

Abstract
RNA interference is gaining attention as a means to explore new molecular pathways and for its potential as a therapeutic; however, its application in immortal and primary T cells is limited due to challenges with efficient delivery in these cells types. Herein, we report the development of guanidinium-rich protein transduction domain mimics (PTDMs) based on a ring-opening metathesis polymerization scaffold that delivers siRNA into Jurkat T cells and human peripheral blood mononuclear cells (hPBMCs). Homopolymer and block copolymer PTDMs with varying numbers of guanidinium moieties were designed and tested to assess the affect cationic charge content and the addition of a segregated, hydrophobic block had on siRNA delivery. Delivery of fluorescently-labeled siRNA into Jurkat T cells illustrates that the optimal cationic charge content, 40 charges per polymer, leads to higher efficiencies, with block copolymers outperforming their homopolymer counterparts. PTDMs also outperformed commercial reagents commonly used for siRNA delivery applications. Select PTDM candidates were further screened to assess the role PTDM structure has on the delivery of biologically active siRNA into primary cells. Specifically, siRNA to hNOTCH1 was delivered to hPBMCs enabling 50–80% knockdown efficiencies, with longer PTDMs showing improved protein reduction. By evaluating the PTDM design parameters for siRNA delivery, more efficient PTDMs were discovered that improved delivery and gene knockdown in T cells. Given the robust delivery of siRNA by these novel PTDMs, their development should aid in the exploration of T cell molecular pathways leading eventually to new therapeutics.
Graphical Abstract
INTRODUCTION
RNA interference (RNAi), discovered almost two decades ago, continues to be an important tool to probe molecular pathways and to potentially treat diseases.1–5 Small interfering RNA (siRNA), which represents one RNAi approach, must be present in the cytosol so that the siRNA guide strand can be incorporated into the RISC complex and degrade its complementary mRNA. This leads to transient, sequence-specific, post-transcriptional gene knockdown,6–8 which is advantageous for discrete biological and clinical applications,1–5 a strategy of critical importance in the context of the immune system and T cells.9–12
T cells orchestrate essential functions during the immune response to pathogens, chronic inflammation, and autoimmune disorders.10 Harnessing the capabilities of RNAi would allow immunologists to explore molecular pathways leading to a better understanding of T cell activation, signaling, and other biological processes, eventually providing new treatment options for autoimmune diseases.9–12 Unfortunately, progress in this area has been severely limited due to the lack of robust delivery technologies for T cell lines and primary cells.10,12–17 Three major strategies are routinely used for siRNA delivery into T cells and primary cells: electroporation, viral vectors, and transfection.12–17 The use of electroporation and viral vectors both have severe drawbacks, which include high cell death and potential mutagenic/immunogenic effects, respectively.14,16 Transfection is generally a safer alternative, with higher cell survival rates; however, many current delivery vehicles exhibit low efficiencies in T cell lines and primary cells.13,15,17 Although designing successful carriers for T cells has been a challenge, highly modular protein transduction domain mimics (PTDMs), sometimes referred to as cell-penetrating peptide mimics (CPPMs), have been used successfully in other more easily transfected cell types and can provide insight for the design of more efficient delivery vehicles.18–20
Inspiration for PTDMs is derived from proteins with translocation abilities, such as the HIV-1 TAT and Antennapedia homeodomain proteins, as well as other protein transduction domains (PTDs) and cell-penetrating peptides (CPPs) that exhibit delivery capabilities.21–23 PTDMs incorporate important features of PTDs and CPPs critical for intracellular delivery24–29, including cationic charge content provided by guanidinium groups, and partial hydrophobicity contributed by the backbone architecture and/or the incorporation of hydrophobic monomers.18,19 Although some PTDs and CPPs, such as CADY and MPG, possess these key features and have already been designed and commercialized for siRNA delivery, PTDMs offer many distinct advantages over their peptide counterparts.30,31 Moving away from a peptide-based architecture provides protection from proteolysis and avoids solid phase peptide synthesis. Additionally, a non-peptidic system offers many more structural options, since it is not restricted to the incorporation of known amino acids.18,19,32 Consequently, different chemistries can be used to design molecules, and chemical compositions can be tuned more widely to improve delivery of specific cargo.18,19,32 This design rationale has proven successful in creating more potent antimicrobial peptide mimics, where the key features, including their facially amphiphilic architecture,33–35 were incorporated into synthetic scaffolds to yield new antimicrobial agents.19,35–37
In the realm of delivery reagents, several groups have demonstrated the utility of synthetic, guanidinium-rich, polymeric scaffolds to deliver siRNA.18,19,38–44 Ring-opening metathesis polymerization (ROMP)20,45–47, polymethacrylamide42,48, arginine-grafted bioreducible polydisulfide40,41, and oligocarbonate38,39,44 scaffolds have all been developed and screened for siRNA delivery. The successful design of PTDMs will further the understanding of the key features necessary for efficient delivery, as well as enable the development of more effective delivery reagents.18,19,32
Related to this long term goal, in 2008, the Tew research group first reported the development of polymeric guanidinium-rich PTDMs based on a ROMP scaffold that mimics PTDs/CPPs, such as TAT49–57 and oligoarginine (R9).19,49,50 Initial studies aimed to understand the effects of PTDM length51, hydrophobicity52, aromaticity53, aromatic π-electronics54, and sequence segregation of cationic/hydrophobic components55 on membrane interactions and cellular uptake.43 In early 2013, we extended this platform to include siRNA delivery. It was demonstrated that our PTDMs delivered FITC-siRNA into Jurkat T cells with efficiencies greater than 90% and achieved 50% knockdown of NOTCH1 protein in human T cells from human peripheral blood mononuclear cells (hPBMCs).20 Due to the initial success of siRNA delivery using these PTDMs, further understanding of the relationship between PTDM structure and siRNA delivery efficiency in T cells was desired.
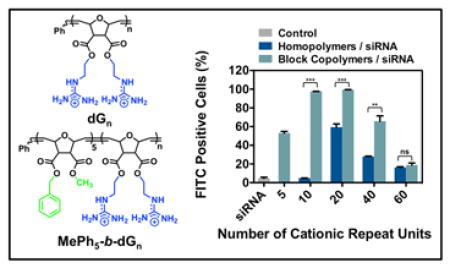
Herein we document our efforts to tune ROMP-based PTDM structures for improved siRNA delivery. Structure activity relationships using Jurkat T cells and hPBMCs were used to probe how polymer charge content and the addition of a segregated, hydrophobic block impacted siRNA internalization and delivery efficiencies. Specifically, two polymer series were developed, homopolymers and block copolymers, with matching cationic charge contents (Figure 1). All block copolymers contained a constant hydrophobic block of five repeat units. It should be noted that guanidinium moieties were incorporated to mimic arginine residues and phenyl moieties were incorporated in the block copolymer PTDMs to mimic phenylalanine residues, both of which have been shown to play critical roles in membrane interactions.27,29,43,51–54,57,58 In this study, we elucidate the essential design parameters for siRNA delivery using our PTDMs, which can be used to develop the next generation of highly efficient transfection reagents.
Figure 1.

Monomer and polymer structures used for this study. A) Monomer structures. B) Polymer structures. C) Table summarizing the polymer nomenclature and the corresponding number of positive charges each polymer contains. Blue represents cationic moieties and green represents hydrophobic moieties.
EXPERIMENTAL SECTION
Monomer Synthesis and Characterization
Monomers were synthesized using a two-step process adapted from Lienkamp et al. with modifications.59 In brief, oxanorbornene anhydride was ring-opened using the desired alcohol and 4-dimethylaminopyridine (DMAP) as a catalyst to yield the half-ester intermediates, which precipitated from solution. The half-ester was then further reacted with the second desired alcohol using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling to yield the monomer. A one-pot synthesis was used for monomers designed to display two of the same functionalities. All monomers were purified using column chromatography (ethyl acetate/dichloromethane (CH2Cl2), 10/90, v/v) and subsequently analyzed by 1H NMR spectroscopy, 13C NMR spectroscopy, and mass spectrometry (MS) to assess their chemical compositions and purity. Detailed synthetic procedures and all characterization data are provided in the supporting information (Section II, Figures S1 and S2).
PTDM Synthesis and Characterization
Homopolymers and block copolymer PTDMs were synthesized by ROMP using Grubbs’ third generation catalyst following previously described methods.20,59 In brief, degassed monomer solutions in CH2Cl2 were introduced to degassed catalyst solutions in CH2Cl2 and allowed to stir. For block copolymers, the hydrophobic monomer was added first followed by the Boc-protected guanidinium monomer. To prevent premature termination of polymerizations, all guanidinium-based monomers were Boc-protected to limit the potential for catalyst coordination. The presence of the Boc groups also allowed for sufficient solubility in organic solvents. Polymers were quenched with ethyl vinyl ether, precipitated, and subsequently deprotected using a 1:1 ratio of CH2Cl2:trifluoroacetic acid (TFA). Excess TFA was removed by azeotropic distillation with methanol. Polymers were then dissolved in water and transferred to Biotech CE dialysis tubing membranes with a MWCO 100–500 g/mol and dialyzed against RO water until the conductivity of the water remained < 0.2 μS (2–3 days). All polymers were characterized by 1H NMR and gel permeation chromatography (GPC) to assess chemical compositions and molecular weight distributions, respectively. Detailed synthetic procedures and all characterization data are provided in the supporting information (Section II, Figures S3–S12).
Cell culture (Cell lines) and FITC-siRNA Internalization
Jurkat T cells and HeLa cells were cultured in RPMI 1640 and DMEM, respectively, supplemented with 10% (v/v) FBS, 100 U/mL non-essential amino acids, 100U/mL sodium pyruvate, 100 U/mL penicillin, and 100 U/mL streptomycin. Jurkat T cells were incubated at 37 °C with 5% CO2 and passaged 24 hours prior to experimentation. On the day of the experiment, PTDMs were mixed with siRNA at an N:P ratio of 8:1 (50 nM siRNA/well) and allowed to incubate at room temperature for 30 minutes then added drop-wise to the cells (4x105 cells/well; 1 mL total volume) in a 12 well plate. Cells were incubated at 37 °C with 5% CO2 in serum-containing media for four hours prior to analysis by flow cytometry. For HeLa cell experiments, cells (5x104 cells/well; 1 mL total volume) in serum containing media were cultured in 12 well plates for 48 hours so that the cells would be 70–90% confluent on the day of the experiment. On the day of the experiment, PTDMs were mixed with siRNA at an N:P ratio of 4:1 (50 nM siRNA/well) and allowed to incubate at room temperature for 30 minutes. The cell media was replaced with fresh, complete media prior to adding the PTDM/siRNA complexes carefully to the top of the sample wells. Cells were incubated at 37 °C with 5% CO2 for four hours in serum-containing media prior to analysis by flow cytometry. Cell viability was assessed using 7-AAD/Annexin V staining.
Primary Cell Enrichment and Stimulation
hPBMCs, purchased from Stemcell Technologies, Inc. in 2.5x106 cells/aliquot (Product # 70047.2) and obtained by the company using institutional review board approved consent forms and protocols, were thawed and plated in a 6 well plate using RPMI 1640 supplemented with 10% (v/v) FBS, 100 U/mL non-essential amino acids, 100 U/mL sodium pyruvate, 100 U/mL penicillin, and 100 U/mL streptomycin, to enrich for the T cell populations. The cells were incubated at 37 °C in a 5% CO2 atmosphere overnight. On the day of the experiment, PTDMs were mixed with siRNA at an N:P ratio of 8:1 (100 nM siRNA per well) and allowed to incubate at room temperature for 30 minutes prior to adding them drop-wise to the cells (1x106 cells/well) in a 24 well plate. Cells were incubated at 37 °C with 5% CO2 in serum-containing media for four hours and subsequently harvested, washed, re-plated in an anti-CD3 and anti-CD28 coated well plate (well plate coated the night before) to stimulate the cells. Cells were incubated at 37 °C with 5% CO2 in serum-containing media for 48 hours prior to harvesting for experiments. For flow cytometry, half the cells were fixed for intracellular staining of NOTCH1 protein and the other half were used for viability staining. Detailed cellular assay procedures as well as viability data and flow cytometry histograms are provided in the supporting information (Section V).
Assessment of NOTCH1 Knockdown in hPBMCs
Following cell harvest and wash steps, cells were re-suspended in 100 μL of the Foxp3 Fix/Perm Cocktail and incubated for 30 min on ice protected from light. After the 30 min incubation, the cells were brought up to 200 μL with the permeabilization wash buffer Cells were washed three times with the permeablization wash buffer. After the third wash step, cells were re-suspended in 50 μL of the permeablization wash buffer, stained with 2 μL of anti-human NOTCH1 PE, and incubated for 30 min on ice protected light. After the 30 min incubation, cells were washed three times with the permeablization wash buffer and then re-suspended in 200 μL of FACS wash buffer and transferred to FCM tubes for analysis. For flow cytometry analysis, the fluorescence signal was collected for 10,000 cells. The cell populations were gated in order to assess the percent of positive cells, which reflected the percentage of the cell population expressing hNOTCH1 protein. The calculated MFI represented the amount of hNOTCH1 protein present in the cells. The percent relative protein expression represents the percent positive cells multiplied by the MFI, normalized to the blank, and multiplied by 100%.
Assessment of hPBMC Viability
Following the wash steps documented above, cells were re-suspended in 200 μL of a 7-AAD stock solution (2.4 mL of binding buffer + 60 μL of 7-AAD stain; Solution was scaled up or down as needed) and transferred to FCM tubes for analysis. For flow cytometry analysis, the fluorescence signal was collected for 10,000 cells. The cell populations were gated in order to assess the percent of positive cells, which reflected the percentage of dead cell in the population.
RESULTS AND DISCUSSION
PTDM Design and Characterization
Proteins and peptides have been utilized extensively for intracellular delivery applications.18,32,60–62 These materials, however, have presented many limitations, including long and costly synthetic procedures, as well as poor stability, all of which can be avoided by leveraging more versatile synthetic platforms. In this report, ROMP with Grubbs’ third generation catalyst was used to synthesize all PTDMs since it is a fast, efficient, and functional group tolerant method that also allows for good control over molecular weights and dispersities.. The living nature of many ROMP polymerizations also enables the synthesis both of homopolymers and block copolymers, and allows the influence of an added hydrophobic block on internalization and delivery efficiencies to be evaluated within the same monomer chemistry.63–71 Additionally, the oxanorbornene-based dual-functional monomer platform is quite versatile, allowing for the incorporation of the same or different functionalities to tune the polymer structures at the monomer level.18 For this particular endeavor, a monomer containing two Boc-protected guanidinium groups (dG; Figure 1) and a second monomer containing a methyl group and a phenyl group (MePh; Figure 1) were synthesized. Guanidinium moieties were selected as the cations because they have been previously shown to yield superior uptake and delivery efficiencies as compared to their ammonium counterparts found in lysine and ornithine.27 The number average molecular weight (Mn) for the homopolymerization of the dG monomer increased linearly with monomer to initiator ratio ([M]/[I]), while maintaining low dispersities. This indicated that it polymerized in a controlled fashion and can be used to synthesize both homopolymers and block copolymers (Figure S12).
Homopolymer and block copolymer PTDMs were synthesized to determine how the cationic charge content, as well as the incorporation of a segregated hydrophobic region, impacted siRNA delivery. To that end, PTDMs with increasing amounts of cationic charge were synthesized, as documented in Figure 1. Since the cationic monomer contains two guanidinium groups, the number of charges is reported as twice the degree of polymerization (Figure 1C). In addition, block copolymer PTDMs were designed with segregated hydrophobic and cationic domains, as shown in Figure 1. These were specifically designed because the incorporation of a hydrophobic domain has been shown to improve internalization and delivery efficiencies through interactions with hydrophobic lipids.31 The performance of the block copolymers was compared to their corresponding homopolymer derivatives, which contained the same charge content, in order to assess the effect of incorporating a hydrophobic block. In both cases, the charge content was varied up to 160 charges (80 repeat units); however, PTDMs with 160 charges had limited solubility and were therefore not used for any biological studies. Overall, this series of PTDMs provided insight into key design parameters for efficient siRNA internalization and delivery.
FITC-siRNA Delivery
Preliminary studies were conducted using fluorescein isothiocyanate (FITC) labeled siRNA (FITC-siRNA), to assess trends in internalization efficiencies in Jurkat T cells resulting from differences in PTDM charge content and the presence or absence of a hydrophobic block. The fluorescent label on the siRNA allowed cell populations to be analyzed using flow cytometry. Jurkat T cells were selected because they are typically difficult to transfect and manipulate.10,12–17 The N:P ratio used for complex formation between PTDMs and siRNA was 8:1 and was established by screening FITC-siRNA internalization efficiencies as a function of N:P ratio in addition to using gel retardation assays to assess siRNA complexation (Figures S13–S22 and Figure S25). This N:P ratio used was consistent with our previous publication.20 Gel retardation assays for PTDM/siRNA complexes at N:P ratios of 0.5:1, 1:1, 2:1, 4:1, 8:1, and 12:1 are included in the supporting information (Figures S13–S21). In brief, it was demonstrated that shorter polymers (40 charges or less) completely complexed siRNA at lower N:P ratios than longer polymers (80 charges or more). In terms of media composition, it was previously reported that our ROMP-based PTDMs do not experience significant reduction in performance when tested in the presence of serum.20 Therefore, all cell data was collected in the presence of serum.20
A summary of FITC-siRNA internalization efficiencies for homopolymer and block copolymer PTDMs is shown in Figure 2, where Figure 2A presents the percentage of the cell population that received FITC-siRNA and Figure 2B presents the median fluorescence intensity (MFI) of the cell populations. In both data sets, PTDM internalization efficiencies increased in a charge-dependent manner up to 40 charges (dG20 and MePh5-b-dG20) and then diminish at higher charge contents. MePh5-b-dG20, with an MFI of 2,300, delivered the most amount of siRNA, which was three times larger than the next best PTDM, MePh5-b-dG10, with an MFI of 800. This demonstrates that there is an optimal guanidinium content necessary for efficient internalization and above that charge content, the carriers are no longer as effective and show reduced performance.
Figure 2.

FITC-siRNA internalization in Jurkat T cells using homopolymer and block copolymer PTDMs. Jurkat T cells (cell density = 4x105 cells/mL) were treated with PTDM/FITC-siRNA complexes with an N:P ratio = 8:1 in complete medium for four hours at 37°C and compared to cells only receiving FITC-siRNA. All data was normalized to an untreated control. A) Percent FITC positive cells. B) Median fluorescence intensity (MFI) of the cell population. Data represents the mean ± SEM of three independent experiments. * p = < 0.05, ** = p < 0.01, *** = p < 0.001, ns = not significant, as calculated by the unpaired two-tailed student t-test. * represents significance between homopolymer and block copolymer PTDMs with the same charge content.
From this data set, it can also be seen that the block copolymer PTDMs significantly outperformed their homopolymer counterparts, particularly at charge contents of 40 or less. Both MePh5-b-dG20 and MePh5-b-dG10 had MFI values that were six times higher than their corresponding homopolymers (dG20 and dG10, respectively). At larger charge contents, MFI values were similarly diminished for both homopolymer and block copolymer PTDMs. The block copolymer PTDM with 80 charges (MePh5-b-dG40) delivered FITC-siRNA to 66% of the cell population, which is approximately double the cell population that its homopolymer PTDM counterpart, dG40, could reach. The observation that block copolymers outperformed their homopolymer counterparts is consistent with other reported guanidinium-rich siRNA transport molecules.39,48 In addition to Figure 2, flow cytometry histograms can be found in the supporting information (Figure S23). PTDMs were also tested for their ability to deliver FITC-siRNA into HeLa cells to demonstrate that they enabled internalization in adherent cells and to establish trends in a second cell type. Block copolymer PTDMs containing 20 and 40 charges (MePh5-b-dG10 and MePh5-b-dG20) were shown to deliver double the amount of siRNA than their homopolymer counterparts. PTDMs with 80 charges (dG40 and MePh5-b-dG40) were able to facilitate the internalization of the most siRNA, with similar MFIs of 4,100 and 4,500, respectively. A significant drop-off in internalization efficiency for the largest charged segment was still observed (Figures S33–S36). All polymer-treated cells showed greater than 90% viability using 7-AAD/Annexin V staining (Figure S26–S27 and Figures S37–38).
In a separate viability study, promising PTDM/siRNA complexes were compared to samples that were treated with PTDMs alone in the same working concentrations used for complex formation. Cells were harvested four hours after treatment and washed. Samples were split and viability was determined on one half of the cells. The other half were re-plated in fresh serum-containing medium and viability was assessed after an additional 24 hours of incubation. This not only assessed PTDM toxicity at the maximum free concentrations, but it also examined longer-term toxicity effects of the treatment. All cells exhibited greater than 90% viability as assessed by 7-AAD/Annexin V staining both at the four-hour and 24-hour time points (Figures S29–S31). In addition, cells were also counted at the 24-hour time point to measure cell proliferation. All cell populations were found to approximately double over the 24-hour period, suggesting that PTDM/siRNA treatment did not impair cell growth (Figure S32).
Based on these results, two block copolymer PTDMs, MePh5-b-dG5 and MePh5-b-dG20, were compared to a range of commercial reagents commonly used for siRNA internalization and delivery, including R9 (peptide, Peptide2.0), DeliverX (peptide, Affymetrix), Xfect (polymer, ClonTech), N-Ter (peptide, Sigma Aldrich), and RNAiMAX (lipid, Life Technologies). In addition, JetPEI, a common polyethyleneimine-based pDNA delivery reagent, was included. All commercial reagents were used as directed by the vendor. A summary of FITC-siRNA internalization efficiencies for block copolymer PTDMs and commercial reagents is shown in Figure 3, where Figure 3A presents the percentage of the cell population that received FITC-siRNA and Figure 3B presents the MFI of the cell populations. MePh5-b-dG5 and MePh5-b-dG20 were both shown to facilitate siRNA internalization in a greater percentage of cells than the commercial reagents (Figure 3A). In addition, MePh5-b-dG20 resulted in a nearly 10 folder greater MFI than the commercial reagents, indicating it was able to deliver quantitatively more cargo inside the cells (Figure 3B, Figure S24). Viability was assessed using 7-AAD / Annexin V staining (Figure S28). All samples had greater than 90% viability, with the exception of samples treated with RNAiMAX, which had viabilities closer to 80%. This data demonstrates the superiority of our PTDMs for siRNA delivery compared to common commercial reagents and establishes these materials as viable delivery vehicles for hard-to-transfect T cell lines, for which options for efficient delivery are limited.
Figure 3.

Comparison of PTDM and commercial reagent FITC-siRNA internalization efficiencies in Jurkat T cells (cell density = 4x105 cells/mL). Cells treated with PTDM/FITC-siRNA complexes with an N:P ratio = 8:1 or with commercial reagent/FITC-siRNA complexes (used as directed) in complete medium for four hours at 37°C and compared to cells only receiving FITC-siRNA. All data was normalized to untreated controls. A) Percent FITC positive cells. B) Median fluorescence intensity (MFI) of the cell population. Data represents the mean ± SEM of three independent experiments. All PTDM data is statistically different from the commercial reagents (p <0.001) as calculated by the unpaired two-tailed student t-test.
Delivery of Biologically Active siRNA to NOTCH1
Based on the FITC-siRNA internalization results from Jurkat T cells, four promising PTDMs (MePh5-b-dG5, MePh5-b-dG10, MePh5-b-dG20, and dG20) were screened for delivery of a biologically active siRNA to NOTCH1. NOTCH1 was selected as the primary target for siRNA knockdown studies because it represents an active gene in T cells. Unlike looking at a reporter gene, which is not required for normal cellular activities, the gene of interest is required for cellular function and represents a more sophisticated and realistic system for knockdown assessment. Although NOTCH1 protein is constitutively expressed in Jurkat T cells, it is a mutation in the NOTCH1 gene together with increased NOTCH1 protein stability that gives the T cell line its immortality, thus making down regulation of NOTCH1 expression difficult to monitor. To overcome this limitation, hPBMCs were used in NOTCH1 down regulation experiments. At the same time, demonstrating robust delivery into primary T cells is of critical practical importance. Surface staining cells at the 48 hour time point to check for CD4 and CD8 expression revealed that approximately 30% of the population was CD8+ and approximately 50% of the population was CD4+. The gating strategy for this determination is shown in Figure S39.
In these experiments, hPBMCs were treated with PTDM/siRNA complexes for four hours in serum-containing medium. A fixed amount of siRNA (100nM) was used for each experiment to assess the effect PTDM structure has on knockdown efficiencies. Specifically, with the block copolymer, the length of the cationic sequence was examined while holding the hydrophobic block constant at five repeat units. After four hours, the cells were transferred to anti-CD3 and anti-CD28-coated well plates for stimulation and allowed to incubate for 48 hours. T cell stimulation will result in an induction in NOTCH1 expression and allow us to assess the effect treatment had on NOTCH1 protein levels in comparison to untreated samples. This time point was selected because it is the point of maximum protein expression, and we expect the cells that successfully received siRNA to NOTCH1 would have decreased protein expression compared to the untreated controls. Flow cytometry was used as the primary method to analyze protein expression, enabling us to stain for protein content and quantify the extent to which protein levels were reduced on a per cell basis. The percent relative protein expression data for these experiments is shown in Figure 4A. In this case, percent relative protein expression is the MFI of the total population multiplied by the percent NOTCH1 positive population all normalized to an untreated blank. For the PTDM with the shortest cationic sequence, MePh5-b-dG5, NOTCH1 expression was reduced to ~50%, which is significant given that NOTCH1 is not a reporter gene. Doubling the cationic length with MePh5-b-dG10 resulted in a lower NOTCH1 expression of ~37%. The lowest NOTCH1 expression obtained, ~16%, was observed for MePh5-b-dG20. This data was consistent with the previous data shown in Figures 2 and 3, which support the suggestion that this PTDM is able to deliver the most siRNA into PBMCs. In fact, it appears that for these experiments with 100 nM siRNA that MePh5-b-dG20 delivers too much siRNA since resolution was lost between the target (light blue bars) and scrambled siRNA (purple bars), a common observation when excess siRNA is present intracellularly.72–75 Although reducing the siRNA concentration may help limit these effects73–75, sequence overlaps with target and off-target mRNAs are also a contributing factor and may be present, even at reduced concentrations.72,76 Better screening for scrambled controls that avoid the target mRNA sequence would be expected to improve the specificity of NOTCH1 knockdown experiments.76 While the primary goal of this study was to explore how the polymer structures impact siRNA delivery efficiencies, future studies will be geared towards optimizing these materials for improved specificity.
Figure 4.

Relative NOTCH1 expression levels in PBMCs and their corresponding viabilities (cell density = 1x106 cells/mL). Cells were treated with PTDM/NOTCH1 siRNA complexes or PTDM/scrambled siRNA with an N:P ratio = 8:1 in complete media for four hours at 37°C. After treatment, cells were washed and then stimulated with plate-bound anti-CD3 and anti-CD28 for 48 hours. All data was normalized to an untreated control (grey bar). A) Relative NOTCH1 levels in PBMCs after 48-hour treatment with PTDM/NOTCH1 siRNA (light blue bars) or PTDM/scrambled siRNA complexes (purple bars). B) Percent viable cells following staining with 7-AAD. Data represents the mean ± SEM of four independent experiments using cells isolated from different donors. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, ns = not significant, as calculated by the unpaired two-tailed student t-test.
When evaluating protein expression, it was critical to assess cell viability to ensure that diminished protein expression was not due to cell death. Since monitoring protein content by flow cytometry requires cells to be fixed, cell samples were split after harvest to determine cell viability on the unfixed population. hPBMC viability was assessed by staining with 7-AAD, a DNA-intercalating dye that is excluded from live cells but taken up by dead and dying cells that have compromised membrane integrity. As shown in Figure 4B, all samples were greater than 80% viable, which suggests that reduced protein expression in the cells is a consequence of PTDM/siRNA treatment and not due to cell death.
In addition to using flow cytometry, a western blot was used to monitor protein reduction in cells treated with siRNA to NOTCH1 or treated with scrambled siRNA as they compared to untreated cells for MePh5-b-dG20 (Figure S43). This data was consistent with the flow cytometry data.
CONCLUSIONS
RNAi is an attractive approach to study gene function and has potential for therapeutic applications involving T cells due to its transient knockdown of target proteins; however, difficulties in delivery to these cell types have limited its use. In efforts to address an unmet need in the area of T cell delivery and to better understand how polymer composition impacts delivery and knockdown efficiencies, we report the development of two guanidinium-rich PTDM series for siRNA delivery. Homopolymer and block copolymer PTDMs with varying cationic charge contents were tested to assess the role of cationic charge content and the addition of a segregated, hydrophobic block in the internalization of FITC-siRNA in Jurkat T cells and knockdown of hNOTCH1 in hPBMCs. FITC-siRNA internalization in Jurkat T cells demonstrated that there was an optimal cationic charge content necessary for efficient delivery (40 charges), which, once surpassed, resulted in diminished delivery capabilities. Cells incubated with PTDMs and their corresponding siRNA complexes exhibited greater than 90% viability using 7-AAD/Annexin-V staining. Block copolymers also significantly outperformed commercial reagents designed for siRNA delivery, making these PTDMs potential alternatives for T cell siRNA transfections. Based on the FITC-siRNA internalization results, select PTDM candidates were screened for knockdown of NOTCH1 in hPBMCs. For this set of PTDMs, protein expression was reduced as the cationic charge content increased, with the block copolymer PTDM MePh5-b-dG20 providing the most reduction in protein expression. Viability data indicated that knockdown was due to PTDM/siRNA treatment and not cell death. Overall, optimization of the cationic charge content led to improved delivery efficiencies, as well as improved knockdown efficiencies. This demonstrates the importance of understanding the essential PTDM design parameters for delivery of siRNA and will help guide the design of the next generation of efficient PTDMs.
Supplementary Material
Acknowledgments
This work was funded by the NIH (T32 GMO8515) and NSF (CHE-0910963 and DMR-1308123). The authors would like to thank Dr. Federica Sgolastra, Dr. Gabriella Gonzalez-Perez, and Dr. Christina Kuksin for invaluable scientific discussions and Ms. Leah Caffrey, Ms. Angie Korpusik, and Ms. Salimar Cordero-Mercado for help with monomer synthesis. The authors would also like to thank Ms. Coralie Backlund and Mr. Nicholas Posey for their feedback on early drafts of this manuscript. Mass spectral data were obtained at the University of Massachusetts Mass Spectrometry Facility, which is supported in part by NSF. Flow cytometry data were obtained using the Flow Cytometry Core Facility at the University of Massachusetts Amherst, which is supported in part by NSF.
Footnotes
Notes:
The authors declare no competing financial interest.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
All detailed synthetic procedures, molecular characterization, biological assays, and cellular viability data are provided in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.de Planque MR, Bonev BB, Demmers JA, Greathouse DV, Koeppe RE, 2nd, Separovic F, Watts A, Killian JA. Biochemistry. 2003;42:5341–5348. doi: 10.1021/bi027000r. [DOI] [PubMed] [Google Scholar]
- 2.Kim J, Lee SH, Choe J, Park TG. J Gene Med. 2009;11:804–812. doi: 10.1002/jgm.1365. [DOI] [PubMed] [Google Scholar]
- 3.Mello CC, Conte D. Nature. 2004;431:338–342. doi: 10.1038/nature02872. [DOI] [PubMed] [Google Scholar]
- 4.Vicentini FTMD, Borgheti-Cardoso LN, Depieri LV, Mano DD, Abelha TF, Petrilli R, Bentley MVLB. Pharm Res. 2013;30:915–931. doi: 10.1007/s11095-013-0971-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitehead KA, Langer R, Anderson DG. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carthew RW. Curr Opin Cell Biol. 2001;13:244–248. doi: 10.1016/s0955-0674(00)00204-0. [DOI] [PubMed] [Google Scholar]
- 7.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 8.Meister G, Tuschl T. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 9.Behlke MA. Mol Ther. 2006;13:644–670. doi: 10.1016/j.ymthe.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mantei A, Rutz S, Janke M, Kirchhoff D, Jung U, Patzel V, Vogel U, Rudel T, Andreou I, Weber M, Scheffold A. European Journal of Immunology. 2008;38:2616–2625. doi: 10.1002/eji.200738075. [DOI] [PubMed] [Google Scholar]
- 11.Rutz S, Scheffold A. Arthritis Res Ther. 2004;6:78–85. doi: 10.1186/ar1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeley M, Long A. Biochem J. 2013;455:133–147. doi: 10.1042/BJ20130950. [DOI] [PubMed] [Google Scholar]
- 13.Goffinet C, Keppler OT. Faseb Journal. 2006;20:500. doi: 10.1096/fj.05-4651fje. + [DOI] [PubMed] [Google Scholar]
- 14.Lai W, Chang CH, Farber DL. J Immunol Methods. 2003;282:93–102. doi: 10.1016/j.jim.2003.07.015. [DOI] [PubMed] [Google Scholar]
- 15.Marodon G, Mouly E, Blair EJ, Frisen C, Lemoine FM, Klatzmann D. Blood. 2003;101:3416–3423. doi: 10.1182/blood-2002-02-0578. [DOI] [PubMed] [Google Scholar]
- 16.McManus MT, Haines BB, Dillon CP, Whitehurst CE, van Parijs L, Chen JZ, Sharp PA. J Immunol. 2002;169:5754–5760. doi: 10.4049/jimmunol.169.10.5754. [DOI] [PubMed] [Google Scholar]
- 17.Zhang YF, Lu HZ, LiWang P, Sili U, Templeton NS. Mol Ther. 2003;8:629–636. doi: 10.1016/s1525-0016(03)00231-4. [DOI] [PubMed] [Google Scholar]
- 18.deRonde BM, Tew GN. Biopolymers. 2015 doi: 10.1002/bip.22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sgolastra F, deRonde BM, Sarapas JM, Som A, Tew GN. Acc Chem Res. 2013 doi: 10.1021/ar400066v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tezgel AO, Gonzalez-Perez G, Telfer JC, Osborne BA, Minter LM, Tew GN. Mol Ther. 2013;21:201–209. doi: 10.1038/mt.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frankel AD, Pabo CO. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 22.Green M, Loewenstein PM. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 23.Joliot A, Pernelle C, Deagostinibazin H, Prochiantz A. Proc Natl Acad Sci U S A. 1991;88:1864–1868. doi: 10.1073/pnas.88.5.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Derossi D, Chassaing G, Prochiantz A. Trends Cell Biol. 1998;8:84–87. [PubMed] [Google Scholar]
- 25.Derossi D, Joliot AH, Chassaing G, Prochiantz A. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 26.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Proc Natl Acad Sci U S A. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 28.Vives E, Brodin P, Lebleu B. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 29.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crombez L, Morris MC, Deshayes S, Heitz F, Divita G. Curr Pharm Design. 2008;14:3656–3665. doi: 10.2174/138161208786898842. [DOI] [PubMed] [Google Scholar]
- 31.Simeoni F, Morris MC, Heitz F, Divita G. Nucleic Acids Res. 2003;31:2717–2724. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanzl EG, Trantow BM, Vargas JR, Wender PA. Acc Chem Res. 2013;46:2944–2954. doi: 10.1021/ar4000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brogden KA. Nat Rev Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 34.Som A, Vemparala S, Ivanov I, Tew GN. Biopolymers. 2008;90:83–93. doi: 10.1002/bip.20970. [DOI] [PubMed] [Google Scholar]
- 35.Gabriel GJ, Som A, Madkour AE, Eren T, Tew GN. Mater Sci Eng R Rep. 2007;57:28–64. doi: 10.1016/j.mser.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuroda K, Caputo GA. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013;5:49–66. doi: 10.1002/wnan.1199. [DOI] [PubMed] [Google Scholar]
- 37.Tew GN, Scott RW, Klein ML, Degrado WF. Acc Chem Res. 2010;43:30–39. doi: 10.1021/ar900036b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooley CB, Trantow BM, Nederberg F, Kiesewetter MK, Hedrick JL, Waymouth RM, Wender PA. J Am Chem Soc. 2009;131:16401–16403. doi: 10.1021/ja907363k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geihe EI, Cooley CB, Simon JR, Kiesewetter MK, Edward JA, Hickerson RP, Kaspar RL, Hedrick JL, Waymouth RM, Wender PA. Proc Natl Acad Sci U S A. 2012;109:13171–13176. doi: 10.1073/pnas.1211361109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim SH, Jeong JH, Kim TI, Kim SW, Bull DA. Mol Pharm. 2009;6:718–726. doi: 10.1021/mp800161e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim SH, Jeong JH, Ou M, Yockman JW, Kim SW, Bull DA. Biomaterials. 2008;29:4439–4446. doi: 10.1016/j.biomaterials.2008.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tabujew I, Freidel C, Krieg B, Helm M, Koynov K, Mullen K, Peneva K. Macromol Rapid Commun. 2014;35:1191–1197. doi: 10.1002/marc.201400120. [DOI] [PubMed] [Google Scholar]
- 43.Tezgel AO, Telfer JC, Tew GN. Biomacromolecules. 2011;12:3078–3083. doi: 10.1021/bm200694u. [DOI] [PubMed] [Google Scholar]
- 44.Wender PA, Huttner MA, Staveness D, Vargas JR, Xu AF. Mol Pharm. 2015;12:742–750. doi: 10.1021/mp500581r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolonko EM, Kiessling LL. J Am Chem Soc. 2008;130:5626. doi: 10.1021/ja8001716. + [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kolonko EM, Pontrello JK, Mangold SL, Kiessling LL. J Am Chem Soc. 2009;131:7327–7333. doi: 10.1021/ja809284s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strong LE, Kiessling LL. J Am Chem Soc. 1999;121:6193–6196. [Google Scholar]
- 48.Treat NJ, Smith D, Teng C, Flores JD, Abel BA, York AW, Huang F, McCormick CL. ACS Macro Lett. 2012;1:100–104. doi: 10.1021/mz200012p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gabriel GJ, Madkour AE, Dabkowski JM, Nelson CF, Nusslein K, Tew GN. Biomacromolecules. 2008;9:2980–2983. doi: 10.1021/bm800855t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hennig A, Gabriel GJ, Tew GN, Matile S. J Am Chem Soc. 2008;130:10338–10344. doi: 10.1021/ja802587j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hennig A, Gabriel GJ, Tew GN, Matile S. J Am Chem Soc. 2008;130:10338–10344. doi: 10.1021/ja802587j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Som A, Tezgel AO, Gabriel GJ, Tew GN. Angew Chem Int Ed Engl. 2011;50:6147–6150. doi: 10.1002/anie.201101535. [DOI] [PubMed] [Google Scholar]
- 53.Som A, Reuter A, Tew GN. Angew Chem Int Ed Engl. 2012;51:980–983. doi: 10.1002/anie.201104624. [DOI] [PubMed] [Google Scholar]
- 54.deRonde BM, Birke A, Tew GN. Chem Eur J. 2015;21:3013–3019. doi: 10.1002/chem.201405381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sgolastra F, Minter LM, Osborne BA, Tew GN. Biomacromolecules. 2014;15:812–820. doi: 10.1021/bm401634r. [DOI] [PubMed] [Google Scholar]
- 56.Osborne BA, Minter LM. Nature Reviews Immunology. 2007;7:64–75. doi: 10.1038/nri1998. [DOI] [PubMed] [Google Scholar]
- 57.White SH, Wimley WC. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 58.Wimley WC, White SH. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 59.Lienkamp K, Madkour AE, Musante A, Nelson CF, Nusslein K, Tew GN. J Am Chem Soc. 2008;130:9836–9843. doi: 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deshayes S, Morris M, Heitz F, Divita G. Adv Drug Deliv Rev. 2008;60:537–547. doi: 10.1016/j.addr.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 61.Elliott G, O’Hare P. Cell. 1997;88:223–233. doi: 10.1016/s0092-8674(00)81843-7. [DOI] [PubMed] [Google Scholar]
- 62.Lindgren M, Langel U. Cell-Penetrating Peptides: Methods and Protocols. 2011;683:3–19. doi: 10.1007/978-1-60761-919-2_1. [DOI] [PubMed] [Google Scholar]
- 63.Love JA, Morgan JP, Trnka TM, Grubbs RH. Angew Chem Int Ed Engl. 2002;41:4035–4037. doi: 10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 64.Cannizzo LF, Grubbs RH. Macromolecules. 21:1961–1967. [Google Scholar]
- 65.Schwab P, France MB, Ziller JW, Grubbs RH. Angew Chem Int Ed Engl. 1995;34:2039–2041. [Google Scholar]
- 66.Singh R, Czekelius C, Schrock RR. Macromolecules. 2006;39:1316–1317. [Google Scholar]
- 67.Bielawski CW, Grubbs RH. Prog Polym Sci. 2007;32:1–29. [Google Scholar]
- 68.Schrock RR, Hoveyda AH. Angew Chem Int Ed Engl. 2003;42:4592–4633. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]
- 69.Trnka TM, Grubbs RH. Acc Chem Res. 2001;34:18–29. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]
- 70.Bielawski CW, Grubbs RH. Angew Chem Int Ed Engl. 2000;39:2903–2906. doi: 10.1002/1521-3773(20000818)39:16<2903::aid-anie2903>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 71.Bielawski CW, Benitez D, Grubbs RH. J Am Chem Soc. 2003;125:8424–8425. doi: 10.1021/ja034524l. [DOI] [PubMed] [Google Scholar]
- 72.Fedorov Y, Anderson EM, Birmingham A, Reynolds A, Karpilow J, Robinson K, Leake D, Marshall WS, Khvorova A. RNA. 2006;12:1188–1196. doi: 10.1261/rna.28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jackson AL, Linsley PS. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 74.Persengiev SP, Zhu X, Green MR. RNA. 2004;10:12–18. doi: 10.1261/rna5160904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Semizarov D, Frost L, Sarthy A, Kroeger P, Halbert DN, Fesik SW. Proc Natl Acad Sci U S A. 2003;100:6347–6352. doi: 10.1073/pnas.1131959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS. RNA. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.