

ABSTRACT
The yeast Saccharomyces cerevisiae can harbor a number of distinct prions. Most of the yeast prion proteins contain a glutamine/asparagine (Q/N) rich region that drives prion formation. Prion-like domains, defined as regions with high compositional similarity to yeast prion domains, are common in eukaryotic proteomes, and mutations in various human proteins containing prion-like domains have been linked to degenerative diseases, including amyotrophic lateral sclerosis. Here, we discuss a recent study in which we utilized two strategies to generate prion activity in non-prion Q/N-rich domains. First, we made targeted mutations in four non-prion Q/N-rich domains, replacing predicted prion-inhibiting amino acids with prion-promoting amino acids. All four mutants formed foci when expressed in yeast, and two acquired bona fide prion activity. Prion activity could be generated with as few as two mutations, suggesting that many non-prion Q/N-rich proteins may be just a small number of mutations from acquiring aggregation or prion activity. Second, we created tandem repeats of short prion-prone segments, and observed length-dependent prion activity. These studies demonstrate the considerable progress that has been made in understanding the sequence basis for aggregation of prion and prion-like domains, and suggest possible mechanisms by which new prion domains could evolve.
KEYWORDS: amyloid, prion, protein aggregation, Sup35, yeast
INTRODUCTION
Prions are self-propagating, infectious protein isoforms that generally result from the structural conversion of soluble proteins into an infectious amyloid form. At least 10 prions have been identified in Saccharomyces cerevisiae.1,2 Most of the yeast prion proteins contain glutamine/asparagine (Q/N) rich prion-forming domains (PFDs) that are responsible for prion activity. The PFDs tend to be long, intrinsically disordered, low complexity regions. Similar prion-like domains (PrLDs) are common in eukaryotic genomes, particularly in RNA binding proteins.3 Growing evidence indicates that aggregation of PrLD-containing proteins may be involved in regulating normal cellular processes, including RNA localization and turnover.4 Mutations in a number of these PrLDs have also been linked to degenerative diseases, including amyotrophic lateral sclerosis, fronto-temporal dementia, and inclusion body myopathy.3,5 This has created increased interest in understanding the basis for aggregation PFDs and PrLDs.
Here we discuss a recent study showing that rational mutations can be designed to generate prion activity in non-prion Q/N-rich domains.6 Two strategies were successfully employed: targeted mutations to increase prion propensity, and duplication of predicted prion-prone segments. These results provide insight into the sequence basis for prion activity, and offer possible mechanisms by which new prion domains could evolve. Additionally, the ability to manipulate prion propensity could provide a useful tool to study the role of PrLDs in both normal physiology and disease.
Using Targeted Mutations to Create Prion Activity
Most yeast PFDs have similar amino acid compositions, with high Q/N content, and few charged and hydrophobic residues.7 Randomizing the order of the amino acids in the PFDs of 2 yeast prion proteins, Sup35 and Ure2, does not block prion activity, demonstrating that amino acid composition, not primary sequence, is the predominant determinant of prion propensity.8,9 Various labs have developed prediction methods to identify new prion proteins based on compositional similarity to known PFDs.5 These methods have been quite effective at identifying new prion candidates, but less effective at distinguishing among the top candidates or predicting the effects of mutation.5
To improve prion prediction, we developed a quantitative mutagenesis method to score the prion propensity of each amino acid in the context of a Q/N-rich PFD.10 These values were used to build PAPA, a prion prediction algorithm that shows a strong ability to distinguish between Q/N-rich domains with and without prion activity.11 Charged residues and prolines each score as strongly prion-inhibiting in PAPA, consistent with their relative rarity in yeast PFDs. However, despite their underrepresentation in yeast PFDs, hydrophobic and aromatic residues score as strongly prion-promoting. Indeed, increasing the number of hydrophobic residues in the Sup35 PFD increases aggregation propensity and prion activity.12
We therefore asked whether we could similarly turn non-prion proteins into prions by replacing prion-inhibiting residues with prion-promoting residues. Alberti et al. previously screened the yeast genome for regions with high compositional similarity to known yeast PFDs, and tested the top 100 prion-like domains in 4 assays for aggregation and prion activity.13 We selected 4 of these PrLDs that showed no detectable aggregation or prion activity, and serially replaced prion-inhibiting amino acids with prion-promoting amino acids until the predicted PAPA score was above 0.10, a threshold associated with high prion activity (Fig. 1).5 We observed that among the Alberti data set, PrLDs with high prion activity were more likely to have long stretches without any prion-inhibiting amino acids; we therefore targeted the mutations to create such stretches.
FIGURE 1.

Sequences of the four PrLDs that were mutated to create prion activity. Predicted prion-promoting residues are indicated in green, while prion-inhibiting residues are indicated in red. Mutations made to create prion activity are indicated above each sequence. For Puf4, the four regions for which tandem repeats were generated are underlined. For Yck1, only the first 90 amino acids of the 117 amino acid PrLD are shown.
When expressed in yeast as GFP fusions, each of the 4 mutant PrLDs efficiently formed foci, while the wild-type PrLDs remained diffuse, indicating that the mutations were sufficient to cause aggregation. However, many non-prion proteins form foci in cells; true prion activity requires the ability to exist in, and convert between, two stable states. To test whether the mutants could support bona fide prion activity, each was inserted in the place of the Sup35 PFD. None of the wild-type PrLD-Sup35 fusions showed any prion activity. By contrast, 2 of the 4 mutant PrLD-Sup35 fusions (from Puf4 and YLR177W) formed prions that were stably propagated over multiple generations, and one other (from Pdc2) formed unstable prions that could be maintained with selection, but were rapidly lost on non-selective medium. Although the original mutants had 5-7 amino acid substitutions, prion activity could be generated in Puf4 with as few as 2 mutations.
These studies highlight the significant strides that we have made in understanding aggregation propensity. However, the fact that all 4 mutants formed foci, while only 2 of 4 formed stable prions, suggests that methods like PAPA may be more effective at predicting aggregation propensity than prion propensity. Previous studies do provide possible explanations for the failure of Pdc2 and Yck1 to form stable prions. Yck1 was the only mutant unable to form the two states that are characteristic of prions; interestingly, it also had the highest ratio of Q to N of the 4 proteins, consistent with previous reports that N-rich proteins are more conducive to prion activity than Q-rich proteins.14 Pdc2, which formed prions that were poorly propagated, had the lowest number of aromatic residues. There are distinct sequence and compositional requirements for the formation of protein aggregates and the ability of these aggregates to propagate as bona fide prions;15-17 aromatic residues seem to promote the stable maintenance of prions by promoting chaperone-dependent fragmentation of prion aggregates, thereby offsetting dilution of aggregate numbers upon cell division.15,16 However, it should be noted that there is a significant range of aromatic content among the Q/N-rich yeast PFDs. Sfp1 has the lowest (1.3%), while Sup35 has the highest (17.8%);7 the Pdc2 PrLD, with 5.5% aromatic residues, is well within this range, suggesting that aromatic content alone is not sufficient to explain its failure to propagate stable prions.
Larger sample sizes would be needed to further examine the basis for the failure of Pdc2 and Yck1, but these data suggest that prediction of prion activity will require more complex methods than are currently available. Nevertheless, our success in generating 2 new PFDs indicates that the sequence requirements for prion propagation are sufficiently broad that many Q/N-rich domains, if mutated to aggregate, will acquire prion activity.
Using Repeat Expansions to Create Prion Activity
Most yeast PFDs are long, low complexity regions of consistent, modest prion propensity. We hypothesized that another way to generate a new PFD would be to make tandem repeats of short sequences fitting this description. To test this hypothesis, we identified 4 short segments (5-9 amino acids; Fig. 1) within Puf4 that consisted primarily of polar amino acids, with a small number of prion-promoting hydrophobic or aromatic residues. We inserted tandem repeats of these sequences into Puf4 and tested for prion activity using the Sup35 fusion assay. At all 4 positions, length dependent prion activity was observed.
We hypothesized that this increase in prion activity was not due to the repeats per se, but instead was simply a result of creating larger aggregation-prone segments. However, while scrambling the repeat segments did not eliminate length-dependent prion activity, prion activity generally required longer lengths for the scrambled constructs. This suggests that the primary sequence of the repeats may contribute modestly to prion activity.
These results demonstrate that while amino acid composition is a dominant determinant of the aggregation propensity of Q/N-rich domains, primary sequence also exerts an effect. The proposed structure of the prion fibers may explain both the composition and primary sequence effects. Q/N-rich prion proteins are thought to adopt a serpentine structure in amyloid fibrils consisting of alternating β-strand and loop segments; individual peptide monomers then stack to form an in-register parallel β-sheet (Fig. 2).2 In this structure, interactions along the length of the fiber are between identical amino acids on adjacent monomers. Thus, insertion of a hydrophobic residue would generate stabilizing hydrophobic interactions along the length of the fiber, while an inserted charged residue would create charge repulsions. Scrambling the sequence would not change these interactions, but instead would simply change their context.18 However, there are additional interactions that could be sensitive to primary sequence. Adjacent strands in the serpentine structure are thought to pack to form a steric zipper;19 these packing interactions should be sensitive to primary sequence. Likewise, charged residues and prolines are thought to be best accommodated in loop segments, so the positioning of these residues may affect whether a peptide can adopt a stable serpentine structure. For both of these reasons, the regular periodicity of repeat segments may facilitate formation of the serpentine structure.
FIGURE 2.
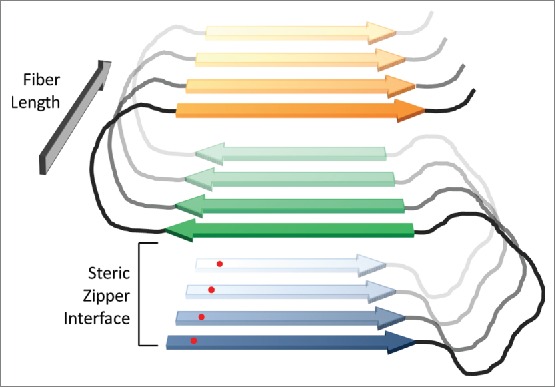
In-register parallel β-sheet structure for prion fibers. In prion fibers, yeast prion proteins are proposed to adopt a serpentine structure, with β-strands (blue, green and orange) separated by loops (black). Protein monomers then stack in-register, forming parallel β-sheets that run the length of the fiber. The fibers are stabilized by both β-sheet interactions along the length of the fiber and steric zipper packing interactions between strands within the plane of the fiber (between the blue and green strands, and the green and orange strands in the figure). Because an individual amino acid (red dot) will align with the corresponding amino acid in the adjacent protein, interactions along the length of the fiber should be largely primary-sequence independent. However, the steric zipper packing interactions should be sensitive to primary sequence.
One prediction algorithm, ArchCandy, has been developed to predict the steric zipper packing interactions within serpentine structures.20 In preliminary analysis with ArchCandy, we were not able to identify any obvious trends that fully explained the observed primary sequence effects. However, it is possible that with larger sample sizes or more sophisticated analysis, this or other similar methods could help explain the effects of primary sequence on prion activity.
EVOLUTION OF NEW PRION DOMAINS
Despite having similar amino acid compositions, the yeast PFDs do not share any obvious sequence homology. This raises the question of how so many yeast proteins appear to have independently evolved these long, compositionally similar PFDs. Our results suggest 2 mechanisms by which new PFDs could evolve.
Tartaglia et al.21 previously proposed that because uncontrolled protein aggregation is generally deleterious, cells are under significant selective pressure to prevent aggregation; however, once a protein's aggregation propensity has evolved to the point that the protein remains soluble under normal physiological conditions, there is little selective pressure to further reduce aggregation propensity. Because most mutations are likely to increase aggregation propensity, the opposing forces of genetic drift and natural selection will tend to result in most proteins being just soluble enough to function under physiological conditions. This “life on the edge” theory is appealing, because it explains why small changes in protein sequence, expression levels, or cellular environment frequently lead to protein aggregation. The theory also offers a potential mechanism to explain how new PFDs could evolve. If a Q/N-rich domain evolved for reasons unrelated to prion formation, the life-on-the-edge theory suggests that natural selection and genetic drift will result in the protein being just a small number of mutations from aggregating. Our results indicate that the ability to propagate as a prion is a sufficiently common feature of Q/N-rich domains that for many Q/N-rich domains, if they were to acquire mutations that resulted in aggregation, they would also acquire prion activity. Some of the yeast PFDs have functions other than prion activity,22-24 making it possible that these functions could have preceded the acquisition of prion activity.
There is debate for specific yeast prions as to whether they are diseases of yeast2 or beneficial epigenetic elements.25 Importantly the life-on-the-edge theory explains how either deleterious or beneficial prions could evolve. In cases where prion formation is beneficial, having many proteins on the cusp of aggregation would increase the probability of evolving new functional prions. In cases where prion formation is deleterious, if the natural selection and genetic drift tend to keep PrLDs on the edge of aggregation, then rare events or changes in expression or cellular environment could push the proteins over the edge, causing prion formation.
The repeat expansion results offer a second mechanism by which new prion domains could evolve. Many prion proteins contain repeat sequences, and expansions of the repeats in both Sup35 and the mammalian prion protein PrP are associated with increased prion activity.26,27 This would seem to suggest that repeats per se promote prion activity; however, this idea is contradicted by the observation that scrambling the Sup35 repeats does not block prion activity.28 We therefore proposed that repeats may be common not because they directly promote prion activity, but because they provide a simple mechanism to generate the long, low-complexity sequences that characterize yeast PFDs.28 Duplication of DNA elements is a common source of genetic diversity,29,30 and can result from errors in DNA repair, recombination or replication, so duplication of prion-like sequences would provide a simple way to rapidly increase prion activity. Trinucleotide repeats represent a simple form of tandem repeat, and are common sources of disease due to repeat instability; it has similarly been proposed that some yeast PFDs may have evolved from poly-Q or poly-N repeats.31
CONCLUSIONS AND CAVEATS
Despite the success in generating new PFDs, these experiments nevertheless carry significant caveats. Importantly, the prediction methods described here are likely specific to Q/N-rich domains. There are 2 basic reasons for this. First, for many amyloid-forming proteins, amyloid-prone segments are buried in the folded state of the protein, so native state stability can be a critical determinant of aggregation propensity.32 This makes predicting the effects of mutations more challenging, as mutations can alter aggregation propensity by affecting either intrinsic aggregation propensity or native state stability. By contrast, Q/N-rich domains tend to be intrinsically disordered, simplifying prediction of aggregation propensity. Indeed, the PAPA algorithm only provides predictions for regions that are predicted to be intrinsically disordered. Second, the sequence features that drive aggregation seem to differ between Q/N-rich and non-Q/N-rich proteins. Aggregation by non-Q/N-rich proteins is frequently driven by short, highly amyloidogenic fragments, and algorithms designed to identify these domains have shown reasonable success at predicting the behavior of non-Q/N-rich domains.33,34 Such fragments may be transiently exposed by local unfolding.35 By contrast, the intrinsic disorder of most Q/N-rich domains likely allows much larger regions to be simultaneously exposed. This may explain why, despite evidence that short aggregation-prone fragments can serve as key nucleating sites in Q/N-rich PFDs,36 their presence is a poor predictor of prion propensity.11 The prediction methods that have had the most success predicting prion activity of Q/N-rich domains have tended to use much larger window sizes.10,13,37
A second caveat is that these experiments were all performed using fusion proteins, and therefore may not reflect the behavior of the PrLDs in their native context. The prion activity of a PrLD-containing protein is dependent not only on the prion propensity of the PrLD, but also on factors such as the sequence context in which the domain occurs, interacting proteins, and the expression level and localization of the protein. This can result in both false positives and false negatives in the Sup35 fusion assay; for example, the PFDs from the yeast prion proteins Cyc8 and Mot3 both fail to form prions when fused to Sup35, and numerous PrLDs that show prion activity when fused to Sup35 have not yet been demonstrated to support prion activity in their native context.13 Therefore, while our experiments reveal two mechanisms by which a Q/N-rich domain could evolve prion activity, these mechanisms would only create a new prion if the domain occurred in the correct context.
Overall, these experiments demonstrate that the prion propensity of Q/N-rich proteins can be rationally manipulated. However, they also reveal areas that are less well understood, including: the basis for primary sequence effects; the sequence features that drive prion activity versus aggregation activity; and how factors other than intrinsic aggregation propensity affect prion activity.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Funding
EDR is supported by the National Institutes of Health (GM105991) and the National Science Foundation (MCB-1517231).
REFERENCES
- 1.Liebman SW, Chernoff YO. Prions in yeast. Genetics 2012; 191:1041-72; PMID:22879407; http://dx.doi.org/ 10.1534/genetics.111.137760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wickner RB, Shewmaker FP, Bateman DA, Edskes HK, Gorkovskiy A, Dayani Y, Bezsonov EE. Yeast prions: structure, biology, and prion-handling systems. Microbiol Mol Biol Rev 2015; 79:1-17; PMID:25631286; http://dx.doi.org/ 10.1128/MMBR.00041-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res 2012; 1462:61-81; PMID:22445064; http://dx.doi.org/ 10.1016/j.brainres.2012.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 2013; 154:727-36; PMID:23953108; http://dx.doi.org/ 10.1016/j.cell.2013.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cascarina SM, Ross ED. Yeast prions and human prion-like proteins: sequence features and prediction methods. Cell Mol Life Sci 2014; 71:2047-63; PMID:24390581; http://dx.doi.org/ 10.1007/s00018-013-1543-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paul KR, Hendrich CG, Waechter A, Harman MR, Ross ED. Generating new prions by targeted mutation or segment duplication. Proc Natl Acad Sci U S A 2015; 112:8584-9; PMID:26100899; http://dx.doi.org/ 10.1073/pnas.1501072112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du Z. The complexity and implications of yeast prion domains. Prion 2011; 5:311-6; PMID:22156731; http://dx.doi.org/ 10.4161/pri.5.4.18304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross ED, Baxa U, Wickner RB. Scrambled Prion Domains Form Prions and Amyloid. Mol Cell Biol 2004; 24:7206-13; PMID:15282319; http://dx.doi.org/ 10.1128/MCB.24.16.7206-7213.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross ED, Edskes HK, Terry MJ, Wickner RB. Primary sequence independence for prion formation. Proc Natl Acad Sci USA 2005; 102:12825-30; PMID:16123127; http://dx.doi.org/ 10.1073/pnas.0506136102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toombs JA, McCarty BR, Ross ED. Compositional determinants of prion formation in yeast. Mol Cell Biol 2010; 30:319-32; PMID:19884345; http://dx.doi.org/ 10.1128/MCB.01140-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toombs JA, Petri M, Paul KR, Kan GY, Ben-Hur A, Ross ED. De novo design of synthetic prion domains. Proc Natl Acad Sci U S A 2012; 109:6519-24; PMID:22474356; http://dx.doi.org/ 10.1073/pnas.1119366109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez Nelson AC, Paul KR, Petri M, Flores N, Rogge RA, Cascarina SM, Ross ED. Increasing prion propensity by hydrophobic insertion. PLoS One 2014; 9:e89286; PMID:24586661; http://dx.doi.org/ 10.1371/journal.pone.0089286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A Systematic Survey Identifies Prions and Illuminates Sequence Features of Prionogenic Proteins. Cell 2009; 137:146-58; PMID:19345193; http://dx.doi.org/ 10.1016/j.cell.2009.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halfmann R, Alberti S, Krishnan R, Lyle N, O'Donnell CW, King OD, Berger B, Pappu RV, Lindquist S. Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins. Mol Cell 2011; 43:72-84; PMID:21726811; http://dx.doi.org/ 10.1016/j.molcel.2011.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexandrov AI, Polyanskaya AB, Serpionov GV, Ter-Avanesyan MD, Kushnirov VV. The effects of amino acid composition of glutamine-rich domains on amyloid formation and fragmentation. PLoS ONE 2012; 7:e46458; PMID:23071575; http://dx.doi.org/ 10.1371/journal.pone.0046458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacLea KS, Paul KR, Ben-Musa Z, Waechter A, Shattuck JE, Gruca M, Ross ED. Distinct amino acid compositional requirements for formation and maintenance of the [PSI(+)] prion in yeast. Mol Cell Biol 2015; 35:899-911; http://dx.doi.org/ 10.1128/MCB.01020-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biol 2004; 2:E86; PMID:15045026; http://dx.doi.org/ 10.1371/journal.pbio.0020086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ross ED, Minton A, Wickner RB. Prion domains: sequences, structures and interactions. Nature Cell Biol 2005; 7:1039-44; PMID:16385730; http://dx.doi.org/ 10.1038/ncb1105-1039 [DOI] [PubMed] [Google Scholar]
- 19.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005; 435:773-8; PMID:15944695; http://dx.doi.org/ 10.1038/nature03680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmed AB, Znassi N, Chateau MT, Kajava AV. A structure-based approach to predict predisposition to amyloidosis. Alzheimers Demen 2015; 11(6):681-90 [DOI] [PubMed] [Google Scholar]
- 21.Tartaglia GG, Pechmann S, Dobson CM, Vendruscolo M. Life on the edge: a link between gene expression levels and aggregation rates of human proteins. Trends Biochem Sci 2007; 32:204-6; PMID:17419062; http://dx.doi.org/ 10.1016/j.tibs.2007.03.005 [DOI] [PubMed] [Google Scholar]
- 22.Hosoda N, Kobayashi T, Uchida N, Funakoshi Y, Kikuchi Y, Hoshino S, Katada T. Translation termination factor eRF3 mediates mRNA decay through the regulation of deadenylation. J Biol Chem 2003; 278:38287-91; PMID:12923185; http://dx.doi.org/ 10.1074/jbc.C300300200 [DOI] [PubMed] [Google Scholar]
- 23.Li X, Rayman JB, Kandel ER, Derkatch IL. Functional role of tia1/pub1 and sup35 prion domains: directing protein synthesis machinery to the tubulin cytoskeleton. Mol Cell 2014; 55:305-18; PMID:24981173; http://dx.doi.org/ 10.1016/j.molcel.2014.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shewmaker F, Mull L, Nakayashiki T, Masison DC, Wickner RB. Ure2p function is enhanced by its prion domain in Saccharomyces cerevisiae. Genetics 2007; 176:1557-65; PMID:17507672; http://dx.doi.org/ 10.1534/genetics.107.074153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newby GA, Lindquist S. Blessings in disguise: biological benefits of prion-like mechanisms. Trends Cell Biol 2013; 23:251-9; PMID:23485338; http://dx.doi.org/ 10.1016/j.tcb.2013.01.007 [DOI] [PubMed] [Google Scholar]
- 26.Liu JJ, Lindquist S. Oligopeptide-repeat expansions modulate 'protein-only' inheritance in yeast. Nature 1999; 400:573-6; PMID:10448860; http://dx.doi.org/ 10.1038/22919 [DOI] [PubMed] [Google Scholar]
- 27.Wadsworth JD, Hill AF, Beck JA, Collinge J. Molecular and clinical classification of human prion disease. Br Med Bull 2003; 66:241-54; PMID:14522862; http://dx.doi.org/ 10.1093/bmb/66.1.241 [DOI] [PubMed] [Google Scholar]
- 28.Toombs JA, Liss NM, Cobble KR, Ben-Musa Z, Ross ED. [PSI+] maintenance is dependent on the composition, not primary sequence, of the oligopeptide repeat domain. PLoS One 2011; 6:e21953; PMID:21760933; http://dx.doi.org/ 10.1371/journal.pone.0021953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kajava AV. Tandem repeats in proteins: from sequence to structure. J Struct Biol 2012; 179:279-88; PMID:21884799; http://dx.doi.org/ 10.1016/j.jsb.2011.08.009 [DOI] [PubMed] [Google Scholar]
- 30.Katti MV, Sami-Subbu R, Ranjekar PK, Gupta VS. Amino acid repeat patterns in protein sequences: their diversity and structural-functional implications. Protein Sci 2000; 9:1203-9; PMID:10892812; http://dx.doi.org/ 10.1110/ps.9.6.1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alexandrov AI, Ter-Avanesyan MD. Could yeast prion domains originate from polyQ/N tracts? Prion 2013; 7:209-14; PMID:23764835; http://dx.doi.org/ 10.4161/pri.24628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol 1998; 8:101-6; PMID:9519302; http://dx.doi.org/ 10.1016/S0959-440X(98)80016-X [DOI] [PubMed] [Google Scholar]
- 33.Maurer-Stroh S, Debulpaep M, Kuemmerer N, Lopez de la Paz M, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, Schymkowitz JW, Rousseau F. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Meth 2010; 7:237-42; PMID:20154676; http://dx.doi.org/ 10.1038/nmeth.1432 [DOI] [PubMed] [Google Scholar]
- 34.Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D, Eisenberg D. The 3D profile method for identifying fibril-forming segments of proteins. Proc Natl Acad Sci U S A 2006; 103:4074-8; PMID:16537487; http://dx.doi.org/ 10.1073/pnas.0511295103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol 2009; 5:15-22; PMID:19088715; http://dx.doi.org/ 10.1038/nchembio.131 [DOI] [PubMed] [Google Scholar]
- 36.Chen B, Bruce KL, Newnam GP, Gyoneva S, Romanyuk AV, Chernoff YO. Genetic and epigenetic control of the efficiency and fidelity of cross-species prion transmission. Mol Microbiol 2010; 76:1483-99; PMID:20444092; http://dx.doi.org/ 10.1111/j.1365-2958.2010.07177.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabate R, Rousseau F, Schymkowitz J, Ventura S. What makes a protein sequence a prion? PLoS Comput Biol 2015; 11:e1004013; PMID:25569335; http://dx.doi.org/ 10.1371/journal.pcbi.1004013 [DOI] [PMC free article] [PubMed] [Google Scholar]