

ABSTRACT
Biochemical similarities have been noted between the natively unstructured region of the cellular prion protein, PrPC, and a GPI-linked glycoprotein called Shadoo (Sho); these proteins are encoded by the Prnp and Sprn genes, respectively. Both proteins are expressed in the adult central nervous system and they share overlapping partners, including each other, in interactome studies. As prior studies have ascribed neuroprotective properties to the N-terminal region of PrPC, specifically the octarepeat region, we investigated Sho's neuroprotective properties. To this end we assessed Sho-null (Sprn0/0) and hemizygous (Sprn0/+) mice in the middle cerebral artery occlusion (MCAO) model versus wild type mice and also vs. transgene-rescued Sprn0/0-TgSprn mice. Sprn0/0 mice had a tendency to greater fragility in reaching endpoint and deficits in parameters including infarct volume and neurogenesis, with a reciprocal trend noted in transgene-rescued mice; however these effects did not reach significance. Loss of both PrPC and Sho immunostaining occurred in parallel to neuronal loss on the ipsilateral side of MCAO-lesioned animals; while focal elevations in immunostaining in the penumbra region were sometimes evident for PrPC, they were not noted for Sho. Our studies argue against discernible neuroprotective action of Sho in the genetic backgrounds used for this MCAO paradigm. Whether or not the positively charged N-terminal regions in Sho and PrPC fulfil different roles in vivo remains to be determined.
KEYWORDS: cerebral ischemia, genetic redundancy, neuroprotection, prion proteins, Sprn
Abbreviations
- BrdU
5-bromo-2′-deoxyuridine
- CNS
Central nervous system
- Dpl
Doppel
- ERKs
extracellular-regulated kinases
- GFAP
Glial fibrillary acidic protein
- Hpc
hippocampal formation
- MCAO
Middle cerebral artery occlusion
- N-ter
N-terminal
- NeuN
Neuron-specific nuclear protein
- NMDA
N-Methyl-D-aspartate
- PrPC
Cellular prion protein
- Sho
Shadoo
INTRODUCTION
The mammalian prion protein family is composed of 3 members: the cellular prion protein (PrPC), doppel (Dpl) and shadoo (Sho); these are all GPI-anchored glycoproteins and are encoded by the Prnp, the Prnd and the Sprn genes, respectively.1-3 In terms of domain organization, Sho resembles the natively unstructured N-terminal (N-ter) region of PrP, possessing a repeated sequence rich in arginine and glycine residues (RGG repeats) followed by a hydrophobic tract rich with 4-residue tandem alanine and valine repeats,4 versus histidine-containing octarepeats followed by a palindromic hydrophobic domain in the case of PrP.
In vitro it has been shown that neurons from Prnp0/0 mice are more sensitive to stress.5 The protective effect of PrPC is apparent under circumstances where cells are exposed to oxidative stress,6,7 elevated concentrations of copper ions,8 protein synthesis inhibitors,9,10 and excitotoxins.11,12 In some experiments it has been shown that the neuroprotective effect of PrP maps to the N-terminal part of the protein.13 In vivo PrPC confers neuroprotection after an ischemic insult.13-20 Prnp0/0 mice are more sensitive to stroke than wild type (wt) mice, having an increased infarct size and a modification of cell signaling cascades including extracellular-regulated kinases (ERKs), the stress-inducible kinase, Jun, and the signal transducer and activator of transcription, STAT.14-16,18 An overload of calcium-dependent signaling has also been proposed.20
The physiological function of Sho has been under study for a shorter period of time than PrPC and remains elusive. Since the neuroprotective action of PrP maps genetically to its N-terminal region, and since Sho N-ter bears a degree of resemblance to the PrP N-ter, a simple extrapolation is that Sho might also have a neuroprotective action. Like PrP, knockout mouse lines have been generated, and these animals remain healthy throughout their lifespan.21 It can be noted that in neuronal cell culture, Shadoo counteracts the neurotoxic effect of Doppel and internally deleted PrP alleles (“ΔPrP”) in a mechanism that may be similar to the one proposed for PrPC.4 Other authors have also shown that Sho protects against glutamate excitotoxicity.12
In the present study we evaluated the role of Sho in transient ischemia. First, we investigated the impact of a middle cerebral artery occlusion (MCAO) stroke model on mice, which are either homozygous (Sprn0/0) or heterozygous (Sprn0/+) for a fully penetrant null allele of the Sprn locus. In addition we have analyzed the early effect of this genetic deficiency by analyzing ischemia in homozygous null mice 24 hrs after the event and the effect of genetic complementation (i.e. restoration of Sho levels) using transgenic TgSprn mice overexpressing Sho on the same null background.
RESULTS
Ischemia with 14 Days Recovery
In a first set of experiments wt, Sprn0/+ and Sprn0/0 animals were subjected to a transient focal cerebral ischemia and were then allowed a total of 14 days of recovery; experimental groups and the results of surgical treatments are presented in Table 1 (Experiment I). Two trends were apparent in this dataset. First, while 20% (1/5) of wt animals did not survive 2 weeks after surgery, the percentage of death in heterozygous and homozygous null animals was increased, being 2/4 and 3/5 (i.e., 50 and 60%, respectively), suggesting a net increase in the fragility of these animals. Secondly, deaths in heterozygous and homozygous null animals occurred earlier than in wt mice (on day 1 and day 2, n = 2, n = 3 in heterozygous and homozygous null, respectively, vs. a single wt animal at day 7).
TABLE 1.
Survival after stroke measured at 24 hrs and 14 days
| (n) | ||||
|---|---|---|---|---|
| Genotype | Experiment I | Planned recovery period (days) | Premature deaths during day 1 | Survival to endpoint, n (%) |
| wt | 5 | 14 | 0 | 41 (80) |
| Sprn0/+ | 4 | 14 | 2 | 2 (50) |
| Sprn0/0 | 5 | 14 | 2 | 22 (40) |
| Experiment II | ||||
| Wt | 14 | 1 | 2 | 12 (86) |
| Sprn0/0 | 18 | 1 | 4 | 14 (78) |
| Sprn0/0-TgSprn+/- | 12 | 1 | 1 | 11 (92) |
1one death on day 7
2one death on day 2, post mortem reveals stroke occupying ~25% of brain area in mid-coronal section
At day 14 survivors were sacrificed and coronal sections of the brain cut and stained with cresyl violet to determine net neuronal loss; in these analyses the Nissl bodies of neurons appear deep blue and a weaker intensity or absence of staining denotes degeneration of neurons (Fig. 1A, D, G). Staining was detected uniformly in the contralateral region of the brain, whereas lighter staining was observed on the ipsilateral side in the region of the stroke appears, notably in the putamen. We observed neurodegeneration in each of the 3 genotypes (i.e. wt (A), Sprn0/+ (D) and Sprn0/0 (G). Adjacent sections of this group of experiment were used for immunohistochemistry with anti-glial fibrillary acidic protein (GFAP) (B, E, H) and anti-neuronal nuclei (NeuN) (C, F, I). The expression of GFAP, a glial marker, increases in the penumbra of the stroke, whereas the level of NeuN, a neuronal marker is decreased in the corresponding area. No change in expression was observed on the contralateral side for either protein.
FIGURE 1.
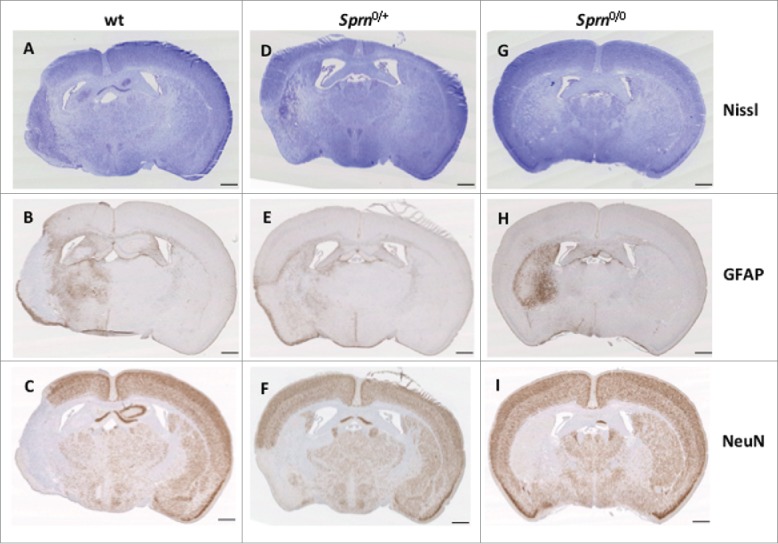
Immunohistochemical staining of 3 mouse genotypes at 14 days post treatment. Representative coronal sections of brains are presented from wt (A, B, C), Sprn0/+ (D, E, F) and Sprn0/0 mice (G, H, I) stained with Nissl (A, D, G), or processed by immunochemistry for GFAP (B, E, H) and NeuN (C, F, I). The ipsilateral side is on the left and contralateral side is on the right. The scale bar represents 1 mm.
Subsequent to surgery, the animal cohorts in Experiment I received injections of BrdU for 5 consecutive days, in order to detect the presence of newborn cells in the penumbra region. At the experimental endpoint (i.e., animals living to day 14) a difference was observed in the area corresponding to the infarct compared to the contralateral side of the brain (where only sparse proliferative cells were present; not shown). Comparing sections from the 3 different genotypes in the ipsilateral side of the brain, we observed a trend for fewer cells in knockout animals versus wt and heterozygotes (Fig. 2A). Quantifying BrdU cells, we found that the highest numbers of proliferating cells and densities for proliferating cells were found in the wt and heterozygous animals cells (Fig. 2B-C) with lower numbers in knockout animals. These labeled cells are mainly neuronal as the BrdU labeling co-localized with NeuN (Fig. 2D), Figures 4-6 - while very little overlap with GFAP expression was noted (Fig. 2D), Figures 1-3. These data suggest that there is a higher rate of neuronal proliferation in the region adjacent to the ischemic core vs. more distant regions, and the proliferation is mainly in neurons 2 weeks after the ischemia event.22
FIGURE 2.
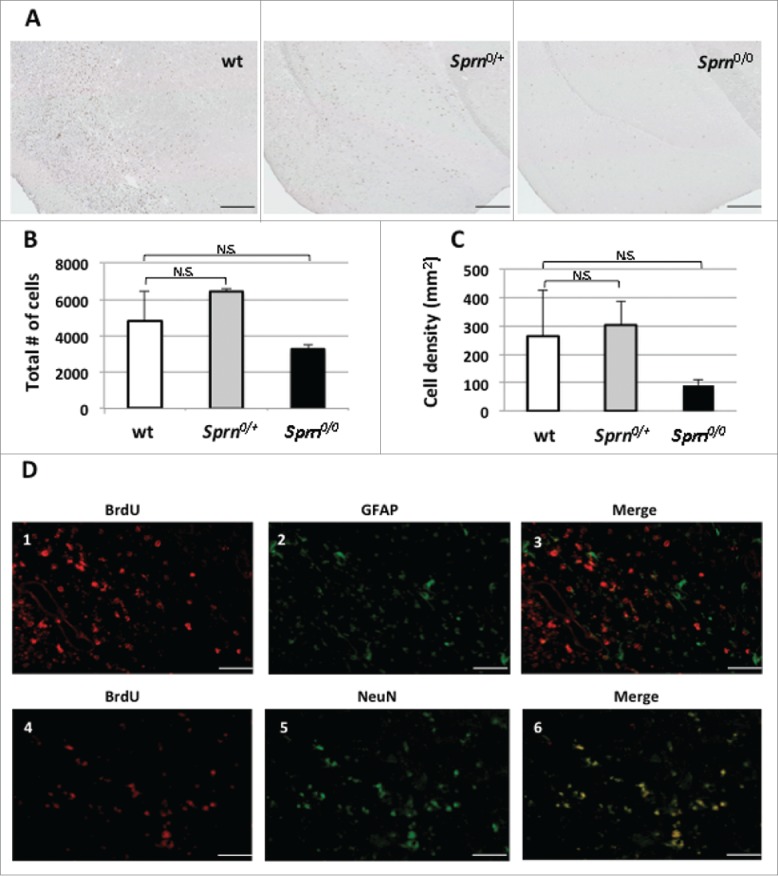
BrdU staining in 3 mouse genotypes 14 days post treatment. Immunohistochemistry of BrdU staining is presented in representative coronal sections of (A) wt, Sprn0/+ and Sprn0/0 mice. Panel B represents a quantification of total number of cells in the ipsilateral hemisphere labeled for BrdU. The genotypes are not significantly different (n.s.), p = 0.39. Panel C represents density of cells per mm2 in penumbra regions adjacent to infarcts. The genotypes are not significantly different (n.s.), p = 0.43. Double immunolabeling for BrdU (D1, D4) and GFAP (D2) and NeuN (D5) are also represented. Merged images are represented in panels D3 and D6. The scale bars represent 0.5mm and 50 μm, respectively. Variation among the 3 genotypes does not reach significance.
FIGURE 4.
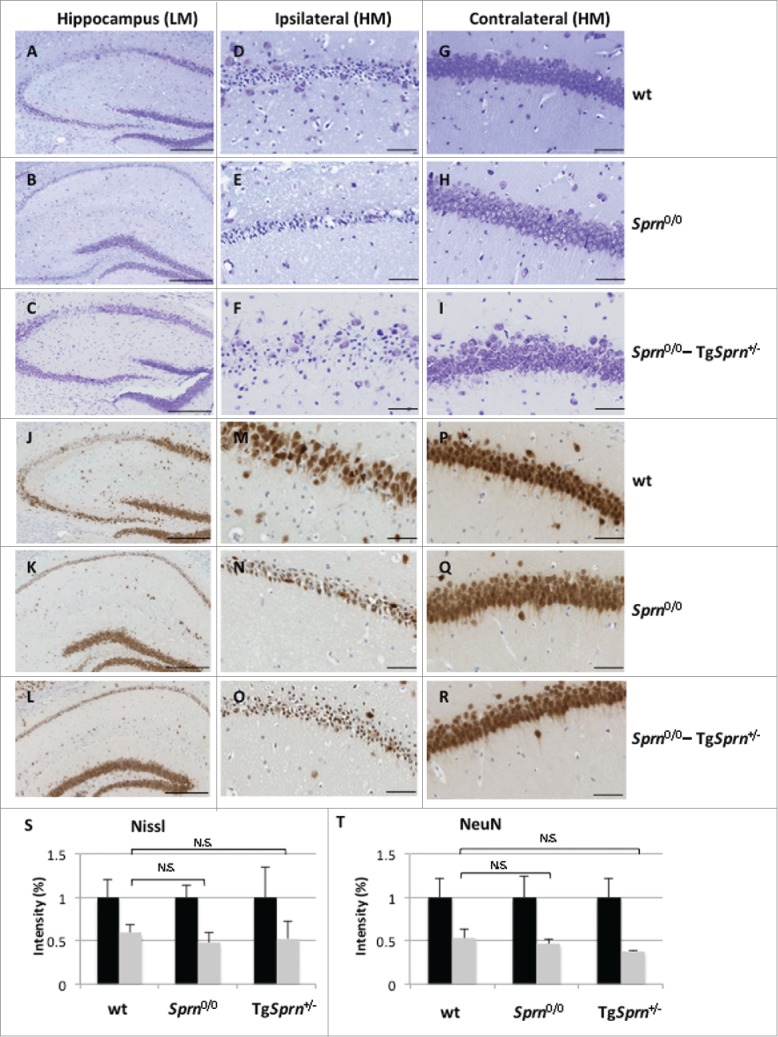
(see next page). Sprn genotypes and damage to the hippocampus. Nissl staining (A-I) and immunohistochemistry for NeuN (J-R) are represented. Low power fields of the hippocampal formation on the ipsilateral side of wt (A, J), Sprn0/0 (B, K) and Sprn0/0-TgSprn+/− mice (C, L) are shown in the left-hand column (scale bar represents 250 μm). The middle column represents a higher magnification of the CA2 region in the ipsilateral side of the infarct, whereas the right-hand column shows the contralateral side of the same region (scale bar represents 50 μm). Panels S and T represent staining intensity for Nissl and NeuN stain, respectively with the contralateral side shown as black bars (normalized to a value of 1.0, 100%) while gray bars represent the ipsilateral side. Sample sizes corresponded to n = 4, 3, 2 for Nissl staining and n = 3, 2, 3 for NeuN staining for wt, Sprn0/0 and Sprn0/0-TgSprn+/− genotypes, respectively. For Panels S and T the ipsilateral values for the 3 genotypes were not significantly different, p = 0.80 and p = 0.43. LM: low magnification, HM: high magnification.
FIGURE 5.
Induction of autophagy. High magnification views of the CA1 region of the hippocampus for the ipsilateral side of the infarct (left panel) and the contralateral side (right panel) are presented, respectively. Immunohistochemical detection of LC3 and GFAP expressions are presented in A-F and G-L, respectively. Region of the hippocampus is represented for wt (A, B, G, H), Sprn0/0 (C, D, I, J) and Sprn0/0-TgSprn+/− (E, F, K, L) (scale bar represents 50 μm).
FIGURE 6.
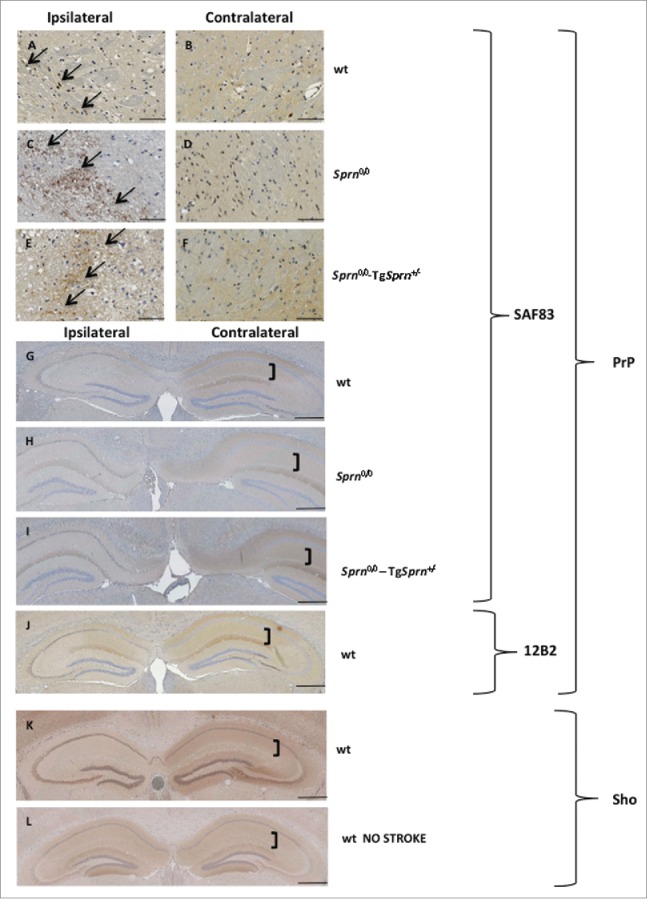
Local patterns of expression of PrP and Sho one day after injury. Patches of cells with PrP immunostaining above background (black arrows) are shown in the penumbra region of wt (A), Sprn0/0 (C) and Sprn0/0-TgSprn+/− (E) mice, as compared to immunostaining in the corresponding contralateral sides (B, D, and F). PrP immunostaining in a low power, bilateral view of the hippocampus is represented for wt (G), Sprn0/0 (H) and Sprn0/0-TgSprn+/− (I) mice. J shows a wt animal with a stroke showing reduced PrP staining on the ipsilateral side with 12B2 antibody. Brackets show the molecular layer. Note reduced PrP staining on the ipsilateral side. Expression of Sho in wt animal exposed to the MCAO procedure (K) is compared to expression in a wt naive animal (L); the effect resembles that seen for PrP. Scale bars represents 1 mm.
FIGURE 3.
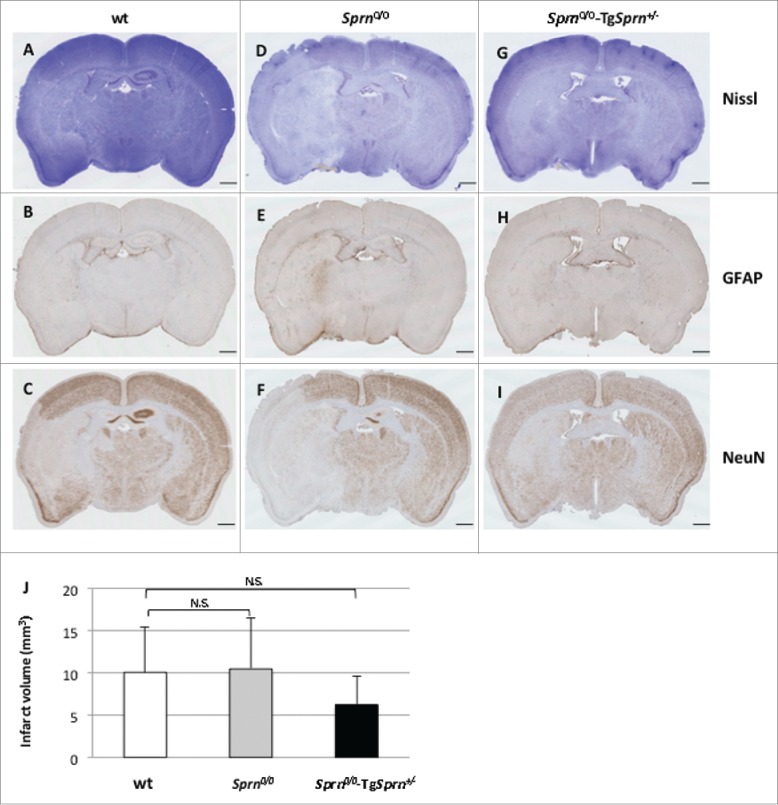
Immunohistochemical staining of 3 mouse genotypes 24hrs post treatment. Nissl staining of cerebral infarction. Representative sections of wt (A, B, C), Sprn0/0 (D, E, F), and Sprn0/0-TgSprn+/− (G, H, I) mice are shown. Normal tissue is stained deep blue while infarcted tissue with loss of neurons appears clearer (A, D, G). Immunohistochemistry is shown representing the effect of brain ischemia on the expression of protein GFAP (B, E, H) and NeuN (C, F, I). J represents a quantification of the size of the infarct volume in the 3 different genotypes (n = 8, 7, 8, respectively). Values between the 3 genotypes were not significantly different (P = 0.57). The scale bar represents 1mm.
Ischemia with 24 Hrs Recovery
Beyond our first study with a 14-day observation period, we embarked on a further set of experiments with an end-point moved forward to 24 hrs post-surgery. Also, in this study we used “transgene rescued” (genetically reconstituted) Sprn0/0-TgSprn+/− mice as well as wt and Sprn0/0 mice. Overexpression of the TgSprn transgene has been previously estimated at 2.5x endogenous in a wt genetic background.23 As expected for a shorter observation period, a larger percentage of animals survived to endpoint. As per experiment I, losses by endpoint were highest in the Sprn0/0 genotype but this figure did not reach significance versus wt or Sprn0/0-TgSprn+/− controls (Table 1: Experiment II). While losses during surgery were 25% for null animals (6/24) vs. 0% for transgene rescued mice, these data did not unequivocally support an inference of increased fragility for null animals as 33% loss was seen for wt animals in the same experiment.
Nissl staining of coronal sections of ischemic brains from experiment II are shown in Fig. 3 wt (A) is compared to Sprn0/0 (D) and to Sprn0/0-TgSprn+/− (G). Infarcted areas, already discernible at 24hrs, were identified by the pallor of the damaged tissue relative to the surrounding healthy tissue. The area of pallid tissue in serial sections was used to calculate infarct volumes, which were lowest in the transgene-rescued Sprn0/0 mice (Fig. 3J). Loss of Nissl bodies is also associated with the increase of expression of GFAP (Fig. 3B, E, H) and decreased expression of NeuN (C, F, I) in the corresponding areas.
Beside the direct infarct location, the hippocampal formation (Hpc) is frequently compromised, with elevated cell death in pyramidal neurons. This is illustrated for 3 genotypes in Figure 4 for the subset of animals with Hpc damage (low power view, A, B, C). On the contralateral side to the lesion, normal neurons are ordered and packed tightly (Fig. 4G-I), and the Nissl substance is uniform in the cytoplasm. On the ipsilateral side neurons are injured, showing shrunken cell bodies and pyknotic nuclei (Fig. 4D-F); they are the witnesses to the damage caused by the MCAO injury. Overt neuronal loss, also evidenced by NeuN staining (Fig. 4J-K, and at higher power M, N versus P, Q), was apparent in wt and Sprn0/0 mice. We quantitated the intensity of staining for Nissl and NeuN staining for ipsilateral sides of the hippocampal formation and expressed this vs. contralateral (100%). A trend for the Sprn0/0 genotype to exhibit a greater % change ipsilateral: contralateral was apparent for both stains (11.3 and 13.5% greater loss of staining, respectively) but did not reach significance (Fig. 4S-T). This effect was attenuated in some, but not all transgene-rescued mice.
Among pathways for degradation of GPI-linked proteins, intersections between the endosomal/lysosomal pathway and especially autophagy have been of interest. To begin to assess the status of autophagic pathways in our MCAO paradigm we examined the LC3 protein involved in autophagy (Fig. 5); these studies used an antibody that recognizes both LC3-I and LC3-II forms of this protein. Interrogation of the Hpc formation in lesioned animals revealed clustered cellular staining punctate labeling corresponding to the increase of LC3-II representing the activation of autophagy, with Figure 5A, C, E representing the ipsilateral side and Figure 5B, D, F representing the contralateral side of the CA1 region of the Hpc. In these ipsilateral/contralateral comparisons we did not notice striking differences in outcome between wt, Sprn0/0 and Sprn0/0-TgSprn+/− genotypes (Fig. 5A, C, E). We also assessed astrocytic reactivity in adjacent sections (Fig. 5G-L) but at these time points we did not notice differences between ipsilateral and contralateral sides to correlate with the induction of autophagy.
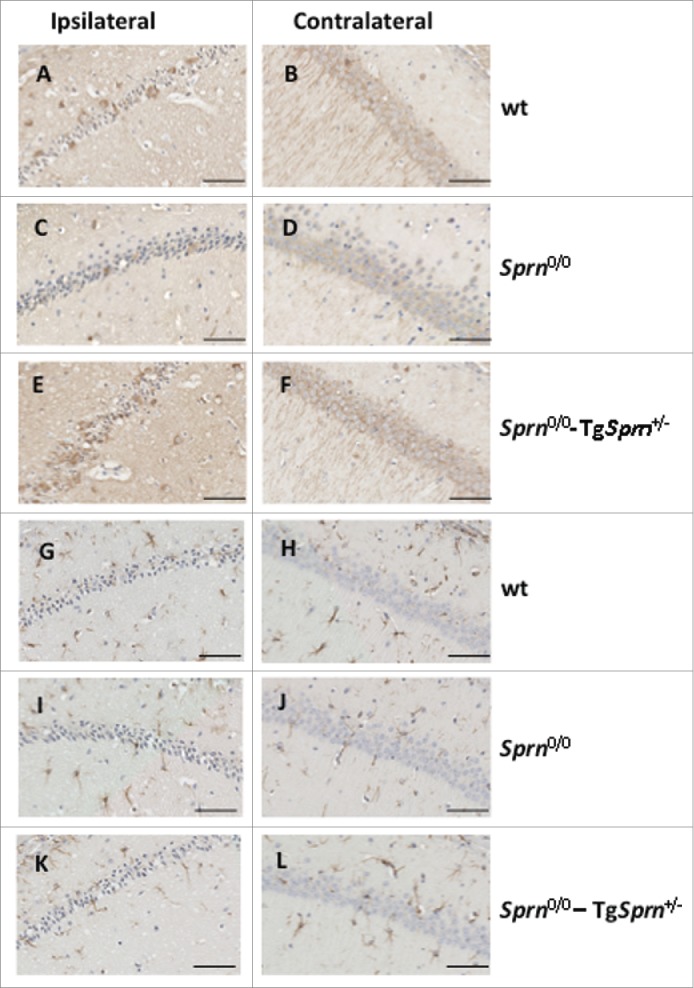
As i) PrP mRNA and PrPC protein expression is described to increase during ischemia,14,15 ii) fields of PrPC expression are well mapped within the orderly architecture of the Hpc, and iii) induction of PrPC expression might conceivably be altered by Sprn genotype, we decided to examine PrPC expression in the Hpc and in the penumbra region adjacent to infarcts using SAF83 (C-terminal) and 12B2 (central region) PrP antibodies. We also examined Sho expression using previously described methods and a 06Sh1 polyclonal antiserum against an N-terminal epitope (Fig. 6).4,21 Patchy staining of PrP in a granular pattern was occasionally evident in the penumbra region on the ipsilateral side of treated animals of all 3 genotypes (Fig. 6A, C, E vs. comparable contralateral fields in B, D, F). A consistent findings was decreased PrPC in the Hpc on the ipsilateral side of the brain (compare Fig. 6G, H, I ipsilateral side vs. contralateral side). This was apparent as a loss of diffuse staining in the stratum lacomusum moleculare of the Hpc versus typical expression maintained on the contralateral side (bracket in figure). This type of loss of PrPC immunostaining was seen in animals irrespective of their Sprn allelic status. While immunostaining for Sho in Sprn0/0 mice was informative in defining the residual noise in our histological procedures (corresponding to some background staining in nuclei), immunostaining for Sho in wt mice subjected to MCAO revealed parallels to findings for PrPC, namely a loss of staining in the ipsilateral Hpc formation (Fig. 6J). Loss of Sho expression was apparent in the stratum radiatum and stratum oriens in the CA1 and CA2 regions, and the molecular layer of the dentate gyrus when compared to a naive wt animal (Fig. 6K). Moreover, these findings correlating loss of PrPC and Sho expression with loss of hippocampal neurons could be extended to the case of animals with the largest lesions, where damage extended to the cortex. In these instances loss of PrPC and Sho immunostaining paralleled the pattern for net neuronal content produced with NeuN staining and Nissl staining (Fig. 7, boundary between healthy and pallid regions denoted by a dashed line). Overall, data in Figures 6 and 7 defining loss of immunostaining offer a parallel to observations of Mittereger et al.13 Lastly, we failed to find focal areas of Sho staining, perhaps indicative of a neuroprotective response, in penumbra regions (not shown).
FIGURE 7.
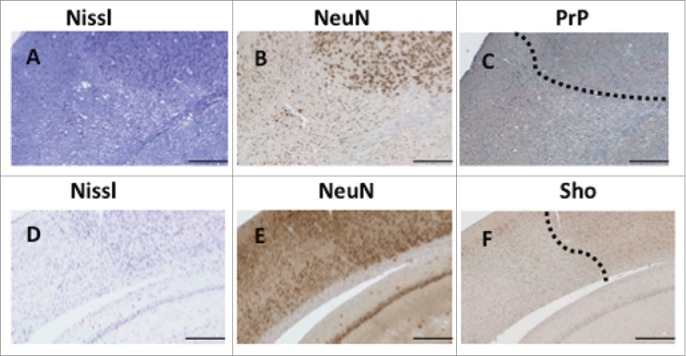
Variation of expression of PrP and Sho. Immunostaining for PrP (C) and Sho (F) in the cortex of lesioned animals is compared to staining of the same region with Nissl dye (A, D) or NeuN antibody (B, E). A dotted line indicates the boundary between normal cell dense regions and pallid (necrotic) regions. Scale bar represents 250 μm.
DISCUSSION
Stroke Models, PrPC and Sho
Around 17 million people die of stroke every year, thus comprising the third leading cause of death in the US.24 Importantly, 76% of people survive their stroke, leading to a major subsequent cost in health care.25,26 To date, there is no neuroprotective agent effective clinically. Thus, it is of major importance to find new approaches to prevent strokes or ameliorate their impact and finding new target genes might be very helpful in this context. With respect to the superfamily of mammalian prion proteins, beyond the neuroprotective properties ascribed to PrPC and its proteolytic fragments in tissue culture paradigms,27,28 there is a prior literature in the context of hypoxic and ischemic injuries. These findings can be broken down in turn, occurring in the context of heightened susceptibilities of Prnp0/0 rodents and describing a potentially compensatory effect of PrPC overexpression.13,16,18-20,29 and/or the ability of hypoxia to induce PrPC expression in the vicinity of an injury or hypoxic cells.14,15,29 Here we have sought to understand the potential role of Sho, as it has both structural similarities with the unstructured domains of PrPC and has also been attributed neuroprotective action.12,30-32 We chose to pursue a MCAO technique as this provides a degree of continuity with earlier studies concerning PrPC.13,16-19 We used the contralateral, uninjured hemisphere of the brain from anaesthetized animals as a negative control. Typically, in cases of severe stroke, the neurons are the primary targets and are selectively vulnerable.33 Our observations here with NeuN staining in the MCAO paradigm are in agreement with this notion and moreover we have shown that the cells regenerating after the ischemia are neurons, as evidenced by the co-localization of BrdU with NeuN.34
Notably, while MCAO is routinely used to induce focal ischemic stroke in mice, the period of occlusion is associated with drastic differences in ischemic cell death in the mouse model. A fivefold increase in infarct volume in C57BL/6 mice has been reported when increasing occlusion duration from 15 to 30 minutes.35 After observing considerable mortality in our 14 d recovery animals (30 minute occlusion, FVB/N inbred strain), 24 hrs recovery and a slightly shorter occlusion period (20 min) was used to reduce ischemia-induced mortality in Experiment 2 using the same inbred strain genetic background. However, this shorter duration of occlusion was associated with an increased variability in infarct size.
Cellular Mechanisms and Allelic Effects in the MCAO Model
During ischemic episodes, N-Methyl-D-aspartate (NMDA) receptors can exert an effect in mediating glutamate excitotoxicity.36 PrPC has been reported to interact physically with the NR2D subunits of NMDA receptors and the Prnp locus to correspondingly modulate excitotoxic effects.37 It is not known if Sho interacts with NMDA receptor subunits but PrPC and Sho have overlapping interactomes in N2a neuroblastoma cells and interact with each other as deduced from formaldehyde cross-linking of living cells32 and in vitro studies,38 so formation of heterodimeric PrP-Sho species which would prevent NMDA receptor hyperactivity is a possibility for mooted protective effects. Genetic studies of PrPC function and turnover can be confounded by use of viral promoters.39 or splice site deletions that affect neighboring loci.2 In the studies here we compared the endogenous wt mouse Sprn allele, a fully penetrant null allele deriving from 129 ES cells with a single-copy of the targeting vector21 and backcrossed to make an FVB/N congenic line and, thirdly, a Sho transgenic line on the same FVB/N strain background driven by the Prnp promoter and with a mild level of overexpression. (2.5x endogenous levels in an Sprn wt background and 1.5x in an Sprn0/0 background.40) In general we observed trends whereby the homozygous null animals were more fragile and compromised in endpoint measures such as infarct volume when compared with wt controls and, reciprocally, genetically reconstituted Sprn0/0-TgSprn+/− mice were more hardy (Table 1). However these effects did not reach significance and the Sprn0/+ heterozygotes included in our first experiment resembled either wt animals or Sprn0/0 animals, depending on the endpoint measure. We also noted, as per prior studies, that Sprn genotype had no discernible effect upon PrPC levels.4,21 The lack of significant effects of Sprn allelic composition on endpoint measures can be reconciled with 2 possibilities that are not mutually exclusive: first, that any effects of Sprn alleles are overshadowed by the activity of PrPC (which is encoded by an undisrupted wt Prnpa locus in these experiments), and secondly that potential effects of Sprn alleles are small and overshadowed by experimental noise (e.g. animal-to-animal variation) encountered in the surgical procedure to occlude arterial blood-flow for a period of 20 minutes. To clarify the first possibility, i.e. that endogenous PrPC obscures the effect of Sho, further studies will need to include the performance of Prnp0/0 and Prnp0/0 + Sprn0/0 mice as important points of reference; indeed it is possible that a tendency for a protective effect of Sho might have reached significance in comparisons Prnp0/0 vs. Prnp0/0 + Sprn0/0 mice (as adult Prnp0/0 + Sprn0/0 mice are known to be viable).21
Neuronal Death, Expression and Localization of Sho and PrPC
The MCAO paradigm used here is capable of producing lesions that can occupy up to 25% of the cortical area in coronal sections, as well as affecting the Hpc (Figs. 1, 3, 4 and S1). These lesions are readily visualized by loss of Nissl staining and by loss of NeuN immunoreactive cell bodies and they are accompanied by an increased LC3-reactive structures indicative of an autophagic response to cellular damage. As stated above, significant genotypic effects upon these damage measures were not attained, but since expression of PrPC in MCAO-treated mice has been described as being elevated within 24 hrs of the procedure,13,19 we explored lesioned animals for this effect and for possibly related effects concerning Sho. We sought focal patches of Sho staining in the penumbra region (as per PrP) but failed to obtain results supportive of this effect. On the other hand, a dramatic loss of net PrPC- and Sho-immunoreactivity was clearly apparent in lesioned hemispheres. We note that re-localization effects have been described under certain conditions in tissue culture - for example, under conditions in which ER import is attenuated, Sho relocates from the cell surface to the mitochondria.12 - raising the question of whether perceived decreases in focal immunostaining in these regions might be balanced by increases in immunostaining in other structures, but within the parameters of the applied MCAO paradigm no such redistribution was apparent, just a net loss of staining.
As PrPC is a neuronal protein expressed on the cell surface and subject to axonal transport, its disappearance from neuronal processes in dying neurons seems plausible and reiterates results obtained by Mitteregger et al.13 for the necrotic zone of lesions, assessed both by immunochemistry and by western blot analyses. In our studies 2 anti-PrP antibodies yielded the same effect and this response was closely paralleled by weakened Sho immunostaining, correlating with pallid staining with Cresyl Violet and weak NeuN immunostaining. From the use of doxycycline-regulated transgenes we know that the half-life of mouse PrPC in the healthy brain is about 24 hrs.41 For the immunostaining experiments presented here (Fig. 6) the experimental endpoint was also 24 hrs, so the dramatic reduction in immunostaining may represent something more than a simple failure by affected neurons to continue ongoing protein synthesis. Selective reduction of PrPC and Sho only the ipsilateral side of affected brains excludes autolysis during tissue preparation as a relevant variable here and suggests other types of protein breakdown (perhaps related to down-regulation effects seen for PrPC and Sho in prion infections4,40,42,43 or a simple consequence of release of proteases during necrosis), while yet different effects involving upregulation may operate in the penumbra zone. This perspective avoids 2 potential contradictions, namely (i) how can a molecule like PrPC with locally falling levels enact the protective properties that have been ascribed to it and (ii) be reported to be up-regulated in tissue homogenates from infarcted and peri-infarcted areas in the same 24 hrs time-frame as used here?29 However, focal accumulations of PrPC inside necrotic areas, as reported by others14 were not a feature of our analyses, a discrepancy perhaps attributable to differing genetic backgrounds (129/Ola versus FVB/NJ).
While there is a caveat that we did not reconfirm protective activities of PrPC described by others, in the MCAO paradigm the endogenous Sprn locus or and Sprn transgene array had limited abilities to modulate endpoints to the point of significance. Also, focal upregulation effects of Sho in penumbra regions were not detected by immunostaining. Collectively these findings suggest that neuroprotective effects of Sho might prove to be different from those of PrPC. In the case of PrPC genetic mapping has attributed different biological functions to different domains and has pointed to a neuroprotective role of the metal-binding octarepeats.13 Future studies to exchange PrPC's positively charged octarepeats with Sho's N-terminal, nucleic acid binding Arg-Gly-Gly repeats44 and modulating the way in which these N-terminal regions are processed to metabolically stable fragments45-47 may enlighten our understanding of neuroprotective potential and associated mechanisms.
MATERIALS AND METHODS
Animals
Animals were housed in groups up to 5 under a 12-h light/dark cycle with food and water ad libitum before and after surgery. All animal protocols were in accordance with the Canadian Council on Animal Care and were approved by the Institutional Animal Care and Use Committees at the University of Alberta. Animal used in this study were from the FVB/NJ background, wt, Sprn0/+, Sprn0/0 and rescued knockout (Sprn0/0-TgSprn+/−). The generation of knockout and transgenic mice have been described elsewhere.21,40 Mice were aged 161-184 days for the experiment with 14 days recovery and 230-415 days for the experiment with one day of recovery.
Stroke Procedure
Two separate experiments were performed. Focal ischemia was induced using a transient middle cerebral artery occlusion (MCAO) technique. Briefly, animals were anaesthetized with 2% isoflurane (70:30, 02:N20) and put into supine position. Body temperature was maintained at 36–37 °C with an electric heating pad with feedback from a rectal probe coupled to a temperature regulator. Aseptic procedures were used for all surgeries. First, the common carotid artery, external carotid artery and internal carotid artery were dissected and exposed. A monofilament (7–0 suture with 5–6 mm long coating tip, diameter 0.15–0.25 mm, Doccol Corporation, USA) was inserted into external carotid artery and advanced to the internal carotid artery, blocking the origin of middle cerebral artery. In the first experiment, the filament was held in place for 30 min before removal and animals were allowed 2 weeks recovery. In the second experiment the filament was removed after 20 min of occlusion and mice were euthanized after 24 hrs recovery.
BrdU Treatment
Animals were injected intraperitoneally with 5-bromo-2′-deoxyuridine (BrdU, Sigma) once a day for 5 days (100 mg/kg) starting one day after ischemic onset. BrdU labeled cells were visualized as described below in the Immunohistochemistry section. After immunohistochemistry, tissue sections were scanned using digital slide scanner Nanozoomer XR (Hamamatsu, SZK, Japan) using identical acquisition parameters. Cell counting was performed using ImageJ software. Threshold adjustments were set in order to count only immunopositive nuclei. BrdU labeled cells were counted in the complete ipsilateral hemisphere (3 sections per animal) and in a penumbra region adjacent to an infarct and in the latter case normalized by area in the sampled region and surface corresponding to the infarct area.
Immunohistochemistry
Animals were euthanized 24 hrs or 14 days after surgery. Mice received an overdose of sodium pentobarbital (120 mg/kg) and were then transcardially perfused with 0.9% saline solution followed by 4% paraformaldehyde. The brains were extracted then postfixed with formalin or methacarn. Paraffin embedded tissue samples were processed and serial 6 μm sections were cut. Immunohistochemistry was performed after deparaffinization and hydration processes. Slides were stained with Nissl, using standard protocol to delineate the infarct. For immunostaining, antigen retrieval in 10 mM citrate buffer pH 6 was performed in a pressure cooker. Endogenous peroxidase activity was blocked by treatment with 3% hydrogen peroxide for 6 min. After washes, sections were incubated overnight with primary antibodies: either neuron-specific nuclear protein antibody (NeuN, Millipore, 1:1000) to label neurons; glial fibrillary acidic protein (GFAP, Becton Dickinson, 1:1000) to label activated astrocytes and visualized with horseradish peroxidase using the DAKO ARK™ kit following the manufacturer's instructions. Sections were counterstained using Mayer haematoxylin, dehydrated and cover-slipped with permanent mounting medium. Prion protein expression was visualized using SAF83 antibody (Cayman, 1:500; the epitope corresponds to β strand 1- α helix 1 - β strand 2 region in the globular domain of PrP) and 12B2 (epitope corresponds to residues 88-92 of mouse PrP, a generous gift from J. Langeveld), while Sho was detected with the N-terminal antibody 06Sh1.4 Autophagy was detected using LC3 (MBL, 1:4000). Newborn cells were detected using an antibody against BrdU (Thermofisher, 1:200). Pretreatment for BrdU was as follows: incubation in 4N HCl for 30 min at room temperature followed by 10 min incubation in 0.125% trypsin. Visualization was performed using DAKO ARK™ Kit. For immunofluorescence, the same procedure was applied using BrdU and GFAP or BrdU and NeuN as primary antibodies. The secondary antibodies used were streptavidin Alexa Fluor® 568 and Alexa Fluor® 594 Goat Anti-Mouse (Invitrogen). All histological and immunohistochemical analyses were determined in a blinded way. The slides were analyzed using digital slide scanner Nanozoomer XR (Hamamatsu, SZK, Japan).
Analysis of Lesion Volumes
Brain sections from 10 equidistant rostrocaudal brain levels, 1 mm apart, were used (examples are presented in Fig. S1). After staining with Nissl, the sections were digitized using a Nanozoomer (Hamamatsu, SZK, Japan). Infarct volume was quantified using image analysis software (ImageJ, NIH, Bethesda, MD),48 using the following formula: CIV = [LHA-(RHA-RIA)]-(thickness of section + distance between sections); in which CIV is corrected infarct volume, LHA is left hemisphere area, RHA is right hemisphere area, and RIA is right hemisphere infarct area.49 This method helps to correct for edema in the ipsilateral hemisphere in order to achieve a more accurate assessment of infarct volume.
Statistical Analysis
All data are presented as mean ± SEM. Statistical analyses were performed using GraphPad Instat version 3.0 (GraphPad Software, San Diego, CA, USA). Statistical significance was set at an α value of P < 0.05. Between-group comparisons were compared using one-way analysis of variance (ANOVA) with assumption of Gaussian distributions.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
ACKNOWLEDGMENTS
We thank Jing Yang, Jennifer Grams, Hyena Jeon and Karly Bergen for animal care and management.
SUPPLEMENTAL MATERIAL
Supplemental Material may be downloaded here: publisher's website
Funding
This work was supported by the Canadian Institutes of Health Research (MOP123525).
REFERENCES
- 1.Carlson GA, Kingsbury DT, Goodman PA, Coleman S, Marshall ST, DeArmond S, Westaway D, Prusiner SB. Linkage of prion protein and scrapie incubation time genes. Cell 1986; 46:503-11; PMID:3015416; http://dx.doi.org/ 10.1016/0092-8674(86)90875-5 [DOI] [PubMed] [Google Scholar]
- 2.Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, et al.. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 1999; 292:797-817; PMID:10525406; http://dx.doi.org/ 10.1006/jmbi.1999.3108 [DOI] [PubMed] [Google Scholar]
- 3.Premzl M, Gamulin V. Comparative genomic analysis of prion genes. BMC Genomics 2007; 8:1; PMID:17199895; http://dx.doi.org/ 10.1186/1471-2164-8-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watts JC, Drisaldi B, Ng V, Yang J, Strome B, Horne P, Sy MS, Yoong L, Young R, Mastrangelo P, et al.. The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. Embo J 2007; 26:4038-50; PMID:17703189; http://dx.doi.org/ 10.1038/sj.emboj.7601830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Matsumoto Y, Yokoyama T, Itohara S, et al.. Prions prevent neuronal cell-line death. Nature 1999; 400:225-6; PMID:10421360; http://dx.doi.org/ 10.1038/22241 [DOI] [PubMed] [Google Scholar]
- 6.Watt NT, Taylor DR, Gillott A, Thomas DA, Perera WS, Hooper NM. Reactive oxygen species-mediated β-cleavage of the prion protein in the cellular response to oxidative stress. J Biol Chem 2005; 280:35914-21; PMID:16120605; http://dx.doi.org/ 10.1074/jbc.M507327200 [DOI] [PubMed] [Google Scholar]
- 7.Dupiereux I, Falisse-Poirrier N, Zorzi W, Watt NT, Thellin O, Zorzi D, Pierard O, Hooper NM, Heinen E, Elmoualij B. Protective effect of prion protein via the N-terminal region in mediating a protective effect on paraquat-induced oxidative injury in neuronal cells. J Neurosci Res 2008; 86:653-9; PMID:17896796; http://dx.doi.org/ 10.1002/jnr.21506 [DOI] [PubMed] [Google Scholar]
- 8.Haigh CL, Brown DR. Prion protein reduces both oxidative and non-oxidative copper toxicity. J Neurochem 2006; 98:677-89; PMID:16787422; http://dx.doi.org/ 10.1111/j.1471-4159.2006.03906.x [DOI] [PubMed] [Google Scholar]
- 9.Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. Embo J 2002; 21:3317-26; PMID:12093733; http://dx.doi.org/ 10.1093/emboj/cdf324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, et al.. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. Embo J 2002; 21:3307-16; PMID:12093732; http://dx.doi.org/ 10.1093/emboj/cdf325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rambold AS, Miesbauer M, Olschewski D, Seidel R, Riemer C, Smale L, Brumm L, Levy M, Gazit E, Oesterhelt D, et al.. Green tea extracts interfere with the stress-protective activity of PrP(C) and the formation of PrP(Sc). J Neurochem 2008; 107:218-29; PMID:18691383; http://dx.doi.org/ 10.1111/j.1471-4159.2008.05611.x [DOI] [PubMed] [Google Scholar]
- 12.Sakthivelu V, Seidel RP, Winklhofer KF, Tatzelt J. Conserved stress-protective activity between prion protein and shadoo. J Biol Chem 2011; 286:8901-8; PMID:21257747; http://dx.doi.org/ 10.1074/jbc.M110.185470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitteregger G, Vosko M, Krebs B, Xiang W, Kohlmannsperger V, Nolting S, Hamann GF, Kretzschmar HA. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol 2007; 17:174-83; PMID:17388948; http://dx.doi.org/ 10.1111/j.1750-3639.2007.00061.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, et al.. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol 2004; 165:227-35; PMID:15215178; http://dx.doi.org/ 10.1016/S0002-9440(10)63291-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shyu WC, Lin SZ, Chiang MF, Ding DC, Li KW, Chen SF, Yang HI, Li H. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J Neurosci 2005; 25:8967-77; PMID:16192387; http://dx.doi.org/ 10.1523/JNEUROSCI.1115-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, Bassetti CL, Hermann DM. Aggravation of ischemic brain injury by prion protein deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol Dis 2005; 20:442-9; PMID:15893468; http://dx.doi.org/ 10.1016/j.nbd.2005.04.002 [DOI] [PubMed] [Google Scholar]
- 17.Weise J, Crome O, Sandau R, Schulz-Schaeffer W, Bahr M, Zerr I. Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci Lett 2004; 372:146-50; PMID:15531106; http://dx.doi.org/ 10.1016/j.neulet.2004.09.030 [DOI] [PubMed] [Google Scholar]
- 18.Weise J, Sandau R, Schwarting S, Crome O, Wrede A, Schulz-Schaeffer W, Zerr I, Bahr M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 2006; 37:1296-300; PMID:16574930; http://dx.doi.org/ 10.1161/01.STR.0000217262.03192.d4 [DOI] [PubMed] [Google Scholar]
- 19.Weise J, Doeppner TR, Muller T, Wrede A, Schulz-Schaeffer W, Zerr I, Witte OW, Bahr M. Overexpression of cellular prion protein alters postischemic Erk1/2 phosphorylation but not Akt phosphorylation and protects against focal cerebral ischemia. Restor Neurol Neurosci 2008; 26:57-64; PMID:18431006 [PubMed] [Google Scholar]
- 20.Sakurai-Yamashita Y, Sakaguchi S, Yoshikawa D, Okimura N, Masuda Y, Katamine S, Niwa M. Female-specific neuroprotection against transient brain ischemia observed in mice devoid of prion protein is abolished by ectopic expression of prion protein-like protein. Neuroscience 2005; 136:281-7; PMID:16198494; http://dx.doi.org/ 10.1016/j.neuroscience.2005.06.095 [DOI] [PubMed] [Google Scholar]
- 21.Daude N, Wohlgemuth S, Brown R, Pitstick R, Gapeshina H, Yang J, Carlson GA, Westaway D. Knockout of the prion protein (PrP)-like Sprn gene does not produce embryonic lethality in combination with PrPC-deficiency. Proc Natl Acad Sci U S A 2012; 109:9035-40; PMID:22619325; http://dx.doi.org/ 10.1073/pnas.1202130109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kokaia Z, Lindvall O. Neurogenesis after ischaemic brain insults. Curr Opin Neurobiol 2003; 13:127-32; PMID:12593991; http://dx.doi.org/ 10.1016/S0959-4388(03)00017-5 [DOI] [PubMed] [Google Scholar]
- 23.Westaway D, Daude N, Wohlgemuth S, Harrison P. The PrP-Like Proteins Shadoo and Doppel. Top Curr Chem 2011; 305:225-56; PMID:21728138; http://dx.doi.org/ 10.1007/128_2011_190 [DOI] [PubMed] [Google Scholar]
- 24.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, et al.. Heart disease and stroke statistics-2015 update: a report from the american heart association. Circulation 2015; 131:e29-e322; PMID:25520374; http://dx.doi.org/ 10.1161/CIR.0000000000000152 [DOI] [PubMed] [Google Scholar]
- 25.Klijn CJ, Hankey GJ. Management of acute ischaemic stroke: new guidelines from the American Stroke Association and European Stroke Initiative. Lancet Neurol 2003; 2:698-701; PMID:14572738; http://dx.doi.org/ 10.1016/S1474-4422(03)00558-1 [DOI] [PubMed] [Google Scholar]
- 26.Joo H, George MG, Fang J, Wang G. A literature review of indirect costs associated with stroke. J Stroke Cerebrovasc Dis 2014; 23:1753-63; PMID:24957313; http://dx.doi.org/ 10.1016/j.jstrokecerebrovasdis.2014.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The α-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 2009; 284:35973-86; PMID:19850936; http://dx.doi.org/ 10.1074/jbc.M109.051086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beland M, Roucou X. Taking advantage of physiological proteolytic processing of the prion protein for a therapeutic perspective in prion and Alzheimer diseases. Prion 2013; 8:106-10; http://dx.doi.org/ 10.4161/pri.27438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitsios N, Saka M, Krupinski J, Pennucci R, Sanfeliu C, Miguel Turu M, Gaffney J, Kumar P, Kumar S, Sullivan M, et al.. Cellular prion protein is increased in the plasma and peri-infarcted brain tissue after acute stroke. J Neurosci Res 2007; 85:602-11; PMID:17149767; http://dx.doi.org/ 10.1002/jnr.21142 [DOI] [PubMed] [Google Scholar]
- 30.Premzl M, Sangiorgio L, Strumbo B, Marshall Graves JA, Simonic T, Gready JE. Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene 2003; 314:89-102; PMID:14527721; http://dx.doi.org/ 10.1016/S0378-1119(03)00707-8 [DOI] [PubMed] [Google Scholar]
- 31.Watts JC, Westaway D. The prion protein family: Diversity, rivalry, and dysfunction. Biochim Biophys Acta 2007; 1772:654-72; PMID:17562432; http://dx.doi.org/ 10.1016/j.bbadis.2007.05.001 [DOI] [PubMed] [Google Scholar]
- 32.Watts JC, Huo H, Bai Y, Ehsani S, Won AH, Shi T, Daude N, Lau A, Young R, Xu L, et al.. Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog 2009; 5:e1000608; PMID:19798432; http://dx.doi.org/ 10.1371/journal.ppat.1000608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellecchia M, Blomgren K, Plesnila N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci 2005; 25:10262-72; PMID:16267234; http://dx.doi.org/ 10.1523/JNEUROSCI.2818-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Yu SP, Mohamad O, Genetta T, Wei L. Sublethal transient global ischemia stimulates migration of neuroblasts and neurogenesis in mice. Transl Stroke Res 2010; 1:184-96; PMID:21792374; http://dx.doi.org/ 10.1007/s12975-010-0016-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx 2005; 2:396-409; PMID:16389304; http://dx.doi.org/ 10.1602/neurorx.2.3.396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch 2010; 460:525-42; PMID:20229265; http://dx.doi.org/ 10.1007/s00424-010-0809-1 [DOI] [PubMed] [Google Scholar]
- 37.Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, et al.. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol 2008; 181:551-65; PMID:18443219; http://dx.doi.org/ 10.1083/jcb.20-0711002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciric D, Richard CA, Moudjou M, Chapuis J, Sibille P, Daude N, Westaway D, Adrover M, Beringue V, Martin D, et al.. Interaction between Shadoo and PrP Affects the PrP-Folding Pathway. J Virol 2015; 89:6287-93; PMID:25855735; http://dx.doi.org/ 10.1128/JVI.03429-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini E, Fioriti L, Chiesa R, Harris DA. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem 2003; 278:21732-43; PMID:12663673; http://dx.doi.org/ 10.1074/jbc.M213247200 [DOI] [PubMed] [Google Scholar]
- 40.Westaway D, Genovesi S, Daude N, Brown R, Lau A, Lee I, Mays CE, Coomaraswamy J, Canine B, Pitstick R, et al.. Downregulation of Shadoo in prion infections traces a pre-clinical event inversely related to PrP(Sc) accumulation. PLoS Pathog 2011; 7:e1002391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Safar JG, DeArmond SJ, Kociuba K, Deering C, Didorenko S, Bouzamondo-Bernstein E, Prusiner SB, Tremblay P. Prion clearance in bigenic mice. J Gen Virol 2005; 86:2913-23; PMID:16186247; http://dx.doi.org/ 10.1099/vir.0.80947-0 [DOI] [PubMed] [Google Scholar]
- 42.Watts JC, Stohr J, Bhardwaj S, Wille H, Oehler A, Dearmond SJ, Giles K, Prusiner SB. Protease-resistant prions selectively decrease shadoo protein. PLoS Pathog 2011; 7:e1002382; PMID:22163178; http://dx.doi.org/ 10.1371/journal.ppat.1002382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mays CE, Kim C, Haldiman T, van der Merwe J, Lau A, Yang J, Grams J, Di Bari MA, Nonno R, Telling GC, et al.. Prion disease tempo determined by host-dependent substrate reduction. J Clin Invest 2014; 124:847-58; PMID:24430187; http://dx.doi.org/ 10.1172/JCI72241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lau A, Mays CE, Genovesi S, Westaway D. RGG Repeats of PrP-like Shadoo Protein Bind Nucleic Acids. Biochemistry 2012; 51:9029-31; PMID:23121093; http://dx.doi.org/ 10.1021/bi3013-95w [DOI] [PubMed] [Google Scholar]
- 45.Mays CE, Coomaraswamy J, Watts JC, Yang J, Ko KW, Strome B, Mercer RC, Wohlgemuth SL, Schmitt-Ulms G, Westaway D. Endoproteolytic Processing of the Mammalian Prion Glycoprotein Family. FEBS J 2014; 281:862-76; PMID:24286250; http://dx.doi.org/ 10.1111/febs.12654 [DOI] [PubMed] [Google Scholar]
- 46.Liang J, Kong Q. α-Cleavage of cellular prion protein. Prion 2012; 6:453-60; PMID:23052041; http://dx.doi.org/ 10.4161/pri.22511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lau A, McDonald A, Daude N, Mays CE, Walter ED, Aglietti R, Mercer RC, Wohlgemuth S, van der Merwe J, Yang J, et al.. Octarepeat region flexibility impacts prion function, endoproteolysis and disease manifestation. EMBO Mol Med 2015; 7(3):339-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9:671-5; PMID:22930834; http://dx.doi.org/ 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsuchiya D, Hong S, Kayama T, Panter SS, Weinstein PR. Effect of suture size and carotid clip application upon blood flow and infarct volume after permanent and temporary middle cerebral artery occlusion in mice. Brain Res 2003; 970:131-9; PMID:12706254; http://dx.doi.org/ 10.1016/S0006-8993(03)02300-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.