

Abstract
Exploring the role of polymer structure for the internalization of biologically relevant cargo, specifically siRNA, is of critical importance to the development of improved delivery reagents. Herein, we report guanidinium-rich protein transduction domain mimics (PTDMs) based on a ring-opening metathesis polymerization scaffold containing tunable hydrophobic moieties that promote siRNA internalization. Structure-activity relationships using Jurkat T cells and HeLa cells were explored to determine how the length of the hydrophobic block and the hydrophobic side chain compositions of these PTDMs impacted siRNA internalization. To explore the hydrophobic block length, two different series of diblock copolymers were synthesized: one series with symmetric block lengths and one with asymmetric block lengths. At similar cationic block lengths, asymmetric and symmetric PTDMs promoted siRNA internalization in the same percentages of the cell population regardless of the hydrophobic block length; however, with twenty repeat units of cationic charge, the asymmetric block length had greater siRNA internalization, highlighting the non-trivial relationships between hydrophobicity and overall cationic charge. To further probe how the hydrophobic side chains impacted siRNA internalization, an additional series of asymmetric PTDMs was synthesized that featured a fixed hydrophobic block length of five repeat units that contained either dimethyl (dMe), methyl phenyl (MePh), or diphenyl (dPh) side chains and varied cationic block lengths. This series was further expanded to incorporate hydrophobic blocks consisting of diethyl (dEt), diisobutyl (diBu), and dicyclohexyl (dCy) based repeat units to better define the hydrophobic window for which our PTDMs had optimal activity. HPLC retention times quantified the relative hydrophobicities of the non-cationic building blocks. PTDMs containing the MePh, diBu, and dPh hydrophobic blocks were shown to have superior siRNA internalization capabilities compared to their more and less hydrophobic counterparts, demonstrating a critical window of relative hydrophobicity for optimal internalization. This better understanding of how hydrophobicity impacts PTDM-induced internalization efficiencies will help guide the development of future delivery reagents.
Graphical Abstract
Introduction
Intracellular delivery of therapeutics, particularly siRNA, continues to be a challenge for the biomedical community.1,2 Transient gene knockdown plays an important role in the exploration of molecular pathways and in the development of more advanced treatment options; the field, however, needs a clearer understanding of how to efficiently and reliably deliver bioactive molecules across cellular membranes, particularly for primary human cells.2–8 Nevertheless, nature is already capable of designing proteins that can perform these functions.9–11 One example, HIV-1 TAT, is partly responsible for the spread of the viral genome of HIV,9,10 and contains a region referred to as a protein transduction domain (PTD) which enables the protein to enter cells.12–14 These regions in proteins are generally cation-rich, containing lysine and arginine residues which aid in cellular uptake. Structure-activity relationships (SARs) related to this protein, as well as others such as the Antennapedia homeodomain protein, led to the development of a field referred to as cell-penetrating peptides (CPPs), which are capable of delivering cargo including small molecules, siRNA, pDNA, and proteins into cells.15–20 Three classical examples of CPPs include TAT49–57, which is an arginine-rich peptide based on the PTD of the HIV-1 TAT protein, while Pep-1 and MPG are lysine-rich primary, amphipathic peptides.13,14,20–24
Although extensive work has been devoted to exploring CPPs for siRNA delivery applications24–27, the extension of design principles learned from these systems to the development of synthetic mimics, referred to as cell-penetrating peptide mimics (CPPMs) or protein transduction domain mimics (PTDMs), offers many distinct advantages.28,29 By breaking out of the synthetic confinement of amino acids, a wider range of chemistries can be used to manipulate key features of CPPs, including hydrophobic segregation as well as cationic charge content.28 This field of mimetic polymer chemistry has already demonstrated a range of polymer scaffolds for the development of siRNA delivery reagents28,30,31, including those based on polyoxanorbornene 28,30,32, polymethacrylamide33, arginine-grafted bioreducible polydisulfide34,35, and oligocarbonate in just the past few years.36–38 Similar design principles were previously used for the creation of antimicrobial peptide mimics, where the facially amphiphilic structures of natural peptides were successfully recapitulated using highly modular synthetic scaffolds.39–43 In order to realize the full potential of these PTDM materials and continue to improve internalization and delivery efficiencies, extensive SARs studies are necessary to elucidate key design parameters.
To this end, our research group has devoted an extensive amount of research into understanding how the structures of ring-opening metathesis (ROMP) based protein mimics influence their membrane interactions29,40,44–48, cellular uptake efficiencies29,49, and siRNA delivery.30,32 We have demonstrated the utility of the platform for the successful internalization of siRNA and for the knockdown of active biological genes in T cells.30,32 Previous SARs established that there was a critical cationic block length required for efficient siRNA delivery, which, not surprisingly, showed some cell-type dependencies.32 Additionally, the incorporation of a fixed-length, segregated, hydrophobic segment into the PTDM platform improved siRNA internalization efficiencies by six fold compared to their homopolymer counterparts with the same relative cationic block lengths.32 From our preliminary studies, a great deal was learned about the cationic block length;32 however, further studies were needed to understand how the amount and type of hydrophobic content influenced siRNA internalization.
Many literature reports demonstrate that adding hydrophobicity, either through direct incorporation or through the use of bulky counter ions, generally improves membrane interactions, cellular uptake, and delivery efficiencies of CPPs and their synthetic mimics.30,32,37,47,48,50–55 Pep-1 and MPG, two common CPPs used for protein and siRNA delivery, respectively, have amphipathic structures where their hydrophobic components improve membrane interactions and internalization efficiencies.23,24 In light of these trends and the wide variety of hydrophobic groups available, it is important to understand how the length of the hydrophobic block within the PTDMs and the nature of the hydrophobic moiety impacts siRNA internalization. At the same time the cationic block length particularly in relation to the corresponding hydrophobic block, should not be overlooked as it also influences PTDM activity.
Structure activity relationships using Jurkat T cells and HeLa cells were used to probe how the length and the nature of the hydrophobic block impacted siRNA internalization. To explore the length of the hydrophobic block, two series of diblock copolymers (n and m, where n is the hydrophobic block length and m is the cationic block length) with symmetric (n = m) or asymmetric (n ≠ m) block lengths were synthesized (Figure 1). In a separate series of PTDMs, the hydrophobic side chain was varied to probe how the relative hydrophobicity impacted siRNA internalization. After initial testing, this set of PTDMs was expanded strategically to incorporate additional hydrophobic side chains to more specifically understand how hydrophobicity influences internalization. Relative hydrophobicities of the hydrophobic building blocks were estimated using HPLC retention times to guide the selection of structures used for analysis. Probing the effects of hydrophobicity on siRNA internalization efficiencies enabled us to better elucidate the essential design principles for our PTDMs.
Figure 1.

Monomer and polymer structures used for this study. A) Monomer structures B) Polymer Structures. Blue represents cationic moieties and green represents hydrophobic moieties.
EXPERIMENTAL SECTION
Monomer Synthesis and Characterization
Monomers were synthesized using a two-step process previously reported by our group.32 In brief, oxanorbornene anhydride was ring-opened using the desired alcohol and 4-dimethylaminopyridine (DMAP) as a catalyst to yield the half-ester intermediates. The intermediates that formed precipitates were isolated using vacuum filtration whereas those that did not were recrystallized from a mixture of chloroform/hexanes (3/1, v/v). The half-esters were then further reacted with a second desired alcohol using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling to yield the difunctional monomer. A one-pot synthesis was used for monomers designed to display two of the same functionalities. All monomers were purified using a CombiFlash system using ethyl acetate/dichloromethane (CH2Cl2) (1/9, v/v) and subsequently analyzed by 1H NMR spectroscopy, 13C NMR spectroscopy, and mass spectrometry (MS) to assess their chemical compositions and purity. Detailed synthetic procedures and all characterization data are provided in the supporting information.
PTDM Synthesis and Characterization
All block copolymer PTDMs were synthesized by ROMP using Grubbs’ third generation catalyst following previously described methods.32 In brief, the monomers and catalyst were dissolved separately in CH2Cl2 and degassed using freeze-pump-thaw methods. To initiate the polymerization, the hydrophobic monomer was first cannulated into the catalyst solution followed by the Boc-protected guanidinium monomer. The cationic monomer was polymerized in protected form in order to prevent premature termination of polymerizations due to catalyst coordination and to allow for sufficient solubility in organic solvents. Polymers were quenched with ethyl vinyl ether, precipitated, and subsequently deprotected using a 1:1 ratio of CH2Cl2:trifluoroacetic acid (TFA). Excess TFA was removed by azeotropic distillation with methanol and polymers were then dialyzed against RO water using dialysis membranes with a MWCO of 100–500 g/mol until the water conductivity was < 0.2 _S. All polymers were characterized by 1H NMR and gel permeation chromatography (GPC) to assess chemical compositions and molecular weight distributions, respectively. Detailed synthetic procedures and all characterization data are provided in the supporting information.
Jurkat T Cell Culture and FITC-siRNA Internalization
Jurkat T cells were cultured in RPMI 1640 supplemented with 10% (v/v) FBS, 100 U/mL non-essential amino acids, 100 U/mL sodium pyruvate, 100 U/mL penicillin, and 100 U/mL streptomycin. These cells were incubated at 37 °C with 5% CO2 and passaged 24 hours prior to experimentation. On the day of the experiment, PTDMs and siRNA were mixed at an N:P ratio of 8:1 (50 nM siRNA/well) and incubated for 30 minutes at room temperature prior to adding them drop-wise to the cells (4×105 cells/well; 1 mL total volume) in a 12 well plate. Cells were incubated at 37 °C with 5% CO2 in serum-containing media for four hours prior to analysis by flow cytometry. Cell viability was assessed using 7-AAD staining.
HeLa Cell Culture and FITC-siRNA Internalization
HeLa cells were cultured in DMEM supplemented with 10% (v/v) FBS, 100 U/mL non-essential amino acids, 100U/mL sodium pyruvate, 100 U/mL penicillin, and 100 U/mL streptomycin. For these experiments, cells (5×104 cells/well; 1 mL total volume) were cultured in 12-well plates using serum-containing media for 48 hours. This led to cell populations that were 70–90% confluent on the day of the experiment. On the day of the experiment, PTDMs were and siRNA were mixed at an N:P ratio of 4:1 (50 nM siRNA/well) and incubated for 30 minutes at room temperature. The cell media was replaced with fresh, complete media prior to adding the PTDM/siRNA complexes carefully to the top of the sample wells. Cells were incubated at 37 °C with 5% CO2 for four hours in serum-containing media prior to analysis by flow cytometry. Cell viability was assessed using 7-AAD staining.
HPLC Assessment of Hydrophobicity
HPLC experiments were performed using an HP 5890 HPLC system equipped with a photodiode array detector using an Agilent Zorbax SB-C8, 80 Å, 4.6 × 150 mm ID (5 μm) column. Samples were eluted using a linear gradient of 100% water with 0.1% TFA to 100% acetonitrile with 0.1% TFA over 60 minutes at a flow rate of 1 mL/min, and were detected using a wavelength of 215 nm. HPLC retention times were compared as a means to assess relative hydrophobicity, with more hydrophobic monomers eluting at higher RTs. These methods were previously reported in the literature.56–58
RESULTS AND DISCUSSION
Initial PTDM Design and Characterization
In this report, we document the use of ring-opening metathesis polymerization (ROMP) for the synthesis of block copolymer (BCP) PTDMs with varying hydrophobic block lengths. ROMP is a facile polymerization method that is functional group tolerant and allows for good control over molecular weights and dispersities.59–66 In addition, we continue to exploit the versatile, dual-functional oxanorbornene-based monomer platform as a means to tailor the overall polymer properties;29,30,32,44,47–49,67,68 in this case with a focus on the hydrophobic block length and side chain structure. The monomers and corresponding polymers for this study are summarized in Figure 1. The guanidinium-rich monomer (dG), shown in its Boc-protected form and meant to mimic arginine residues, was used as the cationic component of the BCP PTDMs. This selection was made based on previous literature that demonstrates the superior performance of guanidinium moieties over their ammonium counterparts, which are reminiscent of lysine and ornithine residues.69
The three initial hydrophobic monomers were designed to contain either two methyl substituents (dimethyl, dMe), one methyl and one phenyl substituent (methyl phenyl, MePh), or two phenyl substituents (diphenyl, dPh) (Figure 1A) and were selected because HPLC retention times indicated that they spanned a range of hydrophobicities. The phenyl-based hydrophobic groups have also demonstrated useful activities in prior studies.30,32,44,47 MePh, which has traditionally been selected by our group as the hydrophobic component of PTDMs for siRNA delivery, was used to synthesize BCPs with symmetric (n = m=5, 10, 20, or 40) or asymmetric (n ≠ m, with n being fixed at five for all polymers in the series and m= 10, 20, or 40) block sizes. This allowed us to explore how the length of the hydrophobic block impacts internalization at a given cationic charge length.
In addition, asymmetric BCPs containing a fixed block length (n = 5) of all three hydrophobic monomers and a cationic charge block of five, ten, or twenty dG units were synthesized as a way to further probe the relationship between the hydrophobic side chain and the length of the charged block for siRNA internalization. Previous work in this area documented the effect that the cationic charge block length and the addition of a hydrophobic block had on siRNA internalization and delivery.32 In that study, BCP PTDMs significantly outperformed their homopolymer counterparts, recapitulating the importance of an added hydrophobic component.32 The current study builds further from these initial findings to explore how the quantity and type of hydrophobic repeat units used impacts siRNA internalization.
FITC-siRNA Internalization: Symmetric vs. Asymmetric
Initially, both the symmetric and asymmetric MePhn-b-dGm BCP PTDMs were studied. A total of seven PTDMs were designed; four contained equal hydrophobic and cationic block lengths (symmetric, n = m = 5, 10, 20, and 40) and three asymmetric (n ≠ m) ones contained a fixed hydrophobic block length (n = 5) but cationic charge blocks of 10, 20, and 40 repeat units. Fluorescein isothiocyanate (FITC) labeled siRNA (FITC-siRNA) was used to establish trends in siRNA internalization for both series in Jurkat T cellsand HeLa cells using complete media. In this report, only FITC-labeled siRNA was used since we previously demonstrated the internalization relationship for our PTDMs between FITC-siRNA in Jurkats and siRNA for NOTCH1 in human peripheral blood mononuclear cells (PBMCs). The N:P ratios for these experiments, which are the ratios of the number of positively charged guanidinium groups in the PTDMs to the number of negatively charged phosphate atoms in the FITC-siRNA, were set at 8:1 for Jurkat T cells and 4:1 for HeLa cells and were previously optimized by our group.32 The selection of these N:P ratios were further supported by gel retardation assays, which demonstrated that all PTDMs fully bound siRNA at N:P ratios of 4:1 or less (Figures S17-S27).
A summary of FITC-siRNA internalization for these four symmetric and three asymmetric PTDMs is shown in Figure 2, where Figures 2A and 2B present the percentage of the cell population with internalized FITC-siRNA and Figures 2C and 2D present the median fluorescence intensity (MFI) of the cell populations.
Figure 2.

FITC-siRNA internalization into Jurkat T cells and HeLa cells using symmetric and asymmetric ROMP-based protein mimics. Jurkat T cells (cell density = 4×105 cells/mL) treated with polymer/FITC-siRNA complexes with an N:P ratio = 8:1 in complete media for four hours at 37°C and compared cells only receiving FITC-siRNA. HeLa cells (cell density = 5×104 cells/mL 48 hours prior to experiment; 70–90% confluent on the day of the experiment) treated with polymer/FITC-siRNA complexes with an N/P ratio = 4/1 in complete media for four hours at 37°C and compared cells only receiving FITC-siRNA. All data was compared to an untreated control. A) Percent positive Jurkat T cells. B) Percent positive HeLa cells. C) MFI of the Jurkat T cell population. D) MFI of the HeLa cell population. Each data point represents the mean ± SEM of three independent experiments. * = p < 0.05 and ns = not significant, as calculated by the unpaired two-tailed student t-test. Statistics represents significance between symmetric and asymmetric block copolymer PTDMs with the same cationic charge block length.
All PTDM-treated cells showed greater than 85% viability using 7-AAD staining (Figure S32 and Figure S38). For both cell types, symmetric and asymmetric PTDMs containing the same cationic block length were able to deliver to the same percentages of the cell populations (Figures 2A and 2B) regardless of the different hydrophobic block lengths. When looking at the MFI data, PTDMs with five, ten, and forty cationic repeat units lead to similar internalization amounts regardless of the hydrophobic block length; however, for PTDMs with twenty cationic repeat units, the asymmetric PTDM MePh5-b-dG20, significantly outperformed its symmetric counterpart (Figures 2C and 2D). For both cell types, MePh5-b-dG20 had double the fluorescence intensity of MePh20-b-dG20. These results demonstrate that the relationship between hydrophobicity and cationic block length is not always trivial and that increasing the hydrophobic block length further does not guarantee superior performance.37 In addition, these results suggest that understanding the interplay between hydrophobic and cationic block lengths as illustrated by MePh5-b-dG20 is necessary for optimal internalization.
FITC-siRNA Internalization: Varying the Hydrophobic Block Side Chains
To further probe the relationship between hydrophobic side chain and cationic block length, PTDMs with a fixed hydrophobic block length of five repeat units of either low (dMe), moderate (MePh), or high hydrophobicity (dPh) were prepared and studied. The relative hydrophobicities of these monomers were determined using HPLC and can be found in Table 1. The given retention times were 14.2 min, 27.8 min, and 36.1 min, respectively, where larger HPLC retention times reflect increased hydrophobicity. Based on previous results and those shown in Figure 2, the hydrophobic block length was held constant (n = 5) while the cationic block length was varied (m = 5, 10, 20). Based on results from the previous section, BCPs with m = 40 were not studied due to insufficient internalization regardless of hydrophobic content. This generated a total of nine PTDMs. FITC-siRNA was again used to establish trends in siRNA internalization in Jurkat T cells and HeLa cells using the same protocols as previously described.
Table 1.
Summary of Monomers and Their Corresponding HPLC Retention Times and LogP Values.
|
|

|
|||
| Monomer Name | Monomer Abbreviation | HPLC Rta (min) | LogPb | |
|
| ||||
| Dimethyl | dMe | 14.2 | 0.05 | |
| Methyl ethyl | MeEt | 17.3 | 0.41 | |
| Diethyl | dEt | 21.1 | 0.77 | |
| Methyl Propyl | MePr | 21.8 | 0.93 | |
| Methyl Butyl | MeBu | 25.8 | 1.38 | |
| Methyl Phenyl | MePh | 27.8 | 1.78 | |
| Dipropyl | dPr | 28.6 | 1.81 | |
| Diisobutyl | diBu | 34.8 | 2.54 | |
| Dibutyl | dBu | 34.9 | 2.70 | |
| Diphenyl | dPh | 36.1 | 3.50 | |
| Dicyclohexyl | dCy | 44.2 | 4.28 | |
|
| ||||
HPLC data collected using a linear gradient from 100% water containing 0.1% TFA to 100% CH3CN containing 0.1% TFA at a flow rate of 1 mL/min. HPLC detection wavelength = 215 nm.
LogP is the octanol/water partition coefficient. All values were calculated using MarvinSketch (ChemAxon Ltd.)
A summary of FITC-siRNA internalization for the PTDMs with variable hydrophobic side chains is shown in Figure 3, where Figures 3A and 3B present the percentage of the cell population with internalized FITC-siRNA and Figures 3C and 3D present the MFIs of the cell populations. All PTDM-treated cells showed greater than 85% viability using 7-AAD staining (Figure S33 and Figure S39).
Figure 3.

FITC-siRNA internalization into Jurkat T cells and HeLa cells using ROMP-based protein mimics with different hydrophobic blocks. Jurkat T cells (cell density = 4×105 cells/mL) treated with polymer/FITC-siRNA complexes with an N:P ratio = 8:1 in complete media for four hours at 37°C and compared cells only receiving FITC-siRNA. HeLa cells (cell density = 5×104 cells/mL 48 hours prior to experiment; 70–90% confluent on the day of the experiment) treated with polymer/FITC-siRNA complexes with an N/P ratio = 4/1 in complete media for four hours at 37°C and compared cells only receiving FITC-siRNA. All data was compared to an untreated control. A) Percent positive Jurkat T cells. B) Percent positive HeLa cells. C) MFI of the Jurkat T cell population. D) MFI of the HeLa cell population. Each data point represents the mean ± SEM of three independent experiments. * = p < 0.05, ** = p < 0.01, *** = p < 0.001 as calculated by the unpaired two-tailed student t-test. Statistics represents significance between dMe-containing and MePh-containing PTDMs.
For both cell types, MePh- and dPh-containing PTDMs with the same cationic block length were able to deliver to the same percentages of the cell populations; however, in all cases, the dMe-containing PTDMs delivered to a significantly smaller percentage of the population, particularly at the lower cationic block lengths. This percentage increased from 20% (dMe5-b-dG5) to 80% (dMe5-b-dG20), supporting the idea that cationic charge blocks larger than five (up to 20) improve FITC-siRNA internalization. In addition, for Jurkat T cells, the MFIs for the MePh- and dPh-containing PTDMs were not statistically different from each other but were significantly higher when compared to the dMe-containing PTDMs. Specifically, MePh5-b-dG10 and dPh5-b-dG10 had four times the fluorescence intensity of dMe5-b-dG10, and at higher cationic block lengths, MePh5-b-dG20 and dPh5-b-dG20 had double the fluorescence intensity of dMe5-b-dG20. This observed trend was similar for HeLa cells when comparing the MePh5-b-dGm series to the dMe5-b-dGm series, with the MePh-containing PTDMs having roughly double the fluorescence intensity regardless of the cationic block length. Taken together, this data suggests that there may be a minimum hydrophobic threshold necessary for better siRNA internalization, but there also seems to be a limit to which increasing the hydrophobicity improves siRNA internalization (see red circles in Figure S42).
Expanding the Hydrophobic Monomer Set
To better understand the required hydrophobicity for optimal internalization and to gain more insight regarding the tunability of the hydrophobic domain, several additional PTDMs were designed. In order to target the appropriate PTDMs, a series of hydrophobic monomers was synthesized and analyzed using HPLC to assess their relative hydrophobicities (Table 1, Figure S1). Additionally, logP values were calculated using MarvinSketch (ChemAxon Ltd).70 Monomers with longer retention times required more organic component in the mobile phase in order to be eluted and are considered to be more hydrophobic. A plot of HPLC retention time (RT) as it relates to logP value can be found in the supporting information (Figure S41). The linear relationship between HPLC RT and logP value supports the use of HPLC RTs as a viable, experimental method for assessing relative monomer hydrophobicity.56–58 It also allows more confidence with calculated logP values for this monomer class going forward. The monomer names in Table 1 reflect the substituents used for the monomer R groups, R1 and R2, which can either be the same or different. The wide variety of hydrophobic monomers synthesized in Table 1 also highlights the versatility of the diester monomer platform. 68
The original hydrophobic monomers used for this study, dMe, MePh, and dPh, had HPLC RTs of 14.2 min, 27.8 min, and 36.1 minutes, respectively. From the eleven monomers in Table 1, three new candidates were selected based on their measured hydrophobicities: dEt falls in the hydrophobic region between dMe and MePh, diBu has similar hydrophobicity to MePh and dPh, and dCy is the most hydrophobic of the eleven monomers. These three new monomers and their corresponding polymers are summarized in Figure 4.
Figure 4.

Additional monomer and polymer structures used for this study. A) Monomer structures B) Polymer Structures. Blue represents cationic moieties and green represents hydrophobic moieties.
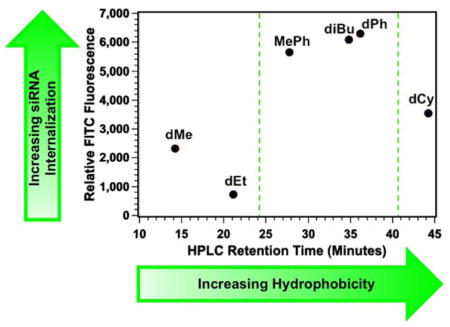
Due to the high internalization of MePh5-b-dG20 into Jurkat T cells, these three additional polymers were synthesized with the same cationic (m = 20) and hydrophobic (n = 5) block lengths. A summary of the FITC-siRNA internalization data in Jurkat T cells can be found in the supporting information (Figure S37) while the MFI values for these PTDMs, plotted as a function of the monomers’ HPLC RTs, is shown in Figure 5. This shows the initial three PTDMs (dMe5-b-dG20, MePh5-b-dG20, and dPh5-b-dG20, red circles) with the three new PTDMs (dEt5-b-dG20, diBu5-b-dG20, and dCy5-b-dG20, blue squares).
Figure 5.

Plot of relative FITC fluorescence in Jurkat T cells as it relates to monomer HPLC retention times. Green dashed lines indicate the hydrophobic window for optimal PTDM performance. Red data points represent hydrophobic monomers initially used. Blue data points represent hydrophobic monomers added after monomer hydrophobicity assessment by HPLC.
From the initial studies, there appeared to be a hydrophobic threshold for optimal internalization (dMeMFI < MePhMFI ~ dPhMFI) shown by the green dashed line near RT = 24 minutes. The new dEt-containing PTDM appears to support this hypothesis given that it delivers similar amounts of FITC-siRNA compared to the dMe-containing PTDM despite being more hydrophobic. Increasing the monomer hydrophobicity with diBu-containing PTDMs beyond the “threshold value” yields internalization values similar to MePh- and dPh-containing PTDMs; however, a further increase in the monomer hydrophobicity to dCy yields a PTDM with decreased FITC-siRNA internalization. This would suggest a window of optimal hydrophobicity from RTs of 27.8 minutes to 36.1 minutes when using these PTDMs for siRNA internalization. Furthermore, polymers that fall within this critical hydrophobic window are comprised of PTDMs with both aromatic and non-aromatic side chains, suggesting that overall hydrophobicity may be more important than monomer side chain structure.47,48
CONCLUSIONS
Understanding the structural components of carrier molecules necessary for siRNA internalization is critical for the development of better delivery reagents. In efforts to better determine appropriate design principles for our ROMP-based PTDMs, several new PTDMs were designed to understand how the length and relative hydrophobicity of the non-charged block as well as the length of the charged block had on FITC-siRNA internalization efficiencies in Jurkat T cells and HeLa cells. Initially, a set of symmetric (n = m) and asymmetric (n ≠ m, with n being fixed at five for all polymers in the series) BCP PTDMs were tested with varied cationic block lengths (m = 5, 10, 20, or 40). At fixed cationic block lengths, the percentage of the cell population receiving FITC-siRNA remained the same regardless of the hydrophobic block length; however, the asymmetric MePh5-b-dG20 had twice the fluorescence intensity of MePh20-b-dG20, demonstrating the complex relationship between hydrophobic and cationic block lengths. In a separate series of polymers, the hydrophobic block length was held constant at five repeat units, but the hydrophobic component was varied from dMe-, to MePh-, to dPh-based repeat units, which represented a range of hydrophobicities. In this series, the dMe-based PTDMs exhibited diminished internalization in comparison to their more hydrophobic counterparts (MePh- and dPh-based PTDMs). This suggested there was a minimum hydrophobicity required for improved internalization. HPLC retention times were used to assess the relative hydrophobicity of new monomers and to select several for the design and preparation of additional PTDMs, (dEt5-b-dG20, diBu5-b-dG20, and dCy5-b-dG20) to more fully explore this optimal hydrophobic window. An approximately 10 minute retention time window between 27 and 37 (or logP values of 1.78 and 3.50) was shown to yield optimal PTDM siRNA internalization. Below this threshold, PTDMs lack sufficient hydrophobicity to promote optimal internalization and the PTDM above this hydrophobicity also showed diminished internalization capabilities. Overall, optimization of PTDM hydrophobicity led to a better understanding of the structural components necessary for siRNA internalization, which will be used in the future to guide the development of superior delivery reagents.
Supplementary Material
Acknowledgments
This work was funded by the NIH (T32 GMO8515) and NSF (CHE-0910963 and DMR-1308123). The authors would like to thank Ms. Leah Caffrey, Ms. Angie Korpusik, and Ms. Salimar Cordero-Mercado for help with monomer synthesis. The authors would also like to thank Ms. Kelly McLeod for feedback on early drafts of this manuscript. Mass spectral data were obtained at the University of Massachusetts Mass Spectrometry Facility, which is supported in part by NSF. Flow cytometry data were obtained using the Flow Cytometry Core Facility at the University of Massachusetts Amherst, which is supported in part by NSF.
Footnotes
The authors declare no competing financial interest.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
ASSOCIATED CONTENT
All detailed synthetic procedures, molecular characterization, biological assays, and cellular viability data are provided in the supporting information. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Lorenzer C, Dirin M, Winkler AM, Baumann V, Winkler J. J Control Release. 2015;203:1–15. doi: 10.1016/j.jconrel.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Whitehead KA, Langer R, Anderson DG. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deshayes S, Morris MC, Divita G, Heitz F. Cell Mol Life Sci. 2005;62:1839–1849. doi: 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fonseca SB, Pereira MP, Kelley SO. Adv Drug Deliv Rev. 2009;61:953–964. doi: 10.1016/j.addr.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Heitz F, Morris MC, Divita G. Brit J Pharmacol. 2009;157:195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park TG, Jeong JH, Kim SW. Adv Drug Deliv Rev. 2006;58:467–486. doi: 10.1016/j.addr.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Snyder EL, Dowdy SF. Pharm Res. 2004;21:389–393. doi: 10.1023/B:PHAM.0000019289.61978.f5. [DOI] [PubMed] [Google Scholar]
- 8.Thomas M, Klibanov AM. Appl Microbiol Biotechnol. 2003;62:27–34. doi: 10.1007/s00253-003-1321-8. [DOI] [PubMed] [Google Scholar]
- 9.Frankel AD, Pabo CO. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 10.Green M, Loewenstein PM. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 11.Joliot A, Pernelle C, Deagostinibazin H, Prochiantz A. Proc Natl Acad Sci U S A. 1991;88:1864–1868. doi: 10.1073/pnas.88.5.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vives E, Brodin P, Lebleu B. Journal of Biological Chemistry. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Journal of Peptide Research. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 14.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindgren M, Langel U. Cell-Penetrating Peptides: Methods and Protocols. 2011;683:3–19. doi: 10.1007/978-1-60761-919-2_1. [DOI] [PubMed] [Google Scholar]
- 16.Siprashvili Z, Reuter JA, Khavari PA. Molecular Therapy. 2004;9:721–728. doi: 10.1016/j.ymthe.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Opalinska JB, Gewirtz AM. Nature Reviews Drug Discovery. 2002;1:503–514. doi: 10.1038/nrd837. [DOI] [PubMed] [Google Scholar]
- 18.Patel LN, Zaro JL, Shen WC. Pharmaceutical Research. 2007;24:1977–1992. doi: 10.1007/s11095-007-9303-7. [DOI] [PubMed] [Google Scholar]
- 19.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Journal of Biological Chemistry. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 20.Morris MC, Depollier J, Mery J, Heitz F, Divita G. Nature Biotechnology. 2001;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- 21.Morris MC, Deshayes S, Heitz F, Divita G. Biology of the Cell. 2008;100:201–217. doi: 10.1042/BC20070116. [DOI] [PubMed] [Google Scholar]
- 22.Morris MC, Vidal P, Chaloin L, Heitz F, Divita G. Nucleic Acids Research. 1997;25:2730–2736. doi: 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris MC, Depollier J, Mery J, Heitz F, Divita G. Nat Biotechnol. 2001;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- 24.Simeoni F, Morris MC, Heitz F, Divita G. Nucleic Acids Res. 2003;31:2717–2724. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boisguerin P, Deshayes S, Gait MJ, O'Donovan L, Godfrey C, Betts CA, Wood MJ, Lebleu B. Adv Drug Deliv Rev. 2015;87:52–67. doi: 10.1016/j.addr.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crombez L, Morris MC, Deshayes S, Heitz F, Divita G. Curr Pharm Design. 2008;14:3656–3665. doi: 10.2174/138161208786898842. [DOI] [PubMed] [Google Scholar]
- 27.Huang YW, Lee HJ, Tolliver LM, Aronstam RS. Biomed Res Int. 2015;2015:834079. doi: 10.1155/2015/834079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.deRonde BM, Tew GN. Biopolymers. 2015;104:265–280. doi: 10.1002/bip.22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sgolastra F, Minter LM, Osborne BA, Tew GN. Biomacromolecules. 2014;15:812–820. doi: 10.1021/bm401634r. [DOI] [PubMed] [Google Scholar]
- 30.Tezgel AO, Gonzalez-Perez G, Telfer JC, Osborne BA, Minter LM, Tew GN. Mol Ther. 2013;21:201–209. doi: 10.1038/mt.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vader P, van der Aa LJ, Storm G, Schiffelers RM, Engbersen JF. Curr Top Med Chem. 2012;12:108–119. doi: 10.2174/156802612798919123. [DOI] [PubMed] [Google Scholar]
- 32.deRonde BM, Torres JA, Minter LM, Tew GN. Biomacromolecules. 2015;16:3172–3179. doi: 10.1021/acs.biomac.5b00795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tabujew I, Freidel C, Krieg B, Helm M, Koynov K, Mullen K, Peneva K. Macromol Rapid Commun. 2014;35:1191–1197. doi: 10.1002/marc.201400120. [DOI] [PubMed] [Google Scholar]
- 34.Kim SH, Jeong JH, Kim TI, Kim SW, Bull DA. Mol Pharm. 2009;6:718–726. doi: 10.1021/mp800161e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim SH, Jeong JH, Ou M, Yockman JW, Kim SW, Bull DA. Biomaterials. 2008;29:4439–4446. doi: 10.1016/j.biomaterials.2008.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooley CB, Trantow BM, Nederberg F, Kiesewetter MK, Hedrick JL, Waymouth RM, Wender PA. J Am Chem Soc. 2009;131:16401–16403. doi: 10.1021/ja907363k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geihe EI, Cooley CB, Simon JR, Kiesewetter MK, Edward JA, Hickerson RP, Kaspar RL, Hedrick JL, Waymouth RM, Wender PA. Proc Natl Acad Sci U S A. 2012;109:13171–13176. doi: 10.1073/pnas.1211361109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wender PA, Huttner MA, Staveness D, Vargas JR, Xu AF. Mol Pharm. 2015;12:742–750. doi: 10.1021/mp500581r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brogden KA. Nat Rev Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 40.Gabriel GJ, Som A, Madkour AE, Eren T, Tew GN. Mater Sci Eng R Rep. 2007;57:28–64. doi: 10.1016/j.mser.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuroda K, Caputo GA. WIREs Nanomed Nanobiotechnol. 2013;5:49–66. doi: 10.1002/wnan.1199. [DOI] [PubMed] [Google Scholar]
- 42.Som A, Vemparala S, Ivanov I, Tew GN. Biopolymers. 2008;90:83–93. doi: 10.1002/bip.20970. [DOI] [PubMed] [Google Scholar]
- 43.Tew GN, Scott RW, Klein ML, Degrado WF. Acc Chem Res. 2010;43:30–39. doi: 10.1021/ar900036b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.deRonde BM, Birke A, Tew GN. Chemistry. 2015;21:3013–3019. doi: 10.1002/chem.201405381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gabriel GJ, Madkour AE, Dabkowski JM, Nelson CF, Nusslein K, Tew GN. Biomacromolecules. 2008;9:2980–2983. doi: 10.1021/bm800855t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hennig A, Gabriel GJ, Tew GN, Matile S. J Am Chem Soc. 2008;130:10338–10344. doi: 10.1021/ja802587j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Som A, Reuter A, Tew GN. Angew Chem Int Ed Engl. 2012;51:980–983. doi: 10.1002/anie.201104624. [DOI] [PubMed] [Google Scholar]
- 48.Som A, Tezgel AO, Gabriel GJ, Tew GN. Angew Chem Int Ed Engl. 2011;50:6147–6150. doi: 10.1002/anie.201101535. [DOI] [PubMed] [Google Scholar]
- 49.Tezgel AO, Telfer JC, Tew GN. Biomacromolecules. 2011;12:3078–3083. doi: 10.1021/bm200694u. [DOI] [PubMed] [Google Scholar]
- 50.Futaki S, Ohashi W, Suzuki T, Niwa M, Tanaka S, Ueda K, Harashima H, Sugiura Y. Bioconjug Chem. 2001;12:1005–1011. doi: 10.1021/bc015508l. [DOI] [PubMed] [Google Scholar]
- 51.Katayama S, Hirose H, Takayama K, Nakase I, Futaki S. J Control Release. 2011;149:29–35. doi: 10.1016/j.jconrel.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 52.Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. J Am Chem Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 53.Takeuchi T, Kosuge M, Tadokoro A, Sugiura Y, Nishi M, Kawata M, Sakai N, Matile S, Futaki S. ACS Chem Biol. 2006;1:299–303. doi: 10.1021/cb600127m. [DOI] [PubMed] [Google Scholar]
- 54.Nishihara M, Perret F, Takeuchi T, Futaki S, Lazar AN, Coleman AW, Sakai N, Matile S. Org Biomol Chem. 2005;3:1659–1669. doi: 10.1039/b501472g. [DOI] [PubMed] [Google Scholar]
- 55.Sakai N, Futaki S, Matile S. Soft Matter. 2006;2:636–641. doi: 10.1039/b606955j. [DOI] [PubMed] [Google Scholar]
- 56.Thaker HD, Cankaya A, Scott RW, Tew GN. ACS Med Chem Lett. 2013;4:481–485. doi: 10.1021/ml300307b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thaker HD, Som A, Ayaz F, Lui D, Pan W, Scott RW, Anguita J, Tew GN. J Am Chem Soc. 2012;134:11088–11091. doi: 10.1021/ja303304j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thaker HD, Sgolastra F, Clements D, Scott RW, Tew GN. J Med Chem. 2011;54:2241–2254. doi: 10.1021/jm101410t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bielawski CW, Benitez D, Grubbs RH. J Am Chem Soc. 2003;125:8424–8425. doi: 10.1021/ja034524l. [DOI] [PubMed] [Google Scholar]
- 60.Bielawski CW, Grubbs RH. Angew Chem Int Ed Engl. 2000;39:2903–2906. doi: 10.1002/1521-3773(20000818)39:16<2903::aid-anie2903>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 61.Bielawski CW, Grubbs RH. Prog Polym Sci. 2007;32:1–29. [Google Scholar]
- 62.Cannizzo LF, Grubbs RH. Macromolecules. 21:1961–1967. [Google Scholar]
- 63.Love JA, Morgan JP, Trnka TM, Grubbs RH. Angew Chem Int Ed Engl. 2002;41:4035–4037. doi: 10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 64.Schwab P, France MB, Ziller JW, Grubbs RH. Angew Chem Int Ed Engl. 1995;34:2039–2041. [Google Scholar]
- 65.Singh R, Czekelius C, Schrock RR. Macromolecules. 2006;39:1316–1317. [Google Scholar]
- 66.Trnka TM, Grubbs RH. Acc Chem Res. 2001;34:18–29. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]
- 67.Lienkamp K, Madkour AE, Kumar KN, Nusslein K, Tew GN. Chem Eur J. 2009;15:11715–11722. doi: 10.1002/chem.200900606. [DOI] [PubMed] [Google Scholar]
- 68.Lienkamp K, Madkour AE, Musante A, Nelson CF, Nusslein K, Tew GN. J Am Chem Soc. 2008;130:9836–9843. doi: 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 70.Magenau AJD, Richards JA, Pasquinelli MA, Savin DA, Mathers RT. Macromolecules. 2015;48:7230–7236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.