

ABSTRACT
Converging observations from disparate lines of inquiry are beginning to clarify the cause of brain iron dyshomeostasis in sporadic Creutzfeldt-Jakob disease (sCJD), a neurodegenerative condition associated with the conversion of prion protein (PrPC), a plasma membrane glycoprotein, from α-helical to a β-sheet rich PrP-scrapie (PrPSc) isoform. Biochemical evidence indicates that PrPC facilitates cellular iron uptake by functioning as a membrane-bound ferrireductase (FR), an activity necessary for the transport of iron across biological membranes through metal transporters. An entirely different experimental approach reveals an evolutionary link between PrPC and the Zrt, Irt-like protein (ZIP) family, a group of proteins involved in the transport of zinc, iron, and manganese across the plasma membrane. Close physical proximity of PrPC with certain members of the ZIP family on the plasma membrane and increased uptake of extracellular iron by cells that co-express PrPC and ZIP14 suggest that PrPC functions as a FR partner for certain members of this family. The connection between PrPC and ZIP proteins therefore extends beyond common ancestry to that of functional cooperation. Here, we summarize evidence supporting the facilitative role of PrPC in cellular iron uptake, and implications of this activity on iron metabolism in sCJD brains.
KEYWORDS: Prion protein, iron, ferrireductase, sCJD, ZIP14, ZIP proteins
INTRODUCTION
Brain iron dyshomeostasis has been reported in several neurodegenerative conditions, including sporadic Creutzfeldt-Jakob disease (sCJD), Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington's disease (HD). The underlying cause has been the subject of much debate.1-3 Recent reports indicating a physiological role of protein(s) implicated in the pathogenesis of these disorders in cellular iron metabolism provide a partial answer to this long-standing question.4 Here, we review evidence supporting the role of PrPC, the protein underlying sCJD pathogenesis, in cellular iron uptake by functioning as a ferrireductase (FR) partner for a member of the Zrt, Irt-like protein (ZIP) family of divalent metal transporters, especially zinc and iron.1,2,5 It is likely that loss of this function combined with gain of abnormal activity by its β-sheet rich PrP-scrapie (PrPSc) isoform causes imbalance of iron homeostasis in sCJD brains, creating a potentially neurotoxic environment.6-8 A complete understanding of this subject would pave the way for the development of disease-specific therapeutic strategies where none exist, and allow a critical assessment of the use of iron chelators as disease-modifying agents.9
Iron is essential for vital metabolic processes, but unbound iron is highly toxic because of its ability to shuttle between 2 oxidation states, ferric (Fe3+) and ferrous (Fe2+) iron. Although ideal as a catalyst, uncontrolled oxidation/reduction of iron is likely to generate toxic hydroxyl radicals by Fenton chemistry. Iron homeostasis is therefore tightly regulated at the cellular and systemic levels by the coordinated action of iron uptake, efflux, and storage proteins that are themselves regulated by iron regulatory proteins and the peptide hormone hepcidin.10 Although iron pools in the brain are separated from systemic circulation by the blood brain barrier (BBB), similar regulatory mechanisms operate within the brain with minor differences.11 Nevertheless, iron accumulates in the aging brain, and the process is hastened by certain neurodegenerative conditions associated with protein aggregation.4 Whether this is an active process mediated by loss or altered function of specific protein(s) or a consequence of neuronal death combined with inefficient clearance of iron-rich cell debris has been difficult to parse out. One of the reasons is the difficulty in developing appropriate experimental models that reproduce brain pathology and associated changes in iron homeostasis faithfully.
In this regard, models of sCJD and other prion disorders offer 2 specific advantages: 1) PrPC is expressed ubiquitously on all cells, providing an opportunity to study its role in iron metabolism in systemic organs that are experimentally accessible and iron metabolism is better understood, and 2) animal models reproduce major pathological features of sCJD faithfully in a relatively short time, providing an opportunity to identify specific changes in brain iron metabolism with disease progression.12,13 This approach has yielded useful information on the physiological function of PrPC in iron metabolism, and possible causes of iron dyshomeostasis in sCJD brains.8,13 Below, we summarize recent literature on the functional role of PrPC in cellular iron uptake, and implications of this activity on brain iron homeostasis in healthy and sCJD affected brains.
IRON TRAFFICKING: A SHORT OVERVIEW
Iron trafficking in the systemic circulation: Of the total body iron, ~80% is utilized for hemoglobin synthesis, and the rest is distributed among systemic organs and the brain. Only ~2 mg of iron are lost daily, and an equivalent amount is absorbed from the intestinal lumen. At the cellular level, iron is taken up by 2 principal mechanisms; transferrin (Tf)-iron via the Tf-receptor (TfR) pathway, and non-Tf bound iron (NTBI) through divalent metal transporters. Heme-associated iron is transported and metabolized by distinct pathways, and will not be discussed here. Under physiological conditions plasma Tf is ~35% saturated with iron, leaving enough buffering capacity for a pathological increase in circulating iron. Nonetheless, circulating NTBI is detected under physiological and several pathological conditions, and is largely sequestered by the liver to prevent cytotoxicity (Fig. 1). For detailed explanation of systemic iron uptake and metabolism the reader is directed to recent reviews on this subject.2,10,14
FIGURE 1.
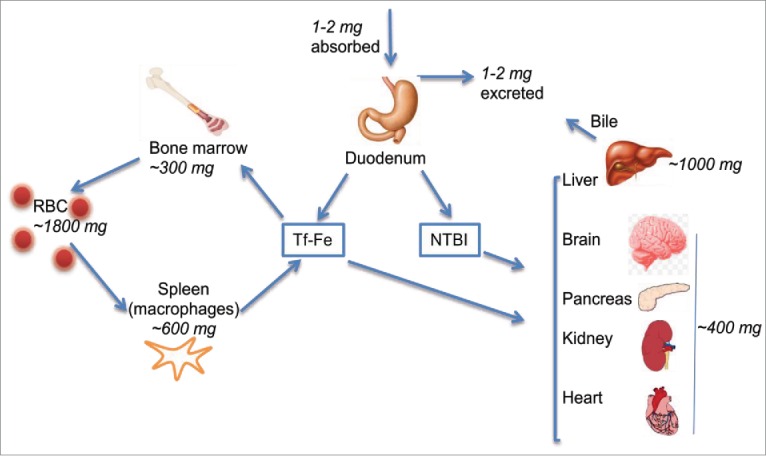
Iron trafficking in the systemic circulation: Systemic iron homeostasis is maintained by regulating uptake from the duodenum. Only 1-2 mg of iron is absorbed daily from the duodenum, and an equivalent amount is lost through epithelial desquamation. Majority of circulating iron is utilized for erythropoiesis, and re-used after recovery from senescent RBCs by splenic macrophages. Iron in the systemic circulation binds plasma Tf or circulates as NTBI in conjugation with small molecular weight compounds. Most organs utilize Tf-Fe preferentially, though some take up significant amounts of NTBI as well. Liver sequesters excess circulating iron and stores in ferritin. Tf-Fe: transferrin iron, NTBI: non-transferrin-bound iron. Numbers indicate the amount of iron in each organ.
Iron trafficking in the brain: Transport of iron from the peripheral circulation to the brain is mediated by the BBB via mechanisms that are still emerging.11 Current understanding of this process is illustrated in Figure 2. It is known that plasma Tf is not transported across the BBB and vice versa. Brain Tf is secreted by the choroid plexus and perhaps oligodendrocytes.15 Unlike serum Tf, brain Tf is ~99% saturated with iron, leaving little buffering capacity for excess iron. Within the brain, each cell type has preference for Tf-iron or NTBI, and express receptors and transporters that facilitate uptake of one or both forms. Neurons utilize both Tf-iron and NTBI, and express TfR for the uptake of Tf-iron and divalent metal transporter 1 (DMT1) and possibly members of the ZIP family for the uptake of NTBI. Evidence for the latter, however, is inconclusive. Astrocytes do not express TfR in vivo and utilize mainly NTBI. Excess intracellular iron is stored in cytosolic ferritin or exported from the cell through the combined action of ferroportin (Fpn) and a ferroxidase (Frx) such as ceruloplasmin (Cp) or hephaestin (Hp) (Fig. 2). Since iron is vital for cell survival, redundancies exist in iron transport pathways such that absence of one does not result in acute depletion or accumulation of iron.
FIGURE 2.
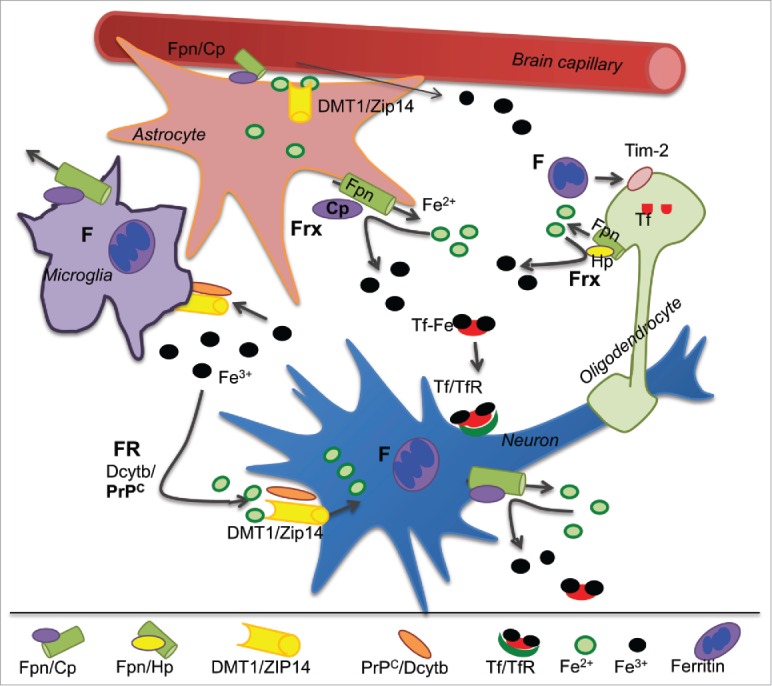
Iron trafficking in the brain: Fe2+ in capillary endothelial cells is exported from the basolateral membrane through Fpn and oxidized to Fe3+ by the Frx Cp expressed on the plasma membrane of astrocytes. Majority of Fe3+ binds to Tf in the brain interstitial fluid. The rest binds to citrate and circulates as NTBI. Astrocytes utilize only NTBI, while neurons take up both Tf-Fe and NTBI. Tf-Fe is internalized through the TfR pathway, and NTBI is reduced to Fe2+ by Dcytb and/or PrPC before transport through DMT1 or ZIP14. Microglia take up NTBI from the extracellular milieu and store iron released from phagocytosed cells in cytosolic ferritin. Oligodendrocytes synthesize and utilize Tf-iron and also take up ferritin iron by the Tim-2 receptor. Frx: ferroxidase, FR: ferrireductase, Cp: ceruloplasmin, Dcytb: duodenal cytochrome b, DMT1: divalent metal transporter 1.
REDOX-CYCLING FOR IRON TRANSPORT
Iron exists in 2 oxidation states, the highly reactive ferrous (Fe2+) form, and the oxidized and relatively stable ferric (Fe3+) form. Both Tf-iron and NTBI are in the Fe3+ form, and require reduction to Fe2+ for transport across biological membranes through metal transporters such as divalent metal transporter 1 (DMT1) and the ZIP family of proteins. This reaction is mediated by FR proteins on the plasma membrane and the endosomal membrane. A major plasma membrane FR is duodenal cytochrome b (Dcytb),16 and the principal endosomal FR is Steap 3.17 The cellular localization of Dcytb and Steap 3 is suited to their ability to function optimally at neutral and acidic pH respectively. Following transport across the membrane, Fe2+ is utilized for metabolic purposes, oxidized and stored in ferritin, or exported from the cell through the combined action of Fpn and Cp or Hp that oxidize Fe2+ to Fe3+ for binding to extracellular Tf. The rest associates with citrate or other small molecular weight compounds that comprise the NTBI pool of iron. Thus, redox-cycling is essential for iron transport, and all iron transporters are linked to an oxido-reductase mechanism (Fig. 3).18
FIGURE 3.
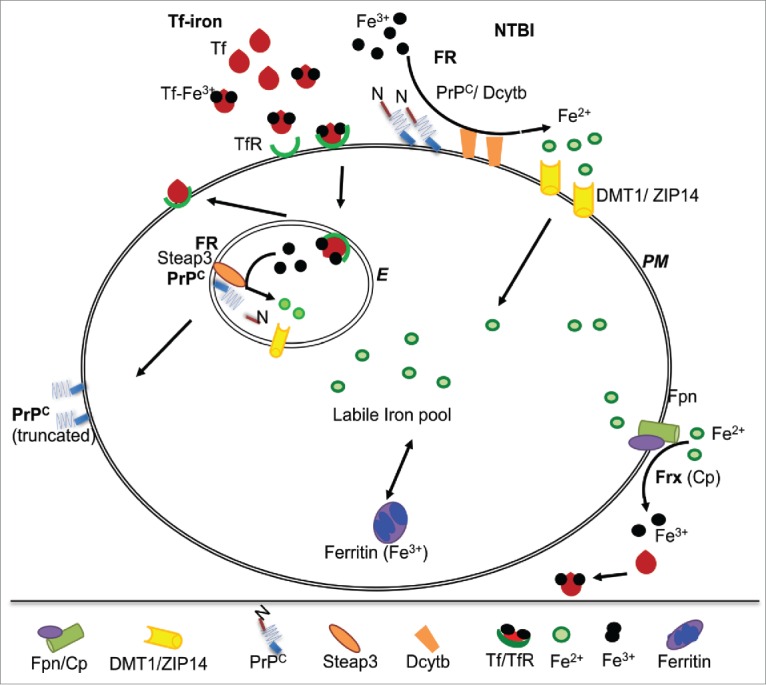
PrPC functions as a FR partner for ZIP14 and DMT1: At the plasma membrane, FR proteins Dcytb and PrPC reduce NTBI to Fe2+ for transport to the cytosol through DMT1 and ZIP14. Fe3+ released from Tf in acidic endosomes is reduced by Steap3 and PrPC for transport to the cytosol through DMT1. PrPC undergoes α-cleavage during this process, releasing its N-terminal FR domain. N-terminally truncated PrPC is transported back to the plasma membrane. PM: plasma membrane, E: endosome.
EVIDENCE SUPPORTING FERRIREDUCTASE ACTIVITY OF PrPC
Initial cues suggesting a functional role of PrPC in iron uptake emerged from studies on PrP-knock-out transgenic mice (PrP−/−) that showed impaired incorporation of supplemental iron administered by the oral or parenteral route in major systemic organs and the brain.19 Reduced levels of iron in PrP−/− mouse brains were also noted using synchroton-based X-ray fluorescence imaging technology.20 However, introduction of PrPC in mice with PrP−/− background reversed their iron deficiency, supporting a non-redundant role of PrPC in iron uptake and metabolism.19 Observations on neuroblastoma cells suggested that PrPC functions as a plasma membrane FR, and the octa-peptide repeat region and linkage to the plasma membrane are essential for this activity.21
The FR activity of PrPC was especially evident in hepatocyte and kidney proximal tubule (PT) cells that absorb significant amounts of NTBI and Tf-iron under physiological conditions, requiring FR activity for uptake and transport. Liver is the main organ that sequesters excess Tf-iron and NTBI from the circulation, and PT cells re-absorb ~99% of Tf-iron and NTBI filtered into the glomerular filtrate, providing excellent models for these studies. Expression of PrPC on HepG2 cells, a hepatocyte cell line, increased uptake of Fe3+, not Fe2+ iron from the extracellular medium, an effect that was reversed by deleting the octa-peptide repeat domain of PrPC.5,21 A similar effect of PrPC was noted on PT cells that form polarized monolayers on filter supports. In these cells, PrPC was localized to the apical plasma membrane and intracellular vesicles, and facilitated trans-cellular transport of NTBI from the apical to the basolateral domain. Again, this activity was abolished by deleting the octa-peptide repeat domain, re-affirming that the facilitative role of PrPC in iron uptake is due to its FR activity.22
In vivo evaluation of FR activity of a specific protein is complicated by the fact that both Tf-iron and NTBI require reduction for transport across biological membranes, and the former pathway is likely to be influenced by factors that alter Tf/TfR recycling. This, experimental models that allow quantification of NTBI uptake in vivo have proved more useful in assessing FR activity of a specific protein in transgenic mouse models. In a typical experiment, circulating NTBI is radiolabeled preferentially with 59Fe by prior saturation of plasma Tf with unlabeled ferric ammonium citrate (FAC), and uptake of 59Fe-NTBI by different organs is monitored over time. Using this approach, it was determined that liver, kidney, heart, and pancreas take up significant amounts of 59Fe-NTBI.23 Evaluation of mouse models using a similar approach revealed significant reduction in the uptake of 59Fe-NTBI by the liver, kidney, and pancreas of PrP-/- mice relative to wild-type controls, supporting a non-redundant role of PrPC as a FR in vivo.5,22
FUNCTIONAL COOPERATION BETWEEN PrPC AND ZIP TRANSPORTERS
Recent reports suggest an evolutionary link between PrPC and the ZIP family of metal transporters that mediate zinc uptake and maintain cellular zinc homeostasis.24 Two members of this family, ZIP8 and ZIP14, transport iron, cadmium, and manganese in addition to zinc, and are expressed on the plasma membrane and the membrane of endocytic vesicles. PrPC and ZIP proteins are expressed on most cells, and certain members of the ZIP family co-immunoprecipitate with PrPC, suggesting close physical proximity on the plasma membrane.25
Interestingly, expression of ZIP14 in PrPC-expressing hepatoma cells increased physiological processing of PrPC at the α-cleavage site, releasing the octa-peptide repeat region including the FR domain. Co-expression of PrPC and ZIP14 increased levels of intracellular iron more than each protein individually, suggesting functional co-operation between the 2 and coupling of FR activity of PrPC with its α-cleavage (Fig. 3).5 Since ~99% of PrPC in the urine is truncated, it is likely that similar processing events occur in PT cells that are known to express PrPC and ZIP transporters on the apical plasma membrane that is bathed in iron-rich glomerular filtrate. It is therefore likely that in addition to mediating iron uptake, PrPC regulates this process by shedding its FR domain. Further investigations are necessary to understand this phenomenon fully.
IMPLICATIONS FOR PRION DISORDERS
Brains of sCJD cases and prion infected mice show deficiency of iron,8 zinc, and manganese.26 The underlying cause of iron deficiency is accumulation in aggregated ferritin in a biologically unavailable form.13 Although it is argued that these changes result from neuronal death, disease-specific alterations in the level(s) of iron modulating proteins in pre-mortem cerebrospinal fluid (CSF) of sCJD and other rapidly progressing dementias suggest otherwise.27 Considering the physical proximity and functional co-operation of PrPC and ZIP proteins, it is likely that loss of function combined with gain of abnormal activity by PrPSc-ZIP aggregates alters homeostasis of iron, zinc, manganese, and other divalent cations in diseased brains. Altered expression of iron regulatory and storage proteins has been reported in the brains of mice inoculated with scrapie intra-cerebrally,28 and spleens of mice where PrPSc was introduced intra-peritoneally.29 Cell lines infected with PrPSc also show altered expression of iron regulatory and storage proteins, supporting the above hypothesis.30 It is noteworthy that prion infectivity is transmitted from cell-to-cell via exosomes that are associated with PrPSc.31 Exosomes also transport other aggregated proteins such as amyloid-β that bind metals with high affinity, and are likely to include PrPSc-ferritin aggregates that resist degradation by lysosomes. Fusion of such vesicles with neighboring cells is likely to alter their metal homeostasis, a phenomenon that is likely to contribute to iron dyshomeostasis and requires further exploration.
METAL CHELATION AS A POTENTIAL DISEASE-MODIFYING STRATEGY
Metal chelation therapy for the management of neurodegenerative conditions has been the focus of several recent reviews. Specific metals targeted by this approach include iron, copper, and zinc. The apparently critical involvement of metals, particularly iron, in both oxidative stress and protein aggregation renders chelation therapy a sensible strategy. An essential feature of such an approach would be the ability to scavenge the free redox-active metal to form a nontoxic metal complex which would be excreted, and secondly, to cap the metal at its labile binding site, preventing Fenton activity and secondary damage to neighboring cells.
Thus far, most trials have been conducted on iron chelators. Deferiprone, desferrioxamine, desferasirox, clioquinol, and certain other compounds have shown promise in cases of PD, AD, and Friedreich's ataxia.32,33 PBT2 (5,7-dichloro-2-(dimethylamino)-methyl-8-hydroxyquinoline) that chelates zinc, copper, and possibly iron has shown cognitive improvement in cases of AD.32 Other trials have shown mixed results, and are summarized in recent reviews.34,35
Limited studies have evaluated the therapeutic potential of metal chelation in sCJD and other variants of human prion disorders. Published reports have focused on reducing copper, zinc, and manganese in diseased brains. Although iron imbalance is clearly a feature of sCJD and experimental scrapie, iron chelation has not been tried, and we hope this review will be instrumental in triggering these studies. However, it is important to consider that chelation of one metal is likely to create imbalance of other metals, creating an equally neurotoxic microenvironment. This scenario is especially likely for metals such as iron, copper, and zinc that share transporters such as the ZIP family of proteins. It is therefore essential to devise strategies that restore metal homeostasis in the diseased brain rather than deplete one or the other metal. As new metal chelators emerge, it may be possible to achieve this goal. This remains a promising area for further research.
FUTURE DIRECTIONS
The functional connection between PrPC and ZIP proteins has significant implications for metal homeostasis under physiological and pathological conditions. Altered function of either partner is likely to influence cellular uptake of iron and other cations transported by ZIP proteins. A better understanding of the functional cooperation between PrPC and members of the ZIP family is likely to impact the therapeutic management of sCJD. Significant areas of future investigation include 1) further characterization of FR activity of PrPC and whether its reductase function extends to copper, 2) a better understanding of the prion-ZIP connection and identification of specific members of the ZIP family that interact with PrPC, 3) functional implications of proteolytic cleavage of PrPC on metal transport, and 4) the impact of conversion of PrPC to PrPSc on PrP-metal interaction and metal homeostasis in neurons and other cell types in the brain. We hope that this review will stimulate research in these areas and improve our understanding of the prion-ZIP connection.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
- 1.Singh N. The role of iron in prion disease and other neurodegenerative diseases. PLoS Pathog 2014; 10:e1004335; PMID:25232824; http://dx.doi.org/ 10.1371/journal.ppat.1004335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh N, Haldar S, Tripathi AK, Horback K, Wong J, Sharma D, Beserra A, Suda S, Anbalagan C, Dev S, et al. Brain iron homeostasis: from molecular mechanisms to clinical significance and therapeutic opportunities. Antioxid Redox Signal 2014; 20:1324-63; PMID:23815406; http://dx.doi.org/ 10.1089/ars.2012.4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Funke C, Schneider SA, Berg D, Kell DB. Genetics and iron in the systems biology of Parkinson disease and some related disorders. Neurochem Int 2013; 62:637-52; PMID:23220386; http://dx.doi.org/ 10.1016/j.neuint.2012.11.015 [DOI] [PubMed] [Google Scholar]
- 4.Wong BX, Duce JA. The iron regulatory capability of the major protein participants in prevalent neurodegenerative disorders. Front Pharmacol 2014; 5:81; PMID:24795635; http://dx.doi.org/ 10.3389/fphar.2014.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tripathi AK, Haldar S, Qian J, Beserra A, Suda S, Singh A, Hopfer U, Chen SG, Garrick MD, Turner JR, et al. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic Biol Med 2015; 84:322-30; PMID:25862412; http://dx.doi.org/ 10.1016/j.freeradbiomed.2015.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hwang D, Lee IY, Yoo H, Gehlenborg N, Cho JH, Petritis B, Baxter D, Pitstick R, Young R, Spicer D, et al. A systems approach to prion disease. Mol Syst Biol 2009; 5:252; PMID:19308092; http://dx.doi.org/ 10.1038/msb.2009.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kell D. Journal club. A systems biologist ponders how disparate ideas can sometimes come together beautifully. Nature 2009; 460:669; PMID:19661875; http://dx.doi.org/ 10.1038/460669e [DOI] [PubMed] [Google Scholar]
- 8.Singh A, Isaac AO, Luo X, Mohan ML, Cohen ML, Chen F, Kong Q, Bartz J, Singh N. Abnormal brain iron homeostasis in human and animal prion disorders. PLoS pathogens 2009; 5:e1000336; PMID:19283067; http://dx.doi.org/ 10.1371/journal.ppat.1000336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinreb O, Mandel S, Youdim MB, Amit T. Targeting dysregulation of brain iron homeostasis in Parkinson disease by iron chelators. Free Radic Biol Med 2013; 62:52-64; PMID:23376471; http://dx.doi.org/ 10.1016/j.freeradbiomed.2013.01.017 [DOI] [PubMed] [Google Scholar]
- 10.Anderson GJ, Vulpe CD. Mammalian iron transport. Cell Mol Life Sci 2009; 66:3241-61; PMID:19484405; http://dx.doi.org/ 10.1007/s00018-009-0051-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simpson IA, Ponnuru P, Klinger ME, Myers RL, Devraj K, Coe CL, Lubach GR, Carruthers A, Connor JR. A novel model for brain iron uptake: introducing the concept of regulation. J Cereb Blood Flow Metab 2015; 35:48-57; PMID:25315861; http://dx.doi.org/ 10.1038/jcbfm.2014.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Head MW, Ironside JW. Review: Creutzfeldt-Jakob disease: prion protein type, disease phenotype and agent strain. Neuropathol Appl Neurobiol 2012; 38:296-310; PMID:22394291; http://dx.doi.org/ 10.1111/j.1365-2990.2012.01265.x [DOI] [PubMed] [Google Scholar]
- 13.Singh A, Qing L, Kong Q, Singh N. Change in the characteristics of ferritin induces iron imbalance in prion disease affected brains. Neurobiol Dis 2012; 45:930-8; PMID:22182691; http://dx.doi.org/ 10.1016/j.nbd.2011.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lane DJ, Merlot AM, Huang ML, Bae DH, Jansson PJ, Sahni S, Kalinowski DS, Richardson DR. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim Biophys Acta 2015; 1853:1130-44; PMID:25661197; http://dx.doi.org/ 10.1016/j.bbamcr.2015.01.021 [DOI] [PubMed] [Google Scholar]
- 15.Skjorringe T, Burkhart A, Johnsen KB, Moos T. Divalent metal transporter 1 (DMT1) in the brain: implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front Mol Neurosci 2015; 8:19; PMID:26106291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Latunde-Dada GO, Simpson RJ, McKie AT. Duodenal cytochrome B expression stimulates iron uptake by human intestinal epithelial cells. J Nutr 2008; 138:991-5; PMID:18492824 [DOI] [PubMed] [Google Scholar]
- 17.Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood 2006; 108:1388-94; PMID:16609065; http://dx.doi.org/ 10.1182/blood-2006-02-003681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kosman DJ. Redox cycling in iron uptake, efflux, and trafficking. J Biol Chem 2010; 285:26729-35; PMID:20522542; http://dx.doi.org/ 10.1074/jbc.R110.113217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh A, Kong Q, Luo X, Petersen RB, Meyerson H, Singh N. Prion protein (PrP) knockout mice show altered iron metabolism: a functional role for PrP in iron uptake and transport. PLoS One 2009; 4:e6115; PMID:19568430; http://dx.doi.org/ 10.1371/journal.pone.0006115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pushie MJ, Pickering IJ, Martin GR, Tsutsui S, Jirik FR, George GN. Prion protein expression level alters regional copper, iron and zinc content in the mouse brain. Metallomics 2011; 3:206-14; PMID:21264406; http://dx.doi.org/ 10.1039/c0mt00037j [DOI] [PubMed] [Google Scholar]
- 21.Singh A, Haldar S, Horback K, Tom C, Zhou L, Meyerson H, Singh N. Prion protein regulates iron transport by functioning as a ferrireductase. J Alzheimer Dis 2013; 35:541-52; PMID:23478311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haldar S, Tripathi A, Qian J, Beserra A, Suda S, McElwee M, Turner J, Hopfer U, Singh N. Prion protein promotes kidney iron uptake via its ferrireductase activity. J Biol Chem 2015; 290(9):5512-22; PMID:25572394; http://dx.doi.org/ 10.1074/jbc.M114.607507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Craven CM, Alexander J, Eldridge M, Kushner JP, Bernstein S, Kaplan J. Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: a rodent model for hemochromatosis. Proc Natl Acad Sci U S A 1987; 84:3457-61; PMID:3472216; http://dx.doi.org/ 10.1073/pnas.84.10.3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehsani S, Mehrabian M, Pocanschi CL, Schmitt-Ulms G. The ZIP-prion connection. Prion 2012; 6:317-21; PMID:22575750; http://dx.doi.org/ 10.4161/pri.20196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ehsani S, Huo H, Salehzadeh A, Pocanschi CL, Watts JC, Wille H, Westaway D, Rogaeva E, St George-Hyslop PH, Schmitt-Ulms G. Family reunion–the ZIP/prion gene family. Prog Neurobiol 2011; 93:405-20; PMID:21163327; http://dx.doi.org/ 10.1016/j.pneurobio.2010.12.001 [DOI] [PubMed] [Google Scholar]
- 26.Ehsani S, Salehzadeh A, Huo H, Reginold W, Pocanschi CL, Ren H, Wang H, So K, Sato C, Mehrabian M, et al. LIV-1 ZIP ectodomain shedding in prion-infected mice resembles cellular response to transition metal starvation. J Mol Biol 2012; 422:556-74; PMID:22687393; http://dx.doi.org/ 10.1016/j.jmb.2012.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haldar S, Beveridge J, Wong J, Singh A, Galimberti D, Borroni B, Zhu X, Blevins J, Greenlee J, Perry G, et al. A low-molecular-weight ferroxidase is increased in the CSF of sCJD cases: CSF ferroxidase and transferrin as diagnostic biomarkers for sCJD. Antioxid Redox Signal 2013; 19:1662-75; PMID:23379482; http://dx.doi.org/ 10.1089/ars.2012.5032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim BH, Jun YC, Jin JK, Kim JI, Kim NH, Leibold EA, Connor JR, Choi EK, Carp RI, Kim YS. Alteration of iron regulatory proteins (IRP1 and IRP2) and ferritin in the brains of scrapie-infected mice. Neurosci Lett 2007; 422:158-63; PMID:17614197; http://dx.doi.org/ 10.1016/j.neulet.2007.05.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huzarewich RL, Medina S, Robertson C, Parchaliuk D, Booth SA. Transcriptional modulation in a leukocyte-depleted splenic cell population during prion disease. J Toxicol Environ Health A 2011; 74:1504-20; PMID:22043911; http://dx.doi.org/ 10.1080/15287394.2011.618979 [DOI] [PubMed] [Google Scholar]
- 30.Fernaeus S, Halldin J, Bedecs K, Land T. Changed iron regulation in scrapie-infected neuroblastoma cells. Brain Res Mol Brain Res 2005; 133:266-73; PMID:15710243; http://dx.doi.org/ 10.1016/j.molbrainres.2004.10.018 [DOI] [PubMed] [Google Scholar]
- 31.Bellingham SA, Guo B, Hill AF. The secret life of extracellular vesicles in metal homeostasis and neurodegeneration. Biol Cell 2015; 107(11):389-418; PMID:26032945 [DOI] [PubMed] [Google Scholar]
- 32.Ward RJ, Dexter DT, Crichton RR. Neurodegenerative diseases and therapeutic strategies using iron chelators. J Trace Elem Med Biol 2015; 31:267-73; PMID:25716300; http://dx.doi.org/ 10.1016/j.jtemb.2014.12.012 [DOI] [PubMed] [Google Scholar]
- 33.Lei P, Ayton S, Appukuttan AT, Volitakis I, Adlard PA, Finkelstein DI, Bush AI. Clioquinol rescues Parkinsonism and dementia phenotypes of the tau knockout mouse. Neurobiol Dis 2015; 81:168-75. Available at http://dx.doi.org/ 10.1016/j.nbd.2015.03.015 [DOI] [PubMed] [Google Scholar]
- 34.Andersen HH, Johnsen KB, Moos T. Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell Mol Life Sci 2014; 71:1607-22; PMID:24218010; http://dx.doi.org/ 10.1007/s00018-013-1509-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salkovic-Petrisic M, Knezovic A, Osmanovic-Barilar J, Smailovic U, Trkulja V, Riederer P, Amit T, Mandel S, Youdim MB. Multi-target iron-chelators improve memory loss in a rat model of sporadic Alzheimer disease. Life sciences 2015; 136:108-19; PMID:26159898; http://dx.doi.org/ 10.1016/j.lfs.2015.06.026 [DOI] [PubMed] [Google Scholar]