSI+] began to intervene into my life, sometimes causing significant trouble! The first such intervention happened around 1987 – we could not disrupt the SUP45 gene and only after significant efforts established that this was due to decreased viability of [PSI+]-bearing cells heterozygous for SUP45 disruption. The second instance was connected to our observation published in 1994 19 that 5′-deletions of the SUP35 gene caused disappearance of [PSI+]. Thus, without any desire, our lab started getting involved in studies of [PSI+]. Some of my colleagues, especially Vitaly Kushnirov who at that time was involved in studies of the SUP35 gene, were skeptical of these efforts, since even though [PSI+] seemed to be linked to SUP35, we could not understand the physical nature of [PSI+]. Fortunately, that same year Reed Wickner published a paper with the prion concept for [PSI+] and from that moment [PSI+] ceased to be only trouble for us and, on the contrary, became an object of our constant and undivided interest. From that moment, our lab's life had changed and had become much more interesting. So, we are very grateful to [PSI+] and to Brian, who described and studied it. Undoubtedly, we also are grateful to our other colleagues, whose work in this area constantly surprised and inspired us. To conclude, I would like to stress the magic of numbers. While the study of the role of chromosomal genes (i.e., DNA) in determination and inheritance of phenotypic traits started with the work of Gregor Mendel published in 1865, the study of proteins as genetic material was initiated by Brian's work published exactly one hundred years later.
Box 5:
Tricia Serio
As a new assistant professor, I had the significant honor of hosting Brian Cox in my laboratory for a few months in the summer of 2006. At one point during his visit, I asked him how he settled on the name [PSI+] for the determinant of the phenotype he first described in 1965. I had always assumed that he had chosen a Greek letter to parallel the [ρ] designation used for the yeast mitochondrial genome, but I was curious about the specific choice of [Ψ]. He replied simply by sighing, and one need only re-read his landmark paper, “Ψ, A Cytoplasmic Suppressor of Super-Suppressors in Yeast,” to immediately understand.
In the course of characterizing a suppressor of multiple yeast auxotrophies, Brian noted a rare phenotypic instability that his curiosity led him to pursue. The underlying complexity turned out to be astounding: a diploid strain carrying one suppressible and one non-suppressible ade2 allele, a spontaneously arising, dominant but inefficient suppressor SUQ5, and a non-Mendelian factor [PSI+]. Fifty years later, knowing full well that the suppressible allele is a nonsense mutation, that SUQ5 is a tRNA suppressor, and that [PSI+] is the prion form of the release factor Sup35, the intricate logic of the crosses and their interpretations are still challenging to follow. And, then I remember that Brian designed these studies without knowledge of the molecular basis of the players. Nonetheless, he proposed the“simplest explanation” of the data, which still stands today: [PSI+] is a cytoplasmically transmitted modulator of the SUQ5 suppressor, which he posited could be a“self-replicating particle.” With the publication of this paper and the understatement,“It will obviously be of interest to establish the nature of this cytoplasmic mutation,” Brian launched a new field that continues to thrive 50 y later.
Every few years I re-introduce the paper in our laboratory journal club. My senior students and post-docs have come to view it as a sign that it is time to finish up their experiments and move on if they make it to a second discussion of the paper. But, I think of it as a way to inspire the next generation to be observant, curious, and thoughtful and to appreciate that even the most impossibly complex systems can be deconstructed with persistence and simple yet elegant experiments.
Acknowledgments
BSC would like to acknowledge the help, skills, insights, wisdom, friendship and in some cases, hospitality in their laboratories, of the following collaborators in the ψ project over the last 50 years. The list is non-hierarchical: Hamish, Shirley, Mick, Chris, Clegg, Cathy, Theresa, Marie, Sheila, Patsy Petal, Hwa Dai, Simon, Lee, Paolo, Frederique, Kerry, Angela, Eric, Nadia, Gemma, Jo, Jintana, Wes, Ricardo, Gloria, Cal, Tricia, Bruce, Mike, Jack, Bob and Ray.
FUNDING
MFT's research on [PSI+] was funded by the BBSRC (ref BB/J000183/1) and a Leverhulme Trust Research Fellowship (ref RF-2012–365). BSC acknowledges the award of 2 Leverhulme Emeritus Fellowships in 2004–2006 and 2012–2014.
REFERENCES
- 1.Cox BS. [PSI], a cytoplasmic suppressor of super-suppressors in yeast. Heredity 1965; 20:505-21; http://dx.doi.org/ 10.1038/hdy.1965.65. [DOI] [Google Scholar]
- 2.Ephrussi B, Hottinguer H, Chimenes AM. Action de l'acriflavine sur les levures. I. La mutation“petitie colonie.” Ann Inst Pasteur 1949; 76:351-67. [Google Scholar]
- 3.Ephrussi B. Nucleo-cytoplasmic relations in micro-organsims 1953; Claredon Press, Oxford. [Google Scholar]
- 4.Aigle M, Lacroute F. Genetical aspects of [URE3], a non-Mendelian, cytoplasmically-inherited mutation in yeast. Mol Gen Genet 1975; 136:327-35; PMID:16095000; http://dx.doi.org/ 10.1007/BF00341717. [DOI] [PubMed] [Google Scholar]
- 5.Lacroute F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J Bacteriol 1971; 106:519-22; PMID:5573734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woods DR, Bevan EA. Studies on the nature of the killer factor produced by Saccharomyces cerevisiae. J Gen Microbiol 1968; 51:115-26; PMID:5653223; http://dx.doi.org/ 10.1099/00221287-51-1-115. [DOI] [PubMed] [Google Scholar]
- 7.Somers JM, Bevan EA. The inheritance of the killer character in yeast. Genet Res 1969; 13:71-83; PMID:5771662; http://dx.doi.org/ 10.1017/S0016672300002743. [DOI] [PubMed] [Google Scholar]
- 8.Young CS, Cox BS. Extrachromosomal elements in a super-suppression system of yeast. II. Relations with other extrachromosomal elements. Heredity 1972; 28:189-99; PMID:4555664; http://dx.doi.org/ 10.1038/hdy.1972.24. [DOI] [PubMed] [Google Scholar]
- 9.Tuite MF, Lund PM, Futcher AB, Dobson MJ, Cox BS, McLaughlin CS. Relationship of the [psi] factor with other plasmids of Saccharomyces cerevisiae. Plasmid 1982; 8:103-11; PMID:6757991; http://dx.doi.org/ 10.1016/0147-619X(82)90048-8. [DOI] [PubMed] [Google Scholar]
- 10.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 1994; 264:566-9; PMID:7909170; http://dx.doi.org/ 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 11.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136-44; PMID:6801762; http://dx.doi.org/ 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 12.Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet; 47:601-23; PMID:24274755; http://dx.doi.org/ 10.1146/annurev-genet-110711-155524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339-47; PMID:8100741; http://dx.doi.org/ 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 14.Cox BS, Tuite MF, McLaughlin CS. The psi factor of yeast: a problem in inheritance. Yeast 1988; 4:159-78; PMID:3059716; http://dx.doi.org/ 10.1002/yea.320040302. [DOI] [PubMed] [Google Scholar]
- 15.Liebman S, Sherman F. Extrachromosomal psi+ determinant suppresses nonsense mutations in yeast. J Bacteriol 1979; 139:1068-71; PMID:225301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tuite MF, Cox BS, McLaughlin CS. In vitro nonsense suppression in [psi+] and [psi-] cell-free lysates of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 1983; 80:2824-8; PMID:6344070; http://dx.doi.org/ 10.1073/pnas.80.10.2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tuite MF, Cox BS, McLaughlin CS. A ribosome-associated inhibitor of in vitro nonsense suppression in [psi-] strains of yeast. FEBS Lett 1987; 225:205-8; PMID:3319694; http://dx.doi.org/ 10.1016/0014-5793(87)81158-4. [DOI] [PubMed] [Google Scholar]
- 18.Doel SM, McCready SJ, Nierras CR, Cox BS. The dominant PNM2- mutation which eliminates the psi factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics 1994; 137:659-70; PMID:8088511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 1994; 137:671-6; PMID:8088512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cox B. Cytoplasmic inheritance. Prion-like factors in yeast. Curr Biol 1994; 4:744-8; PMID:7953567; http://dx.doi.org/ 10.1016/S0960-9822(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 21.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 1996; 273:622-6; PMID:8662547; http://dx.doi.org/ 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 22.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J 1996; 15:3127-34; PMID:8670813. [PMC free article] [PubMed] [Google Scholar]
- 23.Novick A, Weiner M. Enzyme induction as an all-or-none phenomenon. Proc Natl Acad Sci U S A 1957; 43:553-66; PMID:16590055; http://dx.doi.org/ 10.1073/pnas.43.7.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science 1975; 187:226-32; PMID:1111098; http://dx.doi.org/ 10.1126/science.1111098. [DOI] [PubMed] [Google Scholar]
- 25.Holliday R. The inheritance of epigenetic defects. Science 1987; 238:163-70; PMID:3310230; http://dx.doi.org/ 10.1126/science.3310230. [DOI] [PubMed] [Google Scholar]
- 26.Chernoff YO, Derkach IL, Inge-Vechtomov SG. Multicopy SUP35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr Genet 1993; 24:268-70; PMID:8221937; http://dx.doi.org/ 10.1007/BF00351802. [DOI] [PubMed] [Google Scholar]
- 27.Ter-Avanesyan MD, Kushnirov VV, Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Inge-Vechtomov SG, Smirnov VN. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol Microbiol 1993; 7:683-92; PMID:8469113; http://dx.doi.org/ 10.1111/j.1365-2958.1993.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 28.Wilson PG, Culbertson MR. SUF12 suppressor protein of yeast. A fusion protein related to the EF-1 family of elongation factors. J Mol Biol 1988; 199:559-73; PMID:3280807; http://dx.doi.org/ 10.1016/0022-2836(88)90301-4. [DOI] [PubMed] [Google Scholar]
- 29.Kikuchi Y, Shimatake H, Kikuchi A. A yeast gene required for the G1-to-S transition encodes a protein containing an A-kinase target site and GTPase domain. EMBO J 1988; 7:1175-82; PMID:2841115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuite MF, Mundy CR, Cox BS. Agents that cause a high frequency of genetic change from [psi+] to [psi-] in Saccharomyces cerevisiae. Genetics 1981; 98:691-711; PMID:7037537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munday CR. The genetics of translation in yeast. DPhil Thesis, University of Oxford; 1979. [Google Scholar]
- 32.Cox BS, Tuite MF, Mundy CJ. Reversion from suppression to nonsuppression in SUQ5 [psi+] strains of yeast: the classificaion of mutations. Genetics 1980; 95:589-609; PMID:7002720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cox BS. Allosuppressors in yeast. Genet Res 1977; 30:187-205; http://dx.doi.org/ 10.1017/S0016672300017584. [DOI] [Google Scholar]
- 34.Chabelskaya S, Kiktev D, Inge-Vechtomov S, Philippe M, Zhouravleva G. Nonsense mutations in the essential gene SUP35 of Saccharomyces cerevisiae are non-lethal. Mol Genet Genomics 2004; 272:297-307; PMID:15349771; http://dx.doi.org/ 10.1007/s00438-004-1053-1. [DOI] [PubMed] [Google Scholar]
- 35.Eaglestone SS, Ruddock LW, Cox BS, Tuite MF. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI+] of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 2000; 97:240-4; PMID:10618402; http://dx.doi.org/ 10.1073/pnas.97.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995; 268:880-4; PMID:7754373; http://dx.doi.org/ 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 37.Young CSH, Cox BS. Extrachromosomal elements in a super-suppression system of yeast. I. A nuclear gene controlling the inheritance of the extrachromosomal elements. Heredity 1971; 26:413-22; http://dx.doi.org/ 10.1038/hdy.1971.52. [DOI] [Google Scholar]
- 38.Dai H, Tsay SH, Lund PM, Cox BS. Transformation of psi- Saccharomyces cerevisiae to psi+ with DNA co-purified with 3 micron circles. Curr Genet 1986; 11:79-82; PMID:3329046; http://dx.doi.org/ 10.1007/BF00389429. [DOI] [PubMed] [Google Scholar]
- 39.Tuite MF. Genetics of nonsense supressors in yeast. PhD thesis 1978; University of Oxford. [Google Scholar]
- 40.Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of PSI+, a heritable prion-like factor of S.cerevisiae. Cell 1997; 89:811-9; PMID:9182769; http://dx.doi.org/ 10.1016/S0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 41.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. In vitro propagation of the prion-like state of yeast Sup35 protein. Science 1997; 277:381-3; PMID:9219697; http://dx.doi.org/ 10.1126/science.277.5324.381. [DOI] [PubMed] [Google Scholar]
- 42.King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature 2004; 428:319-23; PMID:15029195; http://dx.doi.org/ 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature 2004; 428:323-8; PMID:15029196; http://dx.doi.org/ 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 44.Cox BS, Ness F, Tuite MF. Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics 2003; 165:23-33; PMID:14504215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol 2001; 40:1357-69; PMID:11442834; http://dx.doi.org/ 10.1046/j.1365-2958.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- 46.Jung G, Masison DC. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr Microbiol 2001; 43:7-10; PMID:11375656; http://dx.doi.org/ 10.1007/s002840010251. [DOI] [PubMed] [Google Scholar]
- 47.Winkler J, Tyedmers J, Bukau B, Mogk A. Chaperone networks in protein disaggregation and prion propagation. J Struct Biol 2012; 179:152-60; PMID:22580344; http://dx.doi.org/ 10.1016/j.jsb.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 48.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137:146-58; PMID:19345193; http://dx.doi.org/ 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN+]. Cell 2001; 106:171-82; PMID:11511345; http://dx.doi.org/ 10.1016/S0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 50.Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell 2001; 106:183-94; PMID:11511346; http://dx.doi.org/ 10.1016/S0092-8674(01)00440-8. [DOI] [PubMed] [Google Scholar]
- 51.Cascarina SM, Ross ED. Yeast prions and human prion-like proteins: sequence features and prediction methods. Cell Mol Life Sci 2014; 71:2047-63; PMID:24390581; http://dx.doi.org/ 10.1007/s00018-013-1543-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soto C. Prion hypothesis: the end of the controversy? Trends Biochem Sci 2011; 36:151-8; PMID:21130657; http://dx.doi.org/ 10.1016/j.tibs.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox BS. A recessive lethal super-suppressor mutation in yeast and other psi phenomena. Heredity 1971; 26:211-32; PMID:5286385; http://dx.doi.org/ 10.1038/hdy.1971.28. [DOI] [PubMed] [Google Scholar]
- 54.McGlinchey RP, Kryndushkin D, Wickner RB. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci U S A 2011; 108:5337-41; PMID:21402947; http://dx.doi.org/ 10.1073/pnas.1102762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Halfmann R, Alberti S, Krishnan R, Lyle N, O'Donnell CW, King OD, Berger B, Pappu RV, Lindquist S. Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins. Mol Cell 2011; 43:72-84; PMID:21726811; http://dx.doi.org/ 10.1016/j.molcel.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.MacLea KS, Paul KR, Ben-Musa Z, Waechter A, Shattuck JE, Gruca M, Ross ED. Distinct amino acid compositional requirements for formation and maintenance of the [PSI+] prion in yeast. Mol Cell Biol 2015; 35:899-911; PMID:25547291; http://dx.doi.org/ 10.1128/MCB.01020-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toombs JA, McCarty BR, Ross ED. Compositional determinants of prion formation in yeast. Mol Cell Biol 2010; 30:319-32; PMID:19884345; http://dx.doi.org/ 10.1128/MCB.01140-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parham SN, Resende CG, Tuite MF. Oligopeptide repeats in the yeast protein Sup35p stabilize intermolecular prion interactions. EMBO J 2001; 20:2111-9; PMID:11331577; http://dx.doi.org/ 10.1093/emboj/20.9.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shkundina IS, Kushnirov VV, Tuite MF, Ter-Avanesyan MD. The role of the N-terminal oligopeptide repeats of the yeast Sup35 prion protein in propagation and transmission of prion variants. Genetics 2006; 172:827-35; PMID:16272413; http://dx.doi.org/ 10.1534/genetics.105.048660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biol 2004; 2:E86; PMID:15045026; http://dx.doi.org/ 10.1371/journal.pbio.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Summers DW, Cyr DM. Use of yeast as a system to study amyloid toxicity. Methods 2011; 53:226-31; PMID:21115125; http://dx.doi.org/ 10.1016/j.ymeth.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tenreiro S, Munder MC, Alberti S, Outeiro TF. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. J Neurochem 2013; 127:438-52; PMID:23600759; http://dx.doi.org/ 10.1111/jnc.12271. [DOI] [PubMed] [Google Scholar]
- 63.Marchante R, Rowe M, Zenthon J, Howard MJ, Tuite MF. Structural definition is important for the propagation of the yeast [PSI+] prion. Mol Cell 2013; 50:675-85; PMID:23746351; http://dx.doi.org/ 10.1016/j.molcel.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Asante EA, Smidak M, Grimshaw A, Houghton R, Tomlinson A, Jeelani A, Jakubcova T, Hamdan S, Richard-Londt A, Linehan JM, et al.. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 2015; 522:478-81; PMID:26061765; http://dx.doi.org/ 10.1038/nature14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mead S, Whitfield J, Poulter M, Shah P, Uphill J, Campbell T, Al-Dujaily H, Hummerich H, Beck J, Mein CA, et al.. A novel protective prion protein variant that colocalizes with kuru exposure. N Engl J Med 2009; 361:2056-65; PMID:19923577; http://dx.doi.org/ 10.1056/NEJMoa0809716. [DOI] [PubMed] [Google Scholar]
- 66.DePace AH, Santoso A, Hillner P, Weissman JS. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 1998; 93:1241-52; PMID:9657156; http://dx.doi.org/ 10.1016/S0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 67.Cavalli-Sforza LL, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton University Press, Princeton: 1994. [Google Scholar]
- 68.Sheppard PM. Natutal Selection and Heredity Hutchinson, London: 1958. [Google Scholar]
- 69.Ford EB. Ecological Genetics Chapman & Hall, London: 1964. [Google Scholar]
- 70.Lund PM, Cox BS. Reversion analysis of [psi−] mutations in Saccharomyces cerevisiae. Genet Res 1981; 37:173-82; PMID:7021322; http://dx.doi.org/ 10.1017/S0016672300020140. [DOI] [PubMed] [Google Scholar]
- 71.Cox BS. Psi phenomena in yeast The Early Days of Yeast Genetics 1993; Cold Spring Harbor Laboratory Press: pp219-40. [Google Scholar]